

Abstract
The opioids are commonly used to treat acute and severe pain. Long-term opioid administration eventually reaches a dose ceiling that is attributable to the rapid onset of analgesic tolerance coupled with the slow development of tolerance to the untoward side effects of respiratory depression, nausea and decreased gastrointestinal motility. The need for effective-long term analgesia remains. In order to develop new therapeutics and novel strategies for use of current analgesics, the processes that mediate tolerance must be understood. This review highlights potential pharmacokinetic (changes in metabolite production, metabolizing enzyme expression, and transporter function) and pharmacodynamic (receptor type, location and functionality; alterations in signaling pathways and cross-tolerance) aspects of opioid tolerance development, and presents several pharmacodynamic modeling strategies that have been used to characterize time-dependent attenuation of opioid analgesia.
Key words: opioid, pharmacodynamics, pharmacokinetics, tolerance
INTRODUCTION
Opioid analgesics are commonly used to treat acute and chronic pain associated with surgical interventions or disease states such as cancer. Opioids produce a diverse spectrum of centrally- and peripherally-mediated responses, including respiratory depression, nausea, sedation, euphoria or dysphoria, decreased gastrointestinal motility, and itching (1). Long-term use of opioids can be problematic due to the rapid development of profound tolerance to the analgesic effects coupled with slow development of tolerance to many of the untoward effects of these agents. It is the inability to tolerate these undesirable side effects that eventually limits dose escalations and analgesic efficacy.
The World Health Organization (WHO) has recommended adoption of a three-step “analgesic ladder” to meet the therapeutic challenges presented by opioid tolerance. In this approach, analgesic therapy is initiated with a non-opioid analgesic co-administered, if necessary, with an adjuvant. As the underlying condition progresses and pain becomes more intractable, a weak opioid is substituted for the non-opioid. Eventually, a strong opioid is introduced as a final step (2). While these recommendations are designed to improve quality of life for chronic pain sufferers, the underlying problem of opioid tolerance persists, and the clear and compelling need for effective long-term analgesics, or for effective strategies for applying existing analgesics to control chronic pain, remains. This goal may be achieved through the specific design and development of new agents that are not subject to tolerance development, or through the use of adjunct treatments that can minimize or reverse tolerance. In either case, a comprehensive understanding of the mechanisms underlying tolerance, as well as the kinetics of tolerance development and regeneration of responsivity, is required.
The purpose of this paper is to review the pharmacokinetic and pharmacodynamic mechanisms involved in the development of opioid tolerance. Pharmacokinetic considerations such as the production of metabolites, alterations in metabolic enzyme activity, and modulation of drug transporter expression or function are addressed. Pharmacodynamic processes such as receptor binding type and location, alterations in signal transduction, and cross-tolerance also are considered. Finally, approaches to comprehensive pharmacokinetic and pharmacodynamic (PK-PD) modeling of opioid antinociception and tolerance development are discussed.
ETIOLOGY OF TOLERANCE
Tolerance is defined as a decrease in pharmacologic response following repeated or prolonged drug administration. Tolerance can be separated into two main classifications: innate or acquired. Innate tolerance is a predisposition to exhibit drug sensitivity or insensitivity due to pharmacogenetic makeup. In most situations, innate tolerance is determined upon administration of the initial dose. In contrast, acquired tolerance is a consequence of repeated drug exposure, and can be subdivided into three general types based on the prevailing mechanism: pharmacokinetic, pharmacodynamic, or learned. Pharmacokinetic tolerance occurs when drug disposition or metabolism is altered as a function of time, often a consequence of the drug being an inducer or inhibitor of a specific metabolic enzyme or transporter system, resulting in a time-dependent decrease in presentation of the active moiety to the receptor biophase. Pharmacodynamic tolerance occurs when the intrinsic responsivity of the receptor system diminishes over time.
Acute tolerance is mediated predominantly by pharmacodynamic mechanisms, manifested as a decreased response following a single administration of the agent or during repeat-dosing but over a short time frame. This phenomenon is exemplified by nasally-administered cocaine. Initially, the relationship between cocaine-associated euphoria and blood cocaine concentrations is proportionate. However, at later points in time, the euphoric response decreases despite continued, or even increased, circulating concentration (3). In contrast to acute tolerance, chronic tolerance can be mediated through either pharmacokinetic or pharmacodynamic mechanisms, with an end result of a long-term decrease in drug response in the face of constant systemic exposure. In cases in which chronic tolerance develops, cross-tolerance within the pharmacologic class also may occur. Replacing the initial drug with a comparable agent will result in a lower pharmacologic response compared to that experienced by a drug-naive individual. Cross-tolerance is the principle underlying methadone substitution in the treatment of heroin addicts. Although methadone produces opioid effects in heroin-addicted subjects, euphoria and side effects are minimized by the tolerance produced by long-term heroin exposure. The significance of pharmacokinetic and pharmacodynamic mechanisms in relation to tolerance development will be examined in depth in subsequent sections of this review.
The final class of acquired tolerance is attributed to learning, either behavioral or conditioned. Behavioral tolerance occurs when an individual learns to function despite repeat exposure to a drug. Chronic alcohol abusers, for example, may not exhibit an outward appearance of motor impairment as a consequence of intoxication because of learned motor function adaptations and awareness of their impairment. Conditioned tolerance follows Pavlovian principles in which situational cues are associated with drug administration. Removal of these environmental cues will result in an enhancement in pharmacologic effect. For example, when morphine-tolerant rats are placed in a novel environment and challenged with morphine, antinociceptive tolerance is reduced (4). Analysis of behavioral tolerance has been reviewed in detail elsewhere (5).
MORPHINE AND RELATED AGONISTS
The medicinal value of opium has been acknowledged for centuries. Despite possessing an extensive side-effect profile, morphine, isolated from opium more than 200 years ago, remains the gold standard for treating pain. Structurally similar, semisynthetic morphine-like derivatives as well as structurally distinct opioids have been synthesized in a search for compounds that improve analgesia and minimize side effects. The common, clinically-used opioid analgesics are semisynthetic morphine derivatives, as well as the synthetic phenylpiperidine, anilidopiperidine, and diphenylpropylamine derivatives (Table I). Due to the structural similarities between these drugs and morphine, they primarily bind to the same receptor subtypes (μ-opioid receptors), produce analgesia, and exhibit similar side effect profiles. However, divergence in congener pharmacokinetic–pharmacodynamic parameter values allows the clinician to select a drug based on specific patient needs (1). For example, an ideal drug for a short surgical procedure would have rapid onset and offset of effect, such as the piperidine derivative alfentanil. Overall, opioid selection depends on a number of factors, including the nature of pain, predisposition to opioid responsivity, and previous opioid exposure.
Table I.
Categorization of the Common Opioids Based on Structural Similarities
| Common opioids |
|---|
| Semisynthetic derivatives |
| Morphine-related agonists |
| Hydromorphone |
| Hydrocodone |
| Oxycodone |
| Oxymorphone |
| Codeine |
| Morphine-related partial agonists and antagonists |
| Buprenorphine |
| Naloxone |
| Naltrexone |
| Synthetic derivatives |
| Phenylpiperidines |
| Meperidine |
| Loperamide |
| Diphenylpropylamines |
| Methadone |
| Propoxyphene |
| Piperidines |
| Fentanyl |
| Alfentanil |
| Sufentanil |
| Remifentanil |
PHARMACOKINETIC MEDIATORS OF OPIOID TOLERANCE
Receptor occupancy theory holds that pharmacologic response will be proportional to the fraction of the target receptor population that is occupied at a particular drug concentration (6). As the drug concentration in the vicinity of the receptor increases, the likelihood of drug binding to the receptor and producing an effect also increases. Gibaldi and Levy (7) applied this principle in describing the sigmoidal relationship between tubocurarine-mediated paralysis and dose or blood concentration. For this simplest of pharmacodynamic systems, drug concentrations in the receptor biophase must be in immediate equilibrium with concentrations in blood, and response must be instantaneous, reversible and time-independent. However, the relationship between the magnitude or time course of drug response and the kinetics of drug disposition generally is more complex, evidenced by time-dependent dissociations between pharmacologic response and systemic pharmacokinetics. A variety of factors, including slow equilibration of drug between the target receptor and blood or slowly-elaborated pharmacologic responses post-receptor binding, can cause such kinetic–dynamic dissociation. However, a particularly complicated underlying factor is the development of tolerance.
Metabolic and Distributional Mediators of Tolerance
One source of the extensive variability in drug concentrations, and consequently, response, within a population can be attributed to differences in drug absorption, distribution and metabolism. The opioids undergo significant phase I and II biotransformation, and polymorphisms in the cytochrome P450s (CYPs) and uridine-5′-diphosphate-glucuronosyltranserases (UGTs) will influence individual opioid disposition and response. For example, depending on the allelic combinations of the highly polymorphic CYP2D6, patients are characterized as one of four phenotypes: poor, intermediate, extensive and ultrarapid metabolizers (8). Subjects who exhibit the poor-metabolizer phenotype will not convert codeine to morphine efficiently, and therefore will exhibit a reduced effect compared to patients who can form morphine (9). In addition to the important role these enzymes play in mediating opioid concentrations and responses, it has been suggested that biotransformation processes contribute to morphine tolerance secondary to induced enzymatic expression (10,11). However, there is a lack of evidence to support this observation, as long-term administration of methadone or morphine does not alter the parent drug to metabolite concentration ratios, indicating that autoinduction is unlikely (12,13). Ultimately, metabolite formation could influence tolerance development through pharmacodynamic mechanisms, but there is a lack of evidence supporting time-dependent changes in the formation clearance of opioid metabolites.
In addition to metabolism, drug transport can play a significant role in mediating drug exposure at the target site, and therefore, the time course and magnitude of pharmacologic response. Many of the common opioids are substrates of the efflux transporter P-glycoprotein (P-gp), an ATP-dependant, 170-kDa transmembrane protein encoded by the MDR1 (human) and mdr1a/mdr1b (rodent) genes(s) (14). P-gp is expressed in a number of tissues in which it plays a protective role as a barrier transporter, limiting absorption from the intestines or penetration into organs such as testes or brain, as well as an excretory transport system in the kidney and liver (15). P-gp also functions to modulate the secretion of centrally synthesized opioid peptides and neurotransmitters from the brain to the systemic circulation (16).
Studies with P-gp-deficient mice have revealed that P-gp-mediated efflux attenuates the brain-to-serum ratio (Kp,brain) and antinociceptive effect of fentanyl, methadone and loperamide by ~2-, 7-, and 45-fold, respectively (17). Considering the extent to which P-gp mediates transport of some opioids, upregulation of this barrier transporter could further limit CNS penetration and antinociceptive effects of some opioids. Indeed, exposing transgenic mice expressing human pregnane X receptor (hPXR) to rifampin, an acute P-gp inhibitor and long-term P-gp inducer, upregulated expression of P-gp in brain capillary endothelium and attenuated the antinociceptive effect of the P-gp substrate methadone by 70% (18). Based on the results of this study, if an opioid is both a substrate and inducer of P-gp, chronic treatment could result in higher blood brain barrier (BBB) P-gp expression, with a consequent decrease in CNS penetration and antinociception (i.e., pharmacokinetic tolerance). Although autoinduction of BBB P-gp remains a possible mechanism underlying opioid tolerance, increases in P-gp expression following chronic treatment with morphine and oxycodone appear to be modest (2- and 1.3-fold, respectively) (19, 20). This minor increase in expression (≤2-fold) coupled with a negligible alteration in function (a 1.4-fold reduction in paclitaxel brain distribution) suggests that opioid-mediated P-gp upregulation, at least in experimental animals, does not influence opioid antinociceptive tolerance to an extent that can be distinguished from population PK-PD variability (19).
Metabolite Contributions to Opioid Response and Tolerance
Tolerance also may result from the production of metabolites that accumulate in the systemic circulation over time, penetrate the site of action, and interfere with pharmacologic response whether by competing with the parent compound for receptor binding (e.g., antagonists or partial agonists) or by down-regulating the responsivity of the receptor system or downstream events. The contribution of derived metabolites to morphine pharmacodynamics has received significant attention. In humans, morphine is metabolized in the liver by UGT2B7 to form morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G). Although morphine is metabolized preferentially to M3G (~5:1 M3G to M6G formation ratio), this metabolite does not contribute to antinociceptive response. In contrast, M6G is considered to be an unusual Phase II metabolite in that it is biologically active, and in fact appears to be somewhat more potent (2- to 4-fold) than morphine (21).
Some studies have suggested that, rather than contributing to antinociception, M3G attenuates the response to morphine. In this scenario, time-dependent M3G accumulation could be responsible for the appearance of morphine tolerance. For example, Smith and Smith (22) observed an inverse relationship between morphine-associated antinociception and M3G concentrations. However, careful examination of the influence of M3G pretreatment on morphine antinociception in rats revealed that acute M3G exposure, at concentrations similar to those produced by pharmacologically-relevant doses of morphine, does not attenuate antinociception to a significant extent (23,24). In contrast, M3G concentrations that are far in excess of those produced by pharmacologic doses of the parent drug can evoke neuroexcitatory behavioral responses that oppose the analgesic effects mediated by morphine binding to opioid receptors. Such neuroexcitation would account for the observed M3G-associated decrease in morphine-related antinociception (25). These excitatory responses are independent of the opioid receptor system, as evidenced by the absence of naloxone reversibility, and may be mediated through interactions at the N-methyl-d-aspartate (NMDA) and the γ-amino-butryic acid (GABAA) receptors (25,26). Overall, it appears that M3G may influence antinociception and tolerance, although through a mechanism other than μ-opioid receptor (MOR)-mediated interactions, and it is unlikely that accumulation of this metabolite, as it is formed from morphine, contributes substantially to the development of functional morphine tolerance.
PHARMACODYNAMIC MEDIATORS OF OPIOID TOLERANCE
While inherent inter-individual pharmacokinetic variability can influence analgesia within a population, time-dependent changes in systemic disposition appear to play a minimal role in the development of opioid tolerance. In contrast, a number of pharmacodynamic processes have been characterized as potential mediators of opioid tolerance. These adaptations include, but are not exclusively limited to, genetic predisposition, receptor subtypes, cross-tolerance, receptor affinity, alterations in secondary mediators such as the nitric oxide synthase pathway and transcription alterations, and receptor binding in the central versus the peripheral nervous system.
Mu-Opioid Receptor-Mediated Changes
The biologic effects that are characteristic of opioids are a consequence of agonist binding to opioid receptors with subsequent alterations in signal transduction and ion conductance that reduce neuronal pain transmission. The three major mammalian types of opioid receptors (OR) are designated µ, δ, and κ (27–29). Pharmacologic studies with opioid receptor ligands and receptor knockout mice identified the MOR as the primary mediator of both the therapeutic and side effects associated with opioids such as morphine (30–32). The role of the δ- and κ-opioid receptors in mediating analgesia and tolerance will be discussed more thoroughly in subsequent sections of this review.
The MOR is a seven-transmembrane protein belonging to the G-protein coupled receptor (GPCR) superfamily. As shown in Fig. 1, the binding of an agonist to the MOR results in the replacement of the α-subunit GDP with GTP and the subsequent dissociation of the α- and βγ-subunits from the Gαβγ heterotrimer (33,34). These mechanisms result in acute inhibition of neuronal transmission due to alterations in the conductance of ion channels, the stimulation of protein kinase A (PKA), and the transient inhibition of the adenylyl cyclase (AC) pathway (35). Chronic opioid use enhances the cAMP pathway via the superactivation of AC, alterations in ion channel conductance, an increase in neurotransmitter release, and altered gene expression through changes in cAMP-responsive element binding protein (CREB) phosphorylation and expression (36,37). Overall, antinociception is mediated through a complex sequence of downstream effectors, which can differ following acute versus chronic opioid exposure.
Fig. 1.

Downstream mediators of antinociception following opioid agonist binding to the MOR. a Binding of the agonist changes ion channel conductance and alters the cAMP pathway. b Activation of the NMDA receptor/NO pathway. Chronic opioid exposure alters synaptic cleft ion conductance and glutamate expression, leading to the displacement of the Mg2+ block of the NMDA receptor, an influx of Ca2+ and conversion of l-Arg to NO, a mediator of tolerance. Administering an NMDA receptor antagonist attenuates and delays the onset of tolerance by blocking Ca2+ influx and associated alterations
In addition to alterations in signaling, opioid tolerance is associated with decreases in opioid receptor sensitivity and expression in the plasma membrane. Following receptor binding of some, but not all (e.g., morphine), opioids, the MOR is phosphorylated by G-protein receptor kinase (GRKs), with opioid efficacy related to the extent of phosphorylation (38,39). Receptor phosphorylation is the first step in the desensitization and internalization of the MOR, and precedes association of β-arrestin to the receptor complex, which uncouples the receptor from G proteins. This uncoupling event desensitizes the receptor and attenuates the second messenger signal cascade, reducing agonist efficacy (40). The role of β-arrestin in receptor desensitization was characterized in β-arrestin KO mice that exhibited enhanced and prolonged analgesia with a lack of antinociceptive tolerance (41,42). β-arrestin also can modulate tolerance by recruiting adaptor proteins that link the receptor complex to clathrin, resulting in endocytosis in clathrin-coated pits. Following internalization, the receptor is retained and recycled to the plasma membrane as a resensitized receptor or, upon recognition of an interaction with the GPCR-associated sorting protein (GASP), the receptor will be sent to the lysosome for degradation (33,43). Agonist binding therefore results in diverse cellular adaptations that mediate antinociception and onset of tolerance.
The mechanisms underlying morphine tolerance appear to be different from those of other opioid agonists. Despite significant and prolonged efforts directed towards understanding morphine tolerance, the specific pathway has remained obscure. In contrast to most opioids, morphine-receptor binding does not initiate GRK-mediated MOR phosphorylation that results in receptor internalization (40). This observation is further supported by the absence of increased membrane internalization protein expression (e.g. β-arrestin and GRK) that accompanies most opioids (44). Even in the absence of receptor internalization, the interaction between the morphine-bound receptor and β-arrestin persists, evidenced by a reduction in tolerance in β-arrestin KO mice, suggesting that morphine induces tolerance by sustaining signaling pathways and cellular adaptations such as AC superactivation (41,45). An alternative explanation for the reduction in morphine tolerance observed in β-arrestin KO mice is that, although it is not an internalizing opioid, morphine binds to MORs that exist as heterodimers, with the second OR subtype interacting with β-arrestin and the subsequent internalization of the morphine-heterodimer complex contributes to tolerance (46,47). However, the validity of this hypothesis appears unlikely, as the results were not reproducible in later experiments and depended on the co-administration of morphine with an internalizing opioid for dimer endocytosis, and chronic morphine exposure does not change MOR expression (46,48,49).
Another contributor to morphine tolerance is the slow recovery of resensitized MORs because morphine desensitized receptors do not participate in the traditional pathway of internalization, recycling and reinsertion into the plasma membrane (50). Ultimately, the specific molecular mechanisms governing morphine tolerance have not been fully elucidated, and the possibility that mechanisms of morphine tolerance vary depending on the specific brain and spinal cord region requires further investigation (51).
MOR Receptor Polymorphisms
Analgesic variability within a population can, in part, be attributed to opioid pharmacogenetics. The human MOR has over 100 identified single nucleotide polymorphisms (SNPs), some of which are thought to influence opioid responsiveness, abuse potential, and tolerance (52,53). Of these SNPs, the most well-characterized is the A118G nucleotide substitution, which changes the amino-acid sequence at position 40 from asparginine to aspartic acid, eliminating an extracellular N-linked glycosylation site (54,55). Clinically, this variant reduces the analgesic effects of alfentanil and M6G (56,57). These alterations are not likely a consequence of altered binding affinity, as in vitro studies demonstrated that the N40D substitution did not affect binding of the exogenous opioids morphine, fentanyl, or methadone, nor that of the endogenous opioids Met- and Leu-enkephalin, endomorphin-1 and-2 and d-Ala2, N-methyl-Phe4, Glycol5]enkephalin (DAMGO) (54). Although this study demonstrated a 3-fold increase in binding affinity of the endogenous opioid peptide β-endorphine, subsequent studies did not detect any difference in β-endorphine binding, or receptor endocytosis, desensitization or resensitization in the N40D substitution (54,58,59). Additional work on human MOR polymorphisms characterized amino acid substitutions of N15D, R265H, and S268P. The latter substitution, located in the intracellular domain, exhibited a significant loss of receptor signaling (58). The human MOR intracellular loop polymorphisms reduced the extent of MOR-calcium calmodulin CaM interactions (R265H and S268P) and the R260H and R265H variants exhibited a reduction in basal MOR signaling (60).
Despite these detailed studies, the relationship between MOR polymorphisms and binding affinity, signaling pathways and ion channel conductance is not entirely clear. These alterations may, in part, mediate differential development of opioid tolerance within a population. For example, individuals expressing the N40D variant exhibit decreased analgesia, and may require larger opioid doses to achieve equipotent pain relief. It is conceivable that this dose differential could influence the development of tolerance in these subjects compared to normal MOR expressers. Research focused on MOR polymorphisms is continuing, and should shed light on the specific physiologic implications of these variants, including opioid responsivity and tolerance development.
Multiple Opioid Receptors
While less is known about the specific roles of the two remaining ORs, δ and κ, these proteins are expressed throughout the body and mediate antinociception, although to a lesser extent than the MOR (61–64). The κ-opioid receptor (KOR) exhibits tolerance following prolonged opioid exposure, a consequence of GRK-mediated phosphorylation of the KOR that initiates receptor internalization (65,66). While dephosphorylation is the typical mechanism of recovering receptor sensitivity, the KOR appears to require regeneration and reassembly of the receptor complex for restoration of antinociception (67). The net effect of these processes is a reduction in KOR expression that results in a decreased pharmacologic response following long-term opioid exposure. Similar to the KOR, the δ-opioid receptor (DOR) manifests tolerance to specific DOR-agonists via receptor phosphorylation and internalization (68,69). Despite receiving less attention than the MOR, opioid binding to the DOR and KOR also can result in antinociception and tolerance.
Cross-tolerance
Long-term exposure to one drug often results in the development of tolerance to the effects of other structurally similar drugs in the same pharmacologic class. This phenomenon is termed cross-tolerance. Although opioids frequently exhibit cross-tolerance, it is rarely complete, as evidenced by the utility of opioid rotation from morphine to hydromorphone or oxycodone, particularly in treatment of cancer pain (70,71). However, complete cross-tolerance can develop, as evidenced by the lack of analgesia elicited by high-dose morphine in patients receiving methadone maintenance therapy (72). The more common scenario of incomplete cross- tolerance has been attributed to multiple pharmacologic mechanisms. This preservation of effect may be a function of the inverse relationship between intrinsic opioid efficacy and tolerance, as well as that in situations of tolerance, replacing a low efficacy opioid such as morphine with fentanyl, a high-efficacy opioid with a high receptor reserve, will enhance analgesia (73,74). Furthermore, multiplicity in MOR subtypes may play an important role in determining the magnitude of cross-tolerance. For example, when one opioid is substituted for another, the secondary opioid may bind to a different receptor subtype than the initial compound, thereby limiting the apparent extent of cross-tolerance (75). Alternatively, incomplete cross-tolerance may be attributed to the ability of the opioid receptors to homo- and heterodimerize. For example, substituting an initial opioid with an agonist that binds to a different dimer combination can result in the recovery of analgesia because of incomplete tolerance to the new opioid-receptor combination. (47,76). Further investigation of the interplay between opioid receptors likely will reveal novel mechanisms that mediate antinociception and contribute to opioid tolerance. Clinically, opioid cross-tolerance presents a challenge when a patient switches to a new opioid regimen. Despite this possibility, the lack of complete tolerance generally results in a reduction in pain and fewer side effects compared to the initial, tolerant regimen.
NMDA Receptor Contributions to Opioid Tolerance
In addition to the direct actions of opioids, neurotransmitters such as dopamine, serotonin and glutamine have been implicated as mediators of opioid tolerance and antinociception (77–79). Of particular interest is the N-methyl-D-aspartate (NMDA)-sensitive glutamate receptor (NR). The intertwined relationship between the MOR and NMDA receptor systems is in part attributed to co-localization in central tissues (80,81). Mg2+ is bound to the receptor channel of the NMDA receptor under normal physiologic conditions, and serves to block the entrance of ions such as Ca2+ (Fig. 1). Small changes in synaptic cleft concentrations of glutamate (Glu) and inorganic ions can temporarily displace Mg2+ and allow an influx of Ca2+, which will bind to and activate the neuronal nitric oxide synthase (nNOS), CaM and PSD-95 complex, which converts l-arginine (l-arg) to l-citrulline (l-Cit) and nitric oxide (82,83). Finally, NO mediates the conversion of GTP to cGMP, a mediator of tolerance, via soluble guanylyl cyclase (sGC). Chronic opioid exposure enhances NMDA receptor function by increasing intracellular Ca2+, NO and cGMP levels, which contribute to the development of opioid tolerance. The use of NMDA receptor antagonists, or inhibitors of NOS production from l-arginine to enhance antinociception and delay the onset of tolerance has proven effective (82). Of these approaches, the use of NMDA receptor antagonists has shown preclinical and clinical potential in minimizing opioid tolerance. The potential role of NOS in determining the time course and magnitude of opioid tolerance is considered in detail in the final section of this review.
Initially viewed as a “miracle” drug class, the clinical use of NMDA receptor antagonists has yet to be fully realized. NMDA receptor antagonists are particularly promising drug targets due to a number of modulatory sites with different structural subunit combinations, most commonly the NR1 subunit combined with NR2A-D or NR3A-B subunits (84,85). These modulatory sites result in four general classes of NMDA receptor antagonists: the uncompetitive receptor channel blockers (MK-801, ketamine, PCP, memantine), the competitive glutamate antagonists (LY235959, D-CPPene), the glycine site antagonists [(+)(R)-HA-966], and the polyamine site antagonists (86). Preclinical and clinical models have been used to demonstrate that NMDA receptor antagonists can attenuate tolerance development and enhance antinociception, the latter effect allowing decreases in dose which may delay tolerance onset (87).
The majority of studies characterized the NMDA receptor antagonists as enhancing or having no effect on analgesia, whereas a minority demonstrated attenuation of acute morphine analgesia (88–93). One challenge in this area is the absence of consistent results identifying which opioids exhibit NMDA receptor antagonist-enhanced antinociception. For example, Redwine and Trujillo (94) found that morphine analgesia was enhanced only when co-administered with LY235959 (3 mg/kg); fentanyl analgesia was enhanced in the presence of LY235959 (3 mg/kg), dextromethorphan (30 mg/kg) and (+)(R)-HA-966 (30 mg/kg); and no difference was observed for morphine or fentanyl when co-administered with MK-801 (0.1 and 0.3 mg/kg), memantine (3 and 10 mg/kg), or ifendopril (1 and 3 mg/kg). Additional work clearly is needed to identify the specific interactions between each opioid and NMDA receptor antagonist. For example, NMDA receptor antagonist pharmacokinetics are poorly characterized. However, it is clear that they are capable of penetrating central tissues, evidenced by the appearance of centrally-mediated side effects such as hallucinations, memory impairment, blood pressure elevation, catatonia and anesthesia (95). NMDA receptor antagonists of greater selectivity and lower affinity were developed in response to these undesirable central effects, and promising alternatives may include the use of low affinity channel blockers (e.g., memantine), NR2B subunit selective drugs (ifendopril), or the partial glycine site antagonists (96). Based on these varied observations, further characterization of the underlying pharmacologic mechanisms and pharmacokinetic/pharmacodynamic relationships between the opioids and NMDA receptors remains to be elucidated.
The Role of Peripheral Opioid and NMDA Receptors In Mediating Antinociception
While NMDA receptor antagonists may represent a feasible approach to delay the onset and attenuate the extent of tolerance, these compounds manifest intolerable central side effects that limit use. One alternative strategy that may prove effective in treating pain is targeting non-centrally located NMDA receptors and/or ORs to achieve antinociception while minimizing centrally-mediated side effects and, potentially, the development of tolerance. The feasibility of this approach is, in part, supported by the broad peripheral distribution of these receptors. For example, MORs are present in a number of peripheral tissues, including the small and large intestines, kidney, lung, spleen, testis, ovaries and uterus (97). While these sites are important in mediating a number of physiologic processes, molecular and functional studies suggest that MORs and NMDA receptors localized on unmyelinated, cutaneous sensory axons contribute to antinociception (98). Moreover, the density of ORs and NMDA receptors on peripheral sensory axons increases in conditions of inflammation, suggesting a role in mediating pain perception (99,100). Overall, the physiologic distribution of the ORs and NMDA receptors suggests a role in mediating peripheral antinociception.
The functionality of peripheral ORs was demonstrated when a systemically inactive, subcutaneous dose of morphine into the rat tail exhibited naloxone-reversible antinociception (101,102). Additional studies identified the ability of locally-administered morphine and loperamide to mediate antinociception in a radiant heat tail-flick assay and in an inflammatory model, respectively (103). Furthermore, repeated immersion of the tail in opioid-containing DMSO solutions resulted in local antinociception and tolerance development, the latter being reversible following topical or systemic administration of the NMDA receptor antagonist MK-801 (104–106). These results suggest that peripherally-restricted opioids contribute to antinociception and tolerance, which may be minimized by co-administration with non-centrally penetrating NMDA receptor antagonists.
PHARMACOKINETIC–PHARMACODYNAMIC MODELS OF OPIOID TOLERANCE
“Black-Box” Approach
Although it is clear that complex cellular, mechanistic and homeostatic alterations contribute to tolerance development, PK-PD models, including those that incorporate tolerance as a time-dependent loss of intrinsic activity, often utilize a black-box approach that accounts for changes in the response-concentration relationship over time rather than incorporating specific biologic mechanisms that serve to drive the development of tolerance. Such empirical models tend to exhibit a broad applicability, are useful in characterizing and summarizing the temporal aspects of loss of drug response, and can be used to describe numerous tolerance scenarios (107). One such model developed by Porchet et al. (108) characterized acute tolerance to the cardio-accelerating effects of nicotine by integrating a hypothetical noncompetitive antagonist formed by a first-order process, with nicotine concentrations serving as the driving force for formation. Furthermore, optimal recovery from acute nicotine corresponded with longer intervals between nicotine exposure (108).
The generic approach to modeling tolerance development, which included consideration of the relationship between the development of opioid tolerance and administration strategy, was utilized by Ouellet and Pollack (109) for two morphine treatment regimens: multiple increasing i.v. bolus doses over a 12-h period (24 mg/kg total exposure in seven doses) or one i.v. bolus dose per day (beginning with 1.85 mg/kg on day 1, and increasing to 6 mg/kg maximum) for 13 days. The hypothesis underlying this experimental design was that tolerance development would be attenuated in the once-daily administration group if the kinetics of tolerance offset were rapid (i.e., if significant return to basal responsivity occurred within 24 h after morphine exposure). Morphine disposition did not differ significantly between the 12-h and 13-day treatment groups, indicating that changes in systemic morphine disposition could not account for tolerance development. Concentration-normalized peak analgesic effects remained relatively constant over 12 h, while the 13-day treatment group exhibited a decrease in normalized peak effect from day 1 to 8. In addition, the extent of analgesic tolerance development was similar whether morphine was administered as a multiple i.v. bolus or constant infusion. Taken together, these observations suggested that morphine tolerance development was dependent on systemic drug concentrations (i.e., the extent of exposure), but was independent of the kinetics of morphine administration (i.e., the rate of morphine exposure or the presence of relatively brief “drug holidays” in the administration regimen).
The apparent independence of the rate or extent of tolerance development and the kinetics of drug administration was an unexpected observation, as conventional wisdom suggested that continuous presentation of opioids such as morphine would result in the production of profound tolerance. An explanation for this observation was formulated through comprehensive PK-PD modeling of morphine concentration-time and antinociceptive effect-time data obtained from rats during and following continuous morphine infusion (109). In this experiment, morphine was infused for 12 h at several different rates to achieve one of five preselected steady-state concentrations. Blood morphine concentrations and antinociceptive response (tail flick) were determined at timed intervals during infusion. Separate groups of rats were studied for 18 days after termination of a 12-h morphine infusion, with pharmacologic response compared to that produced 15 min after a single intravenous bolus dose (presumably representing a “no-tolerance” state). The data were fit with a tolerance model similar to that displayed schematically in Fig. 2. This model assumed accumulation of a hypothetical “inhibitor” that was formed from morphine by a first-order process; several different “inhibitor” scenarios (reverse agonist, competitive antagonist, non-competitive antagonist, or partial agonist) were evaluated, with the net effect (E) produced under each scenario shown in Eqs. 1, 2, 3, and 4, respectively:
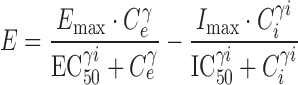 |
1 |
 |
2 |
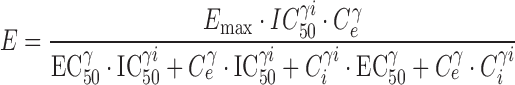 |
3 |
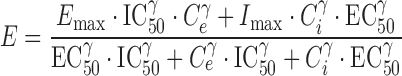 |
4 |
Fig. 2.

Scheme depicting the PK-PD model of tolerance following multiple morphine i.v. bolus doses. The time course of morphine concentrations following multiple i.v. bolus doses was described by a two-compartment model with a central volume of distribution V c, distribution between the central and peripheral compartment according to the rate constants k 12 and k 21, and elimination from the central compartment by the first-order rate constant k 10. The time course of antinociception was described using an approach derived by Porchet et al. (108) where the first-order rate constants of effect onset, k 1e and k 1t, link the central compartment to the effect (E) and tolerance (T) compartments of volumes V e and V t and, effect offset is governed by the first-order rate constants k e0 and k t0 [Adapted from Ouellet and Pollack (118)]
The results of this study indicated that morphine tolerance development was dependent on both morphine concentration and the duration of morphine exposure. As morphine concentrations increased, the net change in antinociceptive response (peak effect minus effect at 12 h) increased; regardless of the steady-state concentration produced, antinociceptive responses at 12 h were similar, resulting in a truncated effect versus concentration relationship at 12 h (Fig. 3). The PK-PD model was capable of describing the time course of loss (Fig. 4) and return (Fig. 5) of pharmacologic effect. When the loss-of-response data were fit alone, it was not possible to distinguish between the four alternative “inhibitor” scenarios. However, the return-of-response data suggested that the kinetics of morphine tolerance behaved as if driven by a partial agonist with a very low intrinsic activity (<10% of the maximum response to morphine) and a high affinity (IC50 ~ 14 ng/ml). In addition, the combined data set allowed estimation of the rate constant associated with tolerance offset (~0.12 day−1), which corresponded to a half-life of 5.7 days.
Fig. 3.

Relationship between antinociceptive response and morphine concentrations in blood during continuous infusion at the time of peak response (circles) or at 12 h into the infusion (triangles). Data are presented as mean for clarity; lines indicate the fit of the Hill equation to the data with the following parameter values: peak effect versus concentration data (E max = 100%, EC50 = 324 ng/ml, γ = 1.92); effect at 12 h versus concentration (E max = 34.8%, EC50 = 262 ng/ml, γ = 1.92) [data were obtained from Ouellet and Pollack (118)]
Fig. 4.

Time course of antinociceptive response during morphine infusion in rats. Infusion rates were selected to produce steady-state concentrations of 200–299 ng/ml (squares), 300–399 ng/ml (inverted triangles), 400–499 ng/ml (diamonds), 500–599 ng/ml (circles), or >600 ng/ml (triangles). Closed circles indicate animals that did not receive morphine. Data are represented as mean for clarity; lines indicate the fit of an integrated PK - PD model (Fig. 2) with the assumption that tolerance is driven by accumulation of a hypothetical partial agonist (Eq. 4) [Adapted from Ouellet and Pollack (118)]
Fig. 5.

Recovery of antinociceptive response to a 2-mg/kg bolus dose of morphine following termination of a 12-h morphine infusion (2 mg kg−1 h−1). Data are presented as mean ± SE; lines indicate model simulations based on an assumption that tolerance is driven by accumulation of a hypothetical reverse agonist (dashed line),competitive antagonist (dotted line), non-competitive antiagonist (dot-dashed line), or partial agonist (solid line) [Adapted from Ouellet and Pollack (118)]
This modeling effort revealed the explanation, if not the underlying mechanism, for the apparent independence between the kinetics of tolerance development and the kinetics of morphine administration, a situation starkly different from that for nicotine. In fully-tolerant animals, a “drug holiday” of nearly 6 days would be required in order to regenerate 50% of the intrinsic responsivity to morphine lost during the development of tolerance. In contrast, the half-life associated with return of responsivity to nicotine (35 min) is consistent with nearly complete recovery and resensitization when doses are separated by three or more hours (108,109).
As these examples demonstrate, “black-box” modeling of tolerance can provide useful information regarding system dynamics, are capable of describing the loss or return of pharmacologic effect over time, and can predict responsivity under differing administration scenarios. By their very nature these models have a limited ability to reflect the specific mechanisms underlying tolerance development in a given system. However, when used appropriately, such modeling efforts can direct further mechanistic experimentation. Subsequent elaboration of mechanistic detail then can be used to inform the PK-PD model of tolerance, which in turn provides additional understanding of system dynamics and an increased capacity to predict system response under differing conditions. An example of this iterative approach to modeling opioid tolerance is provided in the next section of this review.
Mechanistic Approach
When the specific physiologic alterations that lead to time-dependent loss of pharmacologic effect are known, they can be incorporated into mechanistic PK-PD models. As discussed earlier in this review, chronic opioid exposure enhances NMDA receptor function thereby permitting greater Ca2+ influx; Ca2+ will attach to and activate the NOS complex that mediates the conversion of L-Arg to NO. One method of limiting NO production is to coadminister an opioid with an NMDA receptor antagonist or a NOS inhibitor (85,87,110). Furthermore, coadministering exogenous L-Arg with an opioid increased NO levels and attenuated antinociception (84,111,112). These observations suggested an important, and perhaps causative, role of NO in the induction of opioid tolerance.
The significance of NO in mediating the time dependent loss of morphine-associated antinociception was elaborated in a series of experiments by Heinzen and coworkers. Following long-term morphine administration, the extent of antinociceptive tolerance that developed in mice deficient in neuronal nitric oxide synthase (nNOS) was significantly less than that in wild-type mice (30% versus 80% loss of effect, respectively) (113). In rats, infusion of the NO precursor l-arg increased the concentration of NO in brain tissue and decreased the intrinsic antinociceptive activity of morphine (114–116). An integrated PK-PD model was developed (Fig. 6) that was capable of describing the time course of changes in brain NO in response to l-arg infusion (Fig. 7). This model required inclusion of circadian control of NO production to accurately describe the observed data. Furthermore, morphine administration increased the brain concentration of NO, and the temporal pattern of apparent nNOS stimulation by morphine was consistent with the time course of loss of morphine-associated antinociception due to tolerance development (116). Subsequent experiments at the receptor level confirmed that NO alters MOR function (117). Taken together, these observations demonstrate that elevated brain content of NO is both necessary and sufficient to induce functional antinociceptive tolerance to morphine in rats and mice.
Fig. 6.

PK-PD model for l-arginine-associated stimulation of nitric oxide production in rats [From Heinzen and Pollack (115)]
Fig. 7.

Concentration-time profiles for brain NO during (a) saline or administration of l-arginine (b 250 mg kg−1 h−1, c 500 mg kg−1 h−1, or d 1,000 mg kg−1 h−1). Data are mean ± SE. Lines indicate the fit of the PK-PD model (Fig. 6) to the data [From Heinzen and Pollack (115)]
The relationship between morphine concentrations in blood and brain tissue (measured by microdialysis sampling), NO production in brain (measured with an indwelling amperometric probe), and the development of antinociceptive tolerance (measured by electrical stimulation vocalization) was characterized with a comprehensive PK-PD model (116). In this model (Fig. 8), a two-compartment PK model was fit to morphine blood and brain concentrations during and after an 8 h i.v. morphine infusion in rats. A multi-component PD submodel was required to incorporate morphine-associated changes in the production of neuronal NO, morphine associated agonism of the mu-opioid receptor (expressed as an increase in the threshold for electrical stimulation vocalization), and NO-associated decreases in the threshold for electrical stimulation vocalization. This relatively complex PK-PD model was similar in structure to the preceding “black-box” approaches, but utilized an experimentally-determined variable (brain concentration of NO), rather than a hypothetical construct, as the driving force for tolerance development. The model was able to provide a good description of the time course of morphine-associated antinociception during and following infusion in relation to changes in the blood and brain tissue concentrations of morphine (Fig. 9). Despite the complexity of the model, the data obtained were capable of supporting parameter estimates with a reasonable degree of precision (Table II).
Fig. 8.

Scheme depicting the PK-PD model of morphine disposition, NO production and antinociceptive effect. The disposition of morphine administered as a zero-order infusion (k 0) into the blood of volume V BL was described by the first-order rate constants of transfer between the blood and brain, k 12 and k 21, and by the first-order rate constant of elimination k 10 from the blood. Antinociception was mediated by morphine in the brain of volume V BR by acting as an agonist at the MOR and by indirectly stimulating the production of NO, which indirectly inhibited antinociception. The actions of morphine on NO production and antinociception was described using sigmoidal E max models, where E max, E or S is maximum effect or stimulation; EC50, E or S is the concentration that elicits 50% effect or stimulation; γE or S is the shape factor dictating the relationship between concentration and effect or stimulation of NO production (k ON); k OFF is the first-order rate constant of NO degradation; NOBR* is the concentration of NO in the hypothetical compartment that indirectly inhibits effect; I max,E is the maximum inhibitory effect of NO; IC50,E is the concentration of NO that inhibits 50% of the effect; and γI is the shape factor of the inhibitory effect of NO on antinociception [Adapted from Heinzen and Pollack (116)]
Fig. 9.

Blood morphine concentrations (top), brain morphine concentrations (middle), and antinociceptive effect (bottom) during and following an 8-h morphine infusion at 0.3- (circles), 1- (triangles), 2- (squares), or 3- (diamonds) mg kg−1 h−1. Lines indicate the fit of the PK/PD model to data [Adapted from Heinzen and Pollack (116)]
Table II.
Pharmacokinetic/Pharmacodynamic Parameter Estimates and Corresponding Coefficients of Variation Obtained from Simultaneous Modeling of Morphine Disposition, NO Production, and Antinociceptive Effects
| Parameter | Estimate | CV % |
|---|---|---|
| V BL (l/kg) | 2.5 | 25.9 |
| V BR (l/kg) | 26 | 22.6 |
| k 10 (h−1) | 2.17 | 26.3 |
| k 12 (h−1) | 2.15 | 45.2 |
| k 21 (h−1) | 1.77 | 10.8 |
| E max,S (% stim) | 0.19 | 36.8 |
| EC50,S (ng/ml) | 15.2 | 36.7 |
| γ S | 3.1 | 115 |
| k ON | 258 | 126 |
| k OFF | 25.8 | 127 |
| E max,E (% MPE) | 100 | 21.9 |
| EC50,E (ng/ml) | 26.8 | 29.8 |
| γE | 1.04 | 14.8 |
| I max,E (% MPE) | 2726 | 133 |
| IC50,E (% SAL) | 264 | 157 |
| γ I | 5.2 | 52.4 |
| k eo,low dose morphine (h−1) | 1.56 | 19.3 |
| k eo,intermediate dose morphine (h−1) | 1.01 | 15.8 |
| k eo,high dose morphine (h−1) | 0.61 | 21.4 |
Two aspects of the comparison between the “black box” and “mechanistic” modeling approaches for morphine tolerance are noteworthy. First, although both models are capable of describing the time course of changes in pharmacologic response, there are fundamental differences in the interpretation of those models. With the “black box” approach, the hypothetical “inhibitor” of response appeared to be a partial agonist of low intrinsic activity and high affinity; in the “mechanistic” approach, NO (presumably the analog of the hypothetical “inhibitor”) clearly functioned as a reverse agonist. This difference underscores the difficulty in extrapolating specific mechanisms from compartmental modeling when mechanistic information is absent. Secondly, although the “black box” approach did not provide mechanistic detail, it did inform further experimentation that was target towards obtaining the missing mechanistic information. Thus, such modeling efforts can be extraordinarily useful in the design and interpretation of studies aimed as elucidating fundamental mechanisms of kinetic or dynamic behavior. As additional steps in the NO pathway for opioid tolerance development are elaborated, or as alternative pathways for opioid tolerance are elucidated, for the development of opioid tolerance, more detailed mechanistic models can be developed, assuming that the relevant steps in those pathways can be studied in vivo.
CONCLUSIONS
Opioid tolerance is a complex phenomenon mediated by diverse behavioral and cellular adaptations. Many of the specific alterations in intracellular signaling, trafficking and neurotransmitter activity have been thoroughly characterized using in vitro approaches and knockout mice. Despite these advances, there is in general a lack of integrative system approaches characterizing how these cellular adaptations interact within the whole organism. Such an approach will be critical in developing comprehensive understanding of opioid tolerance. As mechanistic information continues to be generated, increasingly sophisticated PK-PD models, with increasing biologic relevance, can be developed. The availability of such models, in turn, will support diverse activities ranging from drug discovery to adjunct therapy in order to minimize or reverse opioid tolerance. The ability to control the tolerance process is a requisite step in devising appropriate pharmacologic strategies for the treatment of chronic pain.
Acknowledgements
This work was supported by National Institutes of Health Grant R01 GM61191. E.R.O. was supported by a predoctoral fellowship from GlaxoSmithKline.
Abbreviations
- AC
adenylyl cyclase
- BBB
blood brain barrier
- CREB
cAMP-responsive element binding protein
- CYP
cytochrome P450
- DOR
δ-opioid receptor
- GABA
γ-amino-butyric acid
- Glu
glutamate
- GPCR
G-protein coupled receptor
- GRK
G-protein receptor kinase
- hPXR
human pregnane X receptor
- KOR
κ-opioid receptor
- Kp,brain
brain-to-serum ratio
- l-Arg
l-arginine
- l-Cit
l-citrulline
- M3G
morphine-3-glucuronide
- M6G
morphine-6-glucuronide
- NMDA
N-methyl-d-aspartate
- nNOS
neuronal nitric oxide synthase
- NR
NMDA receptor
- OR
opioid receptors
- PD
pharmacodynamic
- P-gp
P-glycoprotein
- PK
pharmacokinetic
- PKA
protein kinase A
- PSD-95
post-synaptic density complex
- sGC
soluble guanylyl cyclase
- SNP
single nucleotide polymorphism
- UGT
uridine-5′-diphosphate-glucuronosyltranserase
References
- 1.Inturrisi C. E. Clinical pharmacology of opioids for pain. Clin. J. Pain. 2002;18(4 Suppl):S3–S13. doi: 10.1097/00002508-200207001-00002. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Cancer Pain Relief, Second Edition, World Health Organization: Geneva, Switzerland, 1996, pp. 1–69.
- 3.R. T. Jones. Euphoria vs. cocaine plasma concentrations after nasal administration (20% solution). In C. N. Chiang, R. L. Hawks (eds.), NIDA Research Monograph 99 (Research Findings on Smoking of Abused Substances), 1990, pp. 30–41.
- 4.Siegel S. Morphine analgesic tolerance: its situation specificity supports a Pavlovian conditioning model. Science. 1976;193(4250):323–325. doi: 10.1126/science.935870. [DOI] [PubMed] [Google Scholar]
- 5.van Ree J. M., Gerrits M. A., Vanderschuren L. J. Opioids, reward and addiction: An encounter of biology, psychology, and medicine. Pharmacol. Rev. 1999;51(2):341–396. [PubMed] [Google Scholar]
- 6.E. M. Ross, and T. P. Kenakin. Pharmacodynamics: mechanisms of drug action and the relationship between drug concentration and effect. In J. G. Hardman, and L. E. Limbird (eds.), Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 10th edn., McGraw Hill, 2001, pp. 31–43.
- 7.Gibaldi M., Levy G. Dose-dependent decline of pharmacologic effects of drugs with linear pharmacokinetic characteristics. J. Pharm. Sci. 1972;61(4):567–569. doi: 10.1002/jps.2600610414. [DOI] [PubMed] [Google Scholar]
- 8.Bernard S., Neville K. A., Nguyen A. T., Flockhart D. A. Interethnic differences in genetic polymorphisms of CYP2D6 in the U.S. population: clinical implications. Oncologist. 2006;11(2):126–135. doi: 10.1634/theoncologist.11-2-126. [DOI] [PubMed] [Google Scholar]
- 9.Desmeules J., Gascon M. P., Dayer P., Magistris M. Impact of environmental and genetic factors on codeine analgesia. Eur. J. Clin. Pharmacol. 1991;41(1):23–26. doi: 10.1007/BF00280101. [DOI] [PubMed] [Google Scholar]
- 10.Berkowitz B. A. The relationship of pharmacokinetics to pharmacological activity: morphine, methadone and naloxone. Clin. Pharmacokinet. 1976;1(3):219–230. doi: 10.2165/00003088-197601030-00004. [DOI] [PubMed] [Google Scholar]
- 11.Sawe J. High-dose morphine and methadone in cancer patients. Clinical pharmacokinetic considerations of oral treatment. Clin. Pharmacokinet. 1986;11(2):87–106. doi: 10.2165/00003088-198611020-00001. [DOI] [PubMed] [Google Scholar]
- 12.Fredheim O. M., Borchgrevink P. C., Klepstad P., Kaasa S., Dale O. Long term methadone for chronic pain: a pilot study of pharmacokinetic aspects. Eur. J. Pain. 2007;11(6):599–604. doi: 10.1016/j.ejpain.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Sawe J., Svensson J. O., Rane A. Morphine metabolism in cancer patients on increasing oral doses—no evidence for autoinduction or dose-dependence. Br. J. Clin. Pharmacol. 1983;16(1):85–93. doi: 10.1111/j.1365-2125.1983.tb02148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dagenais C., Graff C. L., Pollack G. M. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochem. Pharmacol. 2004;67(2):269–276. doi: 10.1016/j.bcp.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 15.Matheny C. J., Lamb M. W., Brouwer; K. R., Pollack G. M. Pharmacokinetic and pharmacodynamic implications of P-glycoprotein modulation. Pharmacotherapy. 2001;21(7):778–796. doi: 10.1592/phco.21.9.778.34558. [DOI] [PubMed] [Google Scholar]
- 16.King M., Su W., Chang A., Zuckerman A., Pasternak G. W. Transport of opioids from the brain to the periphery by P-glycoprotein: peripheral actions of central drugs. Nat. Neurosci. 2001;4(3):268–274. doi: 10.1038/85115. [DOI] [PubMed] [Google Scholar]
- 17.J. C. Kalvass, E. O. Olson, and G. M. Pollack. Influence of blood–brain barrier P-glycoprotein on brain penetration and antinociceptive effects of model opioids. AAPS J. 7(S2) (2005).
- 18.Bauer B., Yang X., Hartz A. M., Olson E. R., Zhao R., Kalvass J. C., Pollack G. M., Miller D. S. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol. Pharmacol. 2006;70(4):1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- 19.Hassan H. E., Myers A. L., Lee I. J., Coop A., Eddington N. D. Oxycodone induces overexpression of P-glycoprotein (ABCB1) and affects paclitaxel's tissue distribution in Sprague Dawley rats. J. Pharm. Sci. 2007;96(9):2494–506. doi: 10.1002/jps.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aquilante C. L., Letrent S. P., Pollack G. M., Brouwer K. L. Increased brain P-glycoprotein in morphine tolerant rats. Life. Sci. 2000;66(4):PL47–PL51. doi: 10.1016/s0024-3205(99)00599-8. [DOI] [PubMed] [Google Scholar]
- 21.Wittwer E., Kern S. E. Role of morphine’s metabolites in analgesia: concepts and controversies. AAPS. J. 2006;8(2):E348–E352. doi: 10.1007/BF02854905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith G. D., Smith M. T. Morphine-3-glucuronide: evidence to support its putative role in the development of tolerance to the antinociceptive effects of morphine in the rat. Pain. 1995;62(1):51–60. doi: 10.1016/0304-3959(94)00228-7. [DOI] [PubMed] [Google Scholar]
- 23.Gardmark M., Karlsson M. O., Jonsson F., Hammarlund-Udenaes M. Morphine-3-glucuronide has a minor effect on morphine antinociception. Pharmacodynamic modeling. J. Pharm. Sci. 1998;87(7):813–820. doi: 10.1021/js980056f. [DOI] [PubMed] [Google Scholar]
- 24.Ouellet D. M., Pollack G. M. Effect of prior morphine-3-glucuronide exposure on morphine disposition and antinociception. Biochem. Pharmacol. 1997;53(10):1451–1457. doi: 10.1016/s0006-2952(97)00086-5. [DOI] [PubMed] [Google Scholar]
- 25.Smith M. T. Neuroexcitatory effects of morphine and hydromorphone: evidence implicating the 3-glucuronide metabolites. Clin. Exp. Pharmacol. Physiol. 2000;27(7):524–528. doi: 10.1046/j.1440-1681.2000.03290.x. [DOI] [PubMed] [Google Scholar]
- 26.Bartlett S. E., Cramond T., Smith M. T. The excitatory effects of morphine-3-glucuronide are attenuated by LY274614, a competitive NMDA receptor antagonist, and by midazolam, an agonist at the benzodiazepine site on the GABAA receptor complex. Life. Sci. 1994;54(10):687–694. doi: 10.1016/0024-3205(94)00552-4. [DOI] [PubMed] [Google Scholar]
- 27.Evans C. J., Keith D. E., Jr., Morrison H., Magendzo K., Edwards R. H. Cloning of a delta opioid receptor by functional expression. Science. 1992;258(5090):1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y., Mestek A., Liu J., Hurley J. A., Yu L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993;44(1):8–12. [PubMed] [Google Scholar]
- 29.Li S., Zhu J., Chen C., Chen Y. W., Deriel J. K., Ashby B., Liu-Chen L. Y. Molecular cloning and expression of a rat kappa opioid receptor. Biochem. J. 1993;295(Pt 3):629–633. doi: 10.1042/bj2950629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kieffer B. L. Opioids: first lessons from knockout mice. Trends. Pharmacol. Sci. 1999;20(1):19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- 31.Matthes H. W., Maldonado R., Simonin F., Valverde O., Slowe S., Kitchen I., Befort K., Dierich A., Le Meur M., Dolle P., Tzavara E., Hanoune J., Roques B. P., Kieffer B. L. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383(6603):819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 32.Sora I., Takahashi N., Funada M., Ujike H., Revay R. S., Donovan D. M., Miner L. L., Uhl G. R. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc. Natl. Acad. Sci. U. S. A. 1997;94(4):1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams J. T., Christie M. J., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol. Rev. 2001;81(1):299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- 34.Tso P. H., Wong Y. H. Molecular basis of opioid dependence: role of signal regulation by G-proteins. Clin. Exp. Pharmacol. Physiol. 2003;30(5–6):307–316. doi: 10.1046/j.1440-1681.2003.03835.x. [DOI] [PubMed] [Google Scholar]
- 35.Smart D., Hirst R. A., Hirota K., Grandy D. K., Lambert D. G. The effects of recombinant rat mu-opioid receptor activation in CHO cells on phospholipase C, [Ca2+]i and adenylyl cyclase. Br. J. Pharmacol. 1997;120(6):1165–1171. doi: 10.1038/sj.bjp.0701012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nestler E. J., Aghajanian G. K. Molecular and cellular basis of addiction. Science. 1997;278(5335):58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 37.Taylor D. A., Fleming W. W. Unifying perspectives of the mechanisms underlying the development of tolerance and physical dependence to opioids. J. Pharmacol. Exp. Ther. 2001;297(1):11–18. [PubMed] [Google Scholar]
- 38.Zhang L., Yu Y., Mackin S., Weight F. F., Uhl G. R., Wang J. B. Differential mu opiate receptor phosphorylation and desensitization induced by agonists and phorbol esters. J. Biol. Chem. 1996;271(19):11449–11454. doi: 10.1074/jbc.271.19.11449. [DOI] [PubMed] [Google Scholar]
- 39.Yu Y., Zhang L., Yin X., Sun H., Uhl G. R., Wang J. B. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J. Biol. Chem. 1997;272(46):28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J., Ferguson S. S., Barak L. S., Bodduluri S. R., Laporte S. A., Law P. Y., Caron M. G. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U. S. A. 1998;95(12):7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohn L. M., Gainetdinov R. R., Lin F. T., Lefkowitz R. J., Caron M. G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408(6813):720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 42.Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286(5449):2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 43.Thompson D., Pusch M., Whistler J. L. Changes in G protein-coupled receptor sorting protein affinity regulate postendocytic targeting of G protein-coupled receptors. J. Biol. Chem. 2007;282(40):29178–29185. doi: 10.1074/jbc.M704014200. [DOI] [PubMed] [Google Scholar]
- 44.Narita M., Suzuki M., Narita M., Niikura K., Nakamura A., Miyatake M., Yajima Y., Suzuki T. mu-Opioid receptor internalization-dependent and -independent mechanisms of the development of tolerance to mu-opioid receptor agonists: comparison between etorphine and morphine. Neuroscience. 2006;138(2):609–619. doi: 10.1016/j.neuroscience.2005.11.046. [DOI] [PubMed] [Google Scholar]
- 45.Dang V. C., Williams J. T. Morphine-induced mu-opioid receptor desensitization. Mol. Pharmacol. 2005;68(4):1127–1132. doi: 10.1124/mol.105.013185. [DOI] [PubMed] [Google Scholar]
- 46.He L., Fong J., von Zastrow M., Whistler J. L. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108(2):271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- 47.Jordan B. A., Devi L. A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399(6737):697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey C. P., Couch D., Johnson E., Griffiths K., Kelly E., Henderson G. Mu-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J. Neurosci. 2003;23(33):10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stafford K., Gomes A. B., Shen J., Yoburn B. C. mu-Opioid receptor downregulation contributes to opioid tolerance in vivo. Pharmacol. Biochem. Behav. 2001;69(1–2):233–237. doi: 10.1016/s0091-3057(01)00525-1. [DOI] [PubMed] [Google Scholar]
- 50.Finn A. K., Whistler J. L. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32(5):829–839. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 51.Sim-Selley L. J., Scoggins K. L., Cassidy M. P., Smith L. A., Dewey W. L., Smith F. L., Selley D. E. Region-dependent attenuation of mu opioid receptor-mediated G-protein activation in mouse CNS as a function of morphine tolerance. Br. J. Pharmacol. 2007;151(8):1324–1333. doi: 10.1038/sj.bjp.0707328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaForge K. S., Yuferov V., Kreek M. J. Opioid receptor and peptide gene polymorphisms: potential implications for addictions. Eur. J. Pharmacol. 2000;410(2–3):249–268. doi: 10.1016/s0014-2999(00)00819-0. [DOI] [PubMed] [Google Scholar]
- 53.Kreek M. J., Bart G., Lilly C., LaForge K. S., Nielsen D. A. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol. Rev. 2005;57(1):1–26. doi: 10.1124/pr.57.1.1. [DOI] [PubMed] [Google Scholar]
- 54.Bond C., LaForge K. S., Tian M., Melia D., Zhang S., Borg L., Gong J., Schluger J., Strong J. A., Leal S. M., Tischfield J. A., Kreek M. J., Yu L. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc. Natl. Acad. Sci. U. S. A. 1998;95(16):9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Surratt C. K., Johnson P. S., Moriwaki A., Seidleck B. K., Blaschak C. J., Wang J. B., Uhl G. R. −mu opiate receptor. Charged transmembrane domain amino acids are critical for agonist recognition and intrinsic activity. J. Biol. Chem. 1994;269(32):20548–20553. [PubMed] [Google Scholar]
- 56.Romberg R. R., Olofsen E., Bijl H., Taschner P. E., Teppema L. J., Sarton E. Y., van Kleef J. W., Dahan A. Polymorphism of mu-opioid receptor gene (OPRM1:c.118A>G) does not protect against opioid-induced respiratory depression despite reduced analgesic response. Anesthesiology. 2005;102(3):522–530. doi: 10.1097/00000542-200503000-00008. [DOI] [PubMed] [Google Scholar]
- 57.Oertel B. G., Schmidt R., Schneider A., Geisslinger G., Lotsch J. The mu-opioid receptor gene polymorphism 118A>G depletes alfentanil-induced analgesia and protects against respiratory depression in homozygous carriers. Pharmacogenet. Genomics. 2006;16(9):625–36. doi: 10.1097/01.fpc.0000220566.90466.a2. [DOI] [PubMed] [Google Scholar]
- 58.Befort K., Filliol D., Decaillot F. M., Gaveriaux-Ruff C., Hoehe M. R., Kieffer B. L. A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. J. Biol. Chem. 2001;276(5):3130–3137. doi: 10.1074/jbc.M006352200. [DOI] [PubMed] [Google Scholar]
- 59.Beyer A., Koch T., Schroder H., Schulz S., Hollt V. Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization, and resensitization of the human mu-opioid receptor. J. Neurochem. 2004;89(3):553–560. doi: 10.1111/j.1471-4159.2004.02340.x. [DOI] [PubMed] [Google Scholar]
- 60.Wang D., Quillan J. M., Winans K., Lucas J. L., Sadee W. Single nucleotide polymorphisms in the human mu opioid receptor gene alter basal G protein coupling and calmodulin binding. J. Biol. Chem. 2001;276(37):34624–34630. doi: 10.1074/jbc.M104083200. [DOI] [PubMed] [Google Scholar]
- 61.Millan M. J. Kappa-opioid receptor-mediated antinociception in the rat. I. Comparative actions of mu- and kappa-opioids against noxious thermal, pressure and electrical stimuli. J. Pharmacol. Exp. Ther. 1989;251(1):334–341. [PubMed] [Google Scholar]
- 62.Millan M. J., Czlonkowski A., Lipkowski A., Herz A. Kappa-opioid receptor-mediated antinociception in the rat. II. Supraspinal in addition to spinal sites of action. J. Pharmacol. Exp. Ther. 1989;251(1):342–350. [PubMed] [Google Scholar]
- 63.Heyman J. S., Mulvaney S. A., Mosberg H. I., Porreca F. Opioid delta-receptor involvement in supraspinal and spinal antinociception in mice. Brain. Res. 1987;420(1):100–108. doi: 10.1016/0006-8993(87)90244-7. [DOI] [PubMed] [Google Scholar]
- 64.Wittert G., Hope P., Pyle D. Tissue distribution of opioid receptor gene expression in the rat. Biochem. Biophys. Res. Commun. 1996;218(3):877–881. doi: 10.1006/bbrc.1996.0156. [DOI] [PubMed] [Google Scholar]
- 65.Leighton G. E., Rodriguez R. E., Hill R. G., Hughes J. Kappa-opioid agonists produce antinociception after i.v. and i.c.v. but not intrathecal administration in the rat. Br. J. Pharmacol. 1988;93(3):553–560. doi: 10.1111/j.1476-5381.1988.tb10310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narita M., Khotib J., Suzuki M., Ozaki S., Yajima Y., Suzuki T. Heterologous mu-opioid receptor adaptation by repeated stimulation of kappa-opioid receptor: up-regulation of G-protein activation and antinociception. J. Neurochem. 2003;85(5):1171–1179. doi: 10.1046/j.1471-4159.2003.01754.x. [DOI] [PubMed] [Google Scholar]
- 67.McLaughlin J. P., Myers L. C., Zarek P. E., Caron M. G., Lefkowitz R. J., Czyzyk T. A., Pintar J. E., Chavkin C. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J. Biol. Chem. 2004;279(3):1810–1818. doi: 10.1074/jbc.M305796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Okura T., Varga E. V., Hosohata Y., Navratilova E., Cowell S. M., Rice K., Nagase H., Hruby V. J., Roeske W. R., Yamamura H. I. Agonist-specific down-regulation of the human delta-opioid receptor. Eur. J. Pharmacol. 2003;459(1):9–16. doi: 10.1016/s0014-2999(02)02823-6. [DOI] [PubMed] [Google Scholar]
- 69.Lecoq I., Marie N., Jauzac P., Allouche S. Different regulation of human delta-opioid receptors by SNC-80 [(+)-4-[(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-meth oxybenzyl]-N,N-diethylbenzamide] and endogenous enkephalins. J. Pharmacol. Exp. Ther. 2004;310(2):666–677. doi: 10.1124/jpet.103.063958. [DOI] [PubMed] [Google Scholar]
- 70.de Stoutz N. D., Bruera E., Suarez-Almazor M. Opioid rotation for toxicity reduction in terminal cancer patients. J. Pain. Symptom. Manage. 1995;10(5):378–384. doi: 10.1016/0885-3924(95)90924-c. [DOI] [PubMed] [Google Scholar]
- 71.Riley J., Ross J. R., Rutter D., Wells A. U., Goller K., du Bois R., Welsh K. No pain relief from morphine? Individual variation in sensitivity to morphine and the need to switch to an alternative opioid in cancer patients. Support. Care. Cancer. 2006;14(1):56–64. doi: 10.1007/s00520-005-0843-2. [DOI] [PubMed] [Google Scholar]
- 72.Athanasos P., Smith C. S., White J. M., Somogyi A. A., Bochner F., Ling W. Methadone maintenance patients are cross-tolerant to the antinociceptive effects of very high plasma morphine concentrations. Pain. 2006;120(3):267–275. doi: 10.1016/j.pain.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Duttaroy A., Yoburn B. C. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82(5):1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- 74.Walker E. A., Richardson T. M., Young A. M. Tolerance and cross-tolerance to morphine-like stimulus effects of mu opioids in rats. Psychopharmacology. (Berl) 1997;133(1):17–28. doi: 10.1007/s002130050366. [DOI] [PubMed] [Google Scholar]
- 75.Pasternak G. W. Multiple opiate receptors: deja vu all over again. Neuropharmacology. 2004;47(Suppl 1):312–323. doi: 10.1016/j.neuropharm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 76.Gupta A., Decaillot F. M., Devi L. A. Targeting opioid receptor heterodimers: strategies for screening and drug development. AAPS. J. 2006;8(1):E153–E159. doi: 10.1208/aapsj080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zarrindast M. R., Alaei-Nia K., Shafizadeh M. On the mechanism of tolerance to morphine-induced Straub tail reaction in mice. Pharmacol. Biochem. Behav. 2001;69(3–4):419–424. doi: 10.1016/s0091-3057(01)00519-6. [DOI] [PubMed] [Google Scholar]
- 78.Luccarini P., Perrier L., Degoulange C., Gaydier A. M., Dallel R. Synergistic antinociceptive effect of amitriptyline and morphine in the rat orofacial formalin test. Anesthesiology. 2004;100(3):690–696. doi: 10.1097/00000542-200403000-00033. [DOI] [PubMed] [Google Scholar]
- 79.Sawynok J., Esser M. J., Reid A. R. Antidepressants as analgesics: an overview of central and peripheral mechanisms of action. J. Psychiatry. Neurosci. 2001;26(1):21–29. [PMC free article] [PubMed] [Google Scholar]
- 80.Gracy K. N., Svingos A. L., Pickel V. M. Dual ultrastructural localization of mu-opioid receptors and NMDA-type glutamate receptors in the shell of the rat nucleus accumbens. J. Neurosci. 1997;17(12):4839–4848. doi: 10.1523/JNEUROSCI.17-12-04839.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Commons K. G., van Bockstaele E. J., Pfaff D. W. Frequent colocalization of mu opioid and NMDA-type glutamate receptors at postsynaptic sites in periaqueductal gray neurons. J. Comp. Neurol. 1999;408(4):549–559. [PubMed] [Google Scholar]
- 82.Zhao M., Joo D. T. Subpopulation of dorsal horn neurons displays enhanced N-methyl-D-aspartate receptor function after chronic morphine exposure. Anesthesiology. 2006;104(4):815–825. doi: 10.1097/00000542-200604000-00028. [DOI] [PubMed] [Google Scholar]
- 83.Snyder S. H. Opiate receptors and beyond: 30 years of neural signaling research. Neuropharmacology. 2004;47(Suppl 1):274–285. doi: 10.1016/j.neuropharm.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Kolesnikov Y. A., Pick C. G., Ciszewska G., Pasternak G. W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1993;90(11):5162–5166. doi: 10.1073/pnas.90.11.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Babey A. M., Kolesnikov Y., Cheng J., Inturrisi C. E., Trifilletti R. R., Pasternak G. W. Nitric oxide and opioid tolerance. Neuropharmacology. 1994;33(11):1463–1470. doi: 10.1016/0028-3908(94)90050-7. [DOI] [PubMed] [Google Scholar]
- 86.Kemp J. A., McKernan R. M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002;5(Suppl):1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 87.Herman B. H., Vocci F., Bridge P. The effects of NMDA receptor antagonists and nitric oxide synthase inhibitors on opioid tolerance and withdrawal. Medication development issues for opiate addiction. Neuropsychopharmacology. 1995;13(4):269–293. doi: 10.1016/0893-133X(95)00140-9. [DOI] [PubMed] [Google Scholar]
- 88.Lauretti G. R., Lima I. C., Reis M. P., Prado W. A., Pereira N. L. Oral ketamine and transdermal nitroglycerin as analgesic adjuvants to oral morphine therapy for cancer pain management. Anesthesiology. 1999;90(6):1528–1533. doi: 10.1097/00000542-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 89.Allen R. M., Dykstra L. A. Attenuation of mu-opioid tolerance and cross-tolerance by the competitive N-methyl-D-aspartate receptor antagonist LY235959 is related to tolerance and cross-tolerance magnitude. J. Pharmacol. Exp. Ther. 2000;295(3):1012–1021. [PubMed] [Google Scholar]
- 90.Fischer B. D., Carrigan K. A., Dykstra L. A. Effects of N-methyl-D-aspartate receptor antagonists on acute morphine-induced and l-methadone-induced antinociception in mice. J. Pain. 2005;6(7):425–433. doi: 10.1016/j.jpain.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 91.Medvedev I. O., Malyshkin A. A., Belozertseva I. V., Sukhotina I. A., Sevostianova N. Y., Aliev K., Zvartau E. E., Parsons C. G., Danysz W., Bespalov A. Y. Effects of low-affinity NMDA receptor channel blockers in two rat models of chronic pain. Neuropharmacology. 2004;47(2):175–183. doi: 10.1016/j.neuropharm.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 92.Suzuki M., Kinoshita T., Kikutani T., Yokoyama K., Inagi T., Sugimoto K., Haraguchi S., Hisayoshi T., Shimada Y. Determining the plasma concentration of ketamine that enhances epidural bupivacaine-and-morphine-induced analgesia. Anesth. Analg. 2005;101(3):777–784. doi: 10.1213/01.ane.0000166952.12290.45. [DOI] [PubMed] [Google Scholar]
- 93.Sethna N. F., Liu M., Gracely R., Bennett G. J., Max M. B. Analgesic and cognitive effects of intravenous ketamine-alfentanil combinations versus either drug alone after intradermal capsaicin in normal subjects. Anesth. Analg. 1998;86(6):1250–1256. doi: 10.1097/00000539-199806000-00022. [DOI] [PubMed] [Google Scholar]
- 94.Redwine K. E., Trujillo K. A. Effects of NMDA receptor antagonists on acute mu-opioid analgesia in the rat. Pharmacol. Biochem. Behav. 2003;76(2):361–372. doi: 10.1016/j.pbb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 95.Willetts J., Balster R. L., Leander J. D. The behavioral pharmacology of NMDA receptor antagonists. Trends Pharmacol. Sci. 1990;11(10):423–428. doi: 10.1016/0165-6147(90)90150-7. [DOI] [PubMed] [Google Scholar]
- 96.Chen H. S., Lipton S. A. The chemical biology of clinically tolerated NMDA receptor antagonists. J. Neurochem. 2006;97(6):1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 97.Cosman K. M., Boyle L. L., Porsteinsson A. P. Memantine in the treatment of mild-to-moderate Alzheimer's disease. Expert. Opin. Pharmacother. 2007;8(2):203–214. doi: 10.1517/14656566.8.2.203. [DOI] [PubMed] [Google Scholar]
- 98.Coggeshall R. E., Zhou S., Carlton S. M. Opioid receptors on peripheral sensory axons. Brain. Res. 1997;764(1–2):126–132. doi: 10.1016/s0006-8993(97)00446-0. [DOI] [PubMed] [Google Scholar]
- 99.Carlton S. M., Zhou S., Coggeshall R. E. Evidence for the interaction of glutamate and NK1 receptors in the periphery. Brain. Res. 1998;790(1–2):160–169. doi: 10.1016/s0006-8993(97)01471-6. [DOI] [PubMed] [Google Scholar]
- 100.Carlton S. M., Hargett G. L., Coggeshall R. E. Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin. Neurosci. Lett. 1995;197(1):25–28. doi: 10.1016/0304-3940(95)11889-5. [DOI] [PubMed] [Google Scholar]
- 101.Carlton S. M., Coggeshall R. E. Inflammation-induced changes in peripheral glutamate receptor populations. Brain. Res. 1999;820(1–2):63–70. doi: 10.1016/s0006-8993(98)01328-6. [DOI] [PubMed] [Google Scholar]
- 102.Pol O., Puig M. M. Expression of opioid receptors during peripheral inflammation. Curr. Top. Med. Chem. 2004;4(1):51–61. doi: 10.2174/1568026043451519. [DOI] [PubMed] [Google Scholar]
- 103.Kolesnikov Y. A., Jain S., Wilson R., Pasternak G. W. Peripheral morphine analgesia: synergy with central sites and a target of morphine tolerance. J. Pharmacol. Exp. Ther. 1996;279(2):502–506. [PubMed] [Google Scholar]
- 104.Reichert J. A., Daughters R. S., Rivard R., Simone D. A. Peripheral and preemptive opioid antinociception in a mouse visceral pain model. Pain. 2001;89(2–3):221–227. doi: 10.1016/s0304-3959(00)00365-1. [DOI] [PubMed] [Google Scholar]
- 105.Shannon H. E., Lutz E. A. Comparison of the peripheral and central effects of the opioid agonists loperamide and morphine in the formalin test in rats. Neuropharmacology. 2002;42(2):253–261. doi: 10.1016/s0028-3908(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 106.Kolesnikov Y., Cristea M., Oksman G., Torosjan A., Wilson R. Evaluation of the tail formalin test in mice as a new model to assess local analgesic effects. Brain. Res. 2004;1029(2):217–223. doi: 10.1016/j.brainres.2004.09.058. [DOI] [PubMed] [Google Scholar]
- 107.Gardmark M., Brynne L., Hammarlund-Udenaes M., Karlsson M. O. Interchangeability and predictive performance of empirical tolerance models. Clin. Pharmacokinet. 1999;36(2):145–167. doi: 10.2165/00003088-199936020-00005. [DOI] [PubMed] [Google Scholar]
- 108.Porchet H. C., Benowitz N. L., Sheiner L. B. Pharmacodynamic model of tolerance: application to nicotine. J. Pharmacol. Exp. Ther. 1988;244(1):231–236. [PubMed] [Google Scholar]
- 109.Ouellet D. M., Pollack G. M. Pharmacodynamics and tolerance development during multiple intravenous bolus morphine administration in rats. J. Pharmacol. Exp. Ther. 1997;281(2):713–720. [PubMed] [Google Scholar]
- 110.Lutfy K., Hurlbut D. E., Weber E. Blockade of morphine-induced analgesia and tolerance in mice by MK-801. Brain. Res. 1993;616(1–2):83–88. doi: 10.1016/0006-8993(93)90195-s. [DOI] [PubMed] [Google Scholar]
- 111.Bhargava H. N., Sharma S. S., Bian J. T. Evidence for a role of N-methyl-D-aspartate receptors in L-arginine-induced attenuation of morphine antinociception. Brain. Res. 1998;782(1–2):314–317. doi: 10.1016/s0006-8993(97)01306-1. [DOI] [PubMed] [Google Scholar]
- 112.Bhargava H. N., Bian J. T., Kumar S. Mechanism of attenuation of morphine antinociception by chronic treatment with L-arginine. J. Pharmacol. Exp. Ther. 1997;281(2):707–712. [PubMed] [Google Scholar]
- 113.Heinzen E. L., Pollack G. M. The development of morphine antinociceptive tolerance in nitric oxide synthase-deficient mice. Biochem. Pharmacol. 2004;67(4):735–741. doi: 10.1016/j.bcp.2003.08.046. [DOI] [PubMed] [Google Scholar]
- 114.Heinzen E. L., Pollack G. M. Use of an electrochemical nitric oxide sensor to detect neuronal nitric oxide production in conscious, unrestrained rats. J. Pharmacol. Toxicol. Methods. 2002;48(3):139–146. doi: 10.1016/S1056-8719(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 115.Heinzen E. L., Pollack G. M. Pharmacokinetics and pharmacodynamics of L-arginine in rats: a model of stimulated neuronal nitric oxide synthesis. Brain. Res. 2003;989(1):67–75. doi: 10.1016/s0006-8993(03)03370-5. [DOI] [PubMed] [Google Scholar]
- 116.Heinzen E. L., Pollack G. M. Pharmacodynamics of morphine-induced neuronal nitric oxide production and antinociceptive tolerance development. Brain. Res. 2004;1023(2):175–184. doi: 10.1016/j.brainres.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 117.Heinzen E. L., Booth R. G., Pollack G. M. Neuronal nitric oxide modulates morphine antinociceptive tolerance by enhancing constitutive activity of the mu-opioid receptor. Biochem. Pharmacol. 2005;69(4):679–688. doi: 10.1016/j.bcp.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 118.Ouellet D. M., Pollack G. M. A pharmacokinetic–pharmacodynamic model of tolerance to morphine analgesia during infusion in rats. J. Pharmacokinet. Biopharm. 1995;23(6):531–549. doi: 10.1007/BF02353460. [DOI] [PubMed] [Google Scholar]