

Abstract
This study was performed to prepare and characterize the biotinylated Salmon calcitonin (sCT) for oral delivery and evaluate the hypocalcemic effect of biotinylated-sCTs in rats. Biotinylated sCTs was characterized by using high performance liquid chromatography (HPLC) and MALDITOF-MS. The effect of biotinylation on permeability across Caco-2 cell monolayers was examined. Their hypocalcemic effect was determined in rats. Mono- and di-bio-sCTs were separated by reverse phase HPLC. The molecular weights of mono-bio-sCT and di-bio-sCT were determined to be 3,660.5 and 3,900.2 Da, respectively. The permeability of biotinylated-sCTs across Caco-2 cell monolayers was observed with a significant enhancement compared with sCT. Intrajejunal (ij) administration of mono-bio-sCT and di-bio-sCT resulted in sustained reduction in serum calcium levels, with a maximum reduction (% max(d)) of 21.6% and 30% after 4 h and 6 h of application, respectively. The biotin conjugation of sCT may be a promising strategy for increasing the oral bioavailability of sCT and achieving sustained calcium-lowering effects.
Keywords: biotinylation, Caco-2, MALDITOF-MS, oral delivery, salmon calcitonin
INTRODUCTION
To date, a number of peptides and proteins have been explored for pharmaceutical applications by virtue of their high biological activity and specificity. However, despite the advantages offered by these drugs, their application may suffer because of their high molecular weight, hydrophilicity, and low stability, which are reflected in poor biopharmaceutical properties (1).
Therefore, the development of noninvasive delivery systems for peptides/proteins has a great importance in modern pharmaceutical technology, especially when repeated or routine administration is necessary (2).
Salmon calcitonin (sCT), an endogenous polypeptide hormone composed of 32 amino acids (3,432 Da), the primary structure of sCT are shown in Fig. 1 (3). sCT is currently formulated as either a sterile solution for intramuscular or subcutaneous injection or as a nasal spray (4,5). This drug plays a crucial role in both calcium homeostasis and the treatment of bone disease such as osteoporosis. Therefore, it is very important to develop appropriate oral dosage forms of sCT to increase patient compliance. Moreover, like many other proteins or peptides, the oral bioavailability (BA) of sCT is very low due to enzymatic degradation in the gastrointestinal tract and poor permeation across intestinal epithelial cells (2). The absorption of sCT has been reported to be low (less than 0.1% of BA) after direct administration into the duodenum, ileum, and colon in rats and dogs (6,7).
Fig. 1.

The primary structure of sCT. The sCT has three primary amino groups of N terminus and lysine residues (3)
A number of oral delivery approaches for sCT are described in several reports. One of them is the chemical modification of calcitonin to produce prodrugs and analogues (8,9,10,11). It is plausible that this approach may protect CT against degradation by proteases and other enzymes present at the mucosal barrier (8). Another reported approach uses the drug delivery systems, such as chitosan nanocapsules (12), chitosan-PEG nanocapsules (13), surface-modified lipid nanoparticles (14), thiolated chitosan carrier matrix (15), poly(lactic-co-glycolic acid) nanoparticles (16), and liposome (17).
Here, we describe an alternative, non-invasive approach to facilitate intestinal protein absorption using biotinylation. Biotin is a water-soluble vitamin essential for all known organisms. Intestinal biotin transport is clearly sodium-dependent and the site of maximal biotin absorption is in the jejunum, which has a substantially greater surface area than the ileum and markedly higher than proximal colon (18–22). Biotin can be recognized and absorbed through the Na+-dependent multivitamin transporter (SMVT) systems. SMVT is a transmembrane protein of 635 amino acids with 12 transmembrane domains that mediates the uptake of biotin, lipoic acid, and pantothenate (20). Several papers described the enhanced cell uptake and transport of biotinylated-nonapeptides via SMVT (21,22).
In this study, sCT-biotin conjugates are prepared and the hypocalcemic effect and transport across Caco-2 monolayers of the conjugates are described. The Caco-2 cell monolayer system has become a standard in vitro method for the prediction of oral BA of a compound for over 20 years. Caco-2 cells are derived from a human colorectal carcinoma and, when cultured under the appropriate conditions, will form highly polarized monolayers with tight junctions between individual cells. They express most of the active and facilitative transporters present in the small intestine epithelial barrier including the peptide transporters, glucose transporters, vitamin transporters, and efflux pumps such as p-glycoprotein. The Caco-2 cell monolayer forms a well-developed brush border on the apical side and they express the associated digestive enzymes, including dipeptidyl peptidase IV (DPPIV). They are considered an excellent model of the epithelial barrier of the small intestine (23,24).
METHODS
Materials
Salmon calcitonin (1–32) and sCT enzyme immunosorbent assay (EIA) kit were purchased from Bachem AG (Torrance, CA, USA). (+)-Biotin N-hydroxysuccinimide ester (biotin-NHS), dimethylformamide (DMF), and triethylamine (TEA) were obtained from Sigma Chemical (St. Louis, MO, USA). Calcium assay kit was purchased from Wako Pure Chemical (Calcium-C test, Japan). Caco-2-cells were obtained from the American Tissue Culture Collection (ATCC, Rockville, MD, USA). Male Sprague-Dawley rats (200–250 g) were purchased from the Experimental Animal Lab of Korean FDA (Seoul, Korea). All other chemicals were of analytical grade and were commercially obtained.
Preparation and Characterization of Biotinylated sCTs
The solution of sCT/DMF (0.2 ml, 10 mg/ml) was added to biotin-NHS/DMF-0.2% TEA (0.2 ml, 2 mg/ml). This mixture was shaken gently at ambient temperature for 15 min and subjected to a LiChrospher 100 RP-18 column (4.0 × 250 mm, 5 μm, Merck, Germany) for reversed-phase high performance liquid chromatography (RP-HPLC) analysis (gradient elution, flow-rate of 1.0 ml/min, 34–44% (v/v) acetonitrile (ACN)–0.1% (v/v) trifluoroacetic acid (TFA), 25 min, UV 215 nm). Fractions were collected separately as previously described (9). Thereafter, a proteolytic digestion for identification of the biotinylation sites of separated samples was performed in 0.1 M Tris–HCl buffer (pH 8.5) at 37°C with an enzyme (endoproteinase Lys-C) to substrate ratio of 1:100 (w/w) for 2 h, as previously described (9). The molecular weights of the separated and Lys-C digested samples were determined by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) as previously described (25,26).
Permeability of sCT and Biotinylated sCTs Across Caco-2 Monolayer
Caco-2 cells were grown as previously described (9). Transport studies were conducted on the Caco-2 monolayers after 18–21 days in culture. Prior to the beginning of the experiments, membranes were rinsed twice with transport media (TM) in the apical and basolateral chambers and allowed to equilibrate in the experimental medium for 2 h at 37°C in a CO2 incubator to obtain a constant trans-epithelial electrical resistance (TEER). The resistance was measured using an EVOM epithelial voltohmmeter (World Precision Instruments, USA) and expressed as Ω.cm2. After the equilibration period, the TM on both sides of the Caco-2 cell monolayer was removed by aspiration. A 0.5 mL aliquot of the TM containing sCT and mono/di-bio-sCT (25 μM, sCT eq.) was then added on the apical side and 1.5 mL of the TM without drug was added on the basolateral side. All transport experiments were conducted in the apical to basolateral direction at 37°C in a water bath. The polystyrene inserts in the transwell were moved to other wells containing fresh TM (1.5 mL) every 15 min for 1 h. After moving the inserts at each time point, all aliquots were removed from the basolateral side. The concentration of sCT and derivatives was determined using an EIA kit (Bachem AG).
In Vivo Hypocalcemic Potency of sCT and Biotinylated sCTs
Male Sprague-Dawley rats (200–250 g) were cannulated in the jugular vein after anesthesia by an ip injection of ketamine/xylazine (90/10 mg/kg) a day before the experiment. Rats were fasted for about 18 h before the experiment but had access to water. During the experiment, the animals were randomly divided into four groups (the acetate buffer group and the three sCT, mono-bio-sCT and di-bio-sCT groups; n = 3 each) and anesthetized. To compare hypocalcemic effect, sCT or mono/di-bio-sCT in acetate buffer (10 mM, pH 3.5) in which sCT has the highest stability (10,27) (1 μg/rat) was intravenously (iv) administered. At the predetermined times (0, 15, 30, 45, and 60 min), blood samples were collected and immediately separated by centrifugation (10,000 rpm, 5 min). The plasma was frozen until the calcium assay to determine the hypocalcemic effect of the drug. Total calcium levels were measured using a colorimetric Ca assay based on the o-cresolphthalein complexation method described elsewhere (28).
To determine the effect of the compound via the interjejunal route, the same animal protocol was used as explained in in vivo hypocalcemic potency of sCT and biotinylated sCTs. During the experiment, the animals were randomly divided into four groups and anesthetized. Then, a small midline skin incision was made in the abdomen and a loop of the jejunum, 5 cm distal to the ligament of Treitz, was taken out of the abdominal cavity. To compare hypocalcemic effect, sCT or mono/di-bio-sCT (100 μg/rat) was injected into the jejunal lumen. At the predetermined times (0, 15, 30, 60, 120, 180, 240, 300, 360, and 420 min), blood samples were collected from the jugular vein to obtain plasma. The plasma was frozen until the calcium assay.
Data Analysis
The apparent permeability coefficient (cm/s, Papp) was calculated using the following equation: 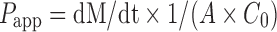 , where dM/dt is the amount of drug appearing in the acceptor compartment in function of time (μmol/s), C0 is the initial concentration in the donor compartment (μM), and A is the surface (cm2) across which the transport occurred. The maximum percentage reduction in serum calcium levels relative to the basal value (% maxd) was calculated. The total decrement in serum calcium levels (D%) was calculated using a modified method by Hirai et al. (29) as follows:
, where dM/dt is the amount of drug appearing in the acceptor compartment in function of time (μmol/s), C0 is the initial concentration in the donor compartment (μM), and A is the surface (cm2) across which the transport occurred. The maximum percentage reduction in serum calcium levels relative to the basal value (% maxd) was calculated. The total decrement in serum calcium levels (D%) was calculated using a modified method by Hirai et al. (29) as follows: 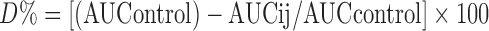 , where AUCij and AUCcontrol were the areas under the serum calcium concentration versus time curves up to 7 h after intrajejunal (ij) administration of samples and placebo, respectively.
, where AUCij and AUCcontrol were the areas under the serum calcium concentration versus time curves up to 7 h after intrajejunal (ij) administration of samples and placebo, respectively.
Statistical Analysis
Statistical data analysis was performed using the ANOVA/LSD post-hoc test. When significant differences were obtained (P < 0.05).
RESULTS
In the present study, prepared biotinylated-sCTs were separated and identified as mono- and di-bio-sCT using RP-HPLC (Fig. 2) and MALDI-TOF-MS, respectively (Fig. 3, Table I). To identify the biotinylation sites of isomers of bio-sCT, the enzymatic digestion method was used. Endoproteinase Lys-C from Lysobacter enzymogenes, which is commercially available lysine specific protease, proved to be useful in the determination of primary structures of proteins (protein sequence analysis). Endoproteinase Lys-C hydrolyzes peptide bonds at the carboxyl side of the lysine residues. Each of the purified biotinylated-sCT derivatives and intact sCT were then subjected to the lysyl endoproteinase, Lys-C, proteolysis to determine the positional substitution of biotin molecules. After incubation, endoproteinase Lys-C with sCT and mono/di-bio-sCTs were directly characterized by MALDI-TOF-MS (Fig. 3, Table I).
Fig. 2.

Reversed-phase gradient HPLC analysis of the reaction mixture obtained by reaction of biotin-NHS with sCT (Retention times, 15.730 min, 21.902 min, and 24.663 min, respectively; linear gradient elution, ACN, 34–44%, 25 min; flow rate, 1 ml/min)
Fig. 3.

MALDI-TOF mass spectra of intact sCT, Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT before/after endoproteinase Lys-C digestion (a and b, respectively). The ion acceleration potential was set at +25 kV under the positive ion linear mode of detection and a saturated solution of α-CHCA in 50% ACN was used as a matrix solution
Table I.
Molecular Masses Determined by MALDI-TOF-MS of sCT, Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT Before/After Lys-C Digestion
| Calculated mass (m/za) | Observed masses (m/za) | |
|---|---|---|
| Before Lys-C digestion | ||
| Product | ||
| sCT | 3,432 | 3,422.21 |
| Lys11-mono-bio-sCT | 3,676 | 3,660.5 |
| Lys11,18-di-bio-sCT | 3,920 | 3,900.2 |
| After Lys-C digestion | ||
| Possible fragments | ||
| Cys1-Lys11 | 1,106.34 | 1,128.04 |
| Leu12-Lys18 | 835.96 | 848.02 |
| Leu19-Pro32 | 1,475.61 | 1,492.9 and 1,495.22 |
| Leu12-Lys18-bio-Pro32 | 2,555.57 | 2,208.87 |
| Cys1-Lys11-bio-Lys18 | 2,186.3 | – |
amass/charge
To determine the effect of biotinylation on permeability of the sCT, an in vitro study was performed using a Caco-2 cell monolayer as an intestinal absorption model. Transport experiments in Caco-2 cells were conducted at pH 7.4 from apical to basolateral direction. TEER values were also monitored to ensure monolayer consistency during the studies. Morphologically and physiologically well-developed Caco-2 cell monolayers with TEER values greater than 400 Ω.cm2 (578.8 ± 39.7) were used. Figure 4 shows the time course of apical-to-basolateral transport of sCT and mono/di-bio-sCTs across the Caco-2 monolayer. The permeability coefficients of sCT and mono/di-bio-sCT were determined at 25 μM, and the apparent permeability (Papp) of sCT was 2.5- and fourfold increased after mono- and di-biotinylation, respectively (Table II).
Fig. 4.

Transport of sCT and mono/di-bio-sCTs across Caco-2 monolayers. sCT and mono/di-bio-sCTs were added to apical side at 25 μM (mean ± SD, n = 3)
Table II.
Effects of Biotinylation on the Apical to Basolateral Permeability of sCT at 15 μM (mean ± SD, n = 3, ANOVA/LSD post-hoc, p < 0.05)
| sCT and biotinylated-sCT derivatives | P app (×107 cm/s, mean ± SD) | Fold increase in P app |
|---|---|---|
| sCT | 1.41 ± 0.29 | 1 |
| Lys11-mono-bio-sCT | 3.58 ± 0.09 | 2.54 |
| Lys11,18-di-bio-sCT | 6.02 ± 0.10 | 4.27 |
Changes in serum calcium levels after iv and ij injection of sCT and mono/di-bio sCTs were examined (Figs. 5 and 6, respectively). Table III lists the maximum decrease in serum calcium levels (% maxd) relative to the basal levels and the total decrement in calcium level (D%) following ij and iv administration at doses of 100 μg and 1 μg, respectively, in rats. Intrajejunal administration of sCT resulted in reduced serum calcium levels, with the maximum decrease (%maxd) of 12.9% at approximately 2 h and a total calcium decrement (D%) of 11.4%. However, the calcium concentrations after ij administration of Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT were continuously decreased, reaching a maximum decrease (%maxd) of 21.6% and 30%, by 4 h and 6 h, respectively.
Fig. 5.

Changes in serum calcium concentrations after intravenous administration of sCT and mono/di-bio sCTs at doses of 1 μg, at 60 min in rats (mean ± SD, n = 3)
Fig. 6.

Changes in serum calcium concentrations after intrajejunal injection of sCT and mono/di-bio sCTs at doses of 100 μg in rats (mean ± SD, n = 3)
Table III.
Serum Calcium Lowering Effect of sCT and Mono/di-bio sCTs Following Intrajejunal or Intravenous Administration at Doses of 100 μg and 1 μg in Rats Respectively (mean ± SD, n = 3, ANOVA/LSD Post-hoc, p < 0.05)
| Treatment | Time to maximum decrease in serum Ca (h) | % maxd in serum Ca | D% in 1 and 7 h for iv and ij application respectively |
|---|---|---|---|
| sCT (iv) | 1 | 12.2 ± 1.3 | 11.4 ± 1.8 |
| Lys11-mono-bio-sCT (iv) | 1 | 17.7 ± 1.7 | 14.5 ± 1.6 |
| Lys11,18-di-bio-sCT (iv) | 1 | 18.2 ± 2.2 | 13.6 ± 1.9 |
| sCT (ij) | 2 | 12.9 ± 2.3 | 11.4 ± 2.6 |
| Lys11-mono-bio-sCT (ij) | 4 | 21.6 ± 2.1 | 19.3 ± 1.9 |
| Lys11,18-di-bio-sCT (ij) | 6 | 30 ± 2.5 | 28.6 ± 2.0 |
DISCUSSION
The use of polypeptides and proteins for systemic treatment of certain diseases is well accepted in medical practice. Most peptide and protein drugs are currently used as parenteral formulations because of their poor oral BA. In addition, during the transit through the intestinal tract, they are chemically and enzymatically inactivated by the high acidity of the stomach and the presence of nutrients and secreted enzymes. Nevertheless, oral delivery is a very attractive option for administration of many proteins (30,31).
In order to benefit from the advantages of oral delivery, biotinylated-sCT derivatives were prepared for oral delivery. The first stage was to prepare and separate the biotinylated-sCT derivatives by using RP-HPLC and MALDI-TOF-MS (Figs. 2, 3 and Table I). In particular, the chemical attachment of functional moieties to the primary amine groups of sCT resulted in position-dependent characteristics. Proteins generally have abundant primary amines in the side chain of lysine residues and the N terminus of each polypeptide that are available as targets for labeling with NHS-activated biotin reagents. sCT contains three potential sites for biotinylation (Fig. 1). Possible biotinylated sites for NHS-activated biotins are primary amines in the side chain of Cys1, Lys11, Lys18 residues. After biotinylation, a heterogeneous mixture of seven possible biotin modified sCTs (one tri-bio-sCT (Cys1, Lys11, Lys18); three di-bio-sCT (Cys1 and Lys11; Cys1 and Lys18; Lys11 and Lys18); three mono-bio-sCT (Cys1 or Lys11 or Lys18) might be obtained. To prepare the Lys-bioconjugated sCTs, Lys-amine specific conjugation reaction conditions conducted in an organic medium, as previously described (5,9,10,28). Figure 3 clearly shows that the primary amine-directed mono-biotinylation of sCT results in the two different isomers of mono- and di-biotin-sCT.
The second stage was to determine biotinylation sites. In addition, sCT also degrades with proteolytic enzymes and loses activity. Luminally secreted serine proteases (trypsin, α-chymotrypsin) seem to be primarily responsible for the digestion of sCT (32). Biotin attachment appears to increase the resistance of the peptide to Lys-C digestion, probably resulting from the steric hindrance of the biotin molecules. Due to the differences in the number and the sites of biotin attachment, different mass spectrometric profiles of Lys-C digested biotinylated-sCT derivatives were observed (Fig. 3, Table I). The MALDI-TOF-MS of digested mono-bio-sCT showed two peaks of the number-averaged molecular weights (Mn) 1,495 and 2,208, indicating that Lys11 was biotinylated. The mass spectrum of di-bio-sCT showed one peak of Mn 3,901, demonstrating that Lys11 and Lys18 were biotinylated.
We examined the ability of mono- and di-bio-sCT to cross the intestinal epithelial membrane using the Caco-2 monolayer system. The intestinal permeability of mono- and di-bio-sCT was higher than that of sCT. The mechanism of sCT permeation is passive diffusion through the mucosa as determined from the Caco-2 monolayer. Due to the probable result of passive diffusion, the transport of sCT across the Caco-2 cell monolayer was not concentration-dependent (33). The permeability of mono- and di-biotinylated-sCT was increased 2.5- and fourfold, respectively. Similar to the results reported by Ramanathan et al. (21,22), the biotinylation of a Tat nonapeptide altered its transport pathway resulting in a significant improvement in its absorptive permeability by enhancing nonspecific passive and carrier-mediated uptake by means of SMVT. In addition, Chae et al. (34) described the preparation and characterization of the bioconjugated glucagon-like peptide-1 (GLP-1) (Lys26,34-di-bio-(GLP-1) and Lys26-bio-Lys34-bio-PEG-GLP-1). They found that Lys26,34-di-bio-(GLP-1) and Lys26-bio-Lys34-bio-PEG-GLP-1 show enhanced intestinal absorption via SMVT, better proteolytic stabilities, and higher oral hypoglycemic effect than native GLP-1. SMVT is expressed in the placenta, intestine, heart, brain, lung, liver, kidney, and pancreas and has been cloned from rats, rabbits, and humans. Three distinct variants (II, III, and IV) of the placental SMVT (I) have been identified in rat small intestine (20,24). Nonspecific passive transport increased presumably due to the enhanced hydrophobicity of the peptide resulting from the addition of biotin (21,22).
Intrajejunal administration of sCT resulted in reduced serum calcium levels, with the maximum decrease (%maxd) of 12.9% at approximately 2 h and a total calcium decrement (D%) of 11.4%. In contrast, the calcium concentrations after ij administration of Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT were continuously decreased, reaching a maximum decrease (%maxd) of 21.6% and 30%, by 4 h and 6 h, respectively, and remained lower than the initial value up to7 h. The total decrement in serum calcium (D%) was measured to be approximately 19.3% and 28.6%, respectively. Therefore, the hypocalcemic effects of Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT were approximately twofold–2.5-fold greater than that of sCT, respectively. The iv injection of sCT, Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT resulted in hypocalcemic effects, with the total decrement (D%) of 11.4%, 14.5%, and 13.6%, respectively. This result showed maintenance of the bioactivity of sCT after biotinylation.
Pharmaceutical strategies have been proposed to maximize oral sCT bioavailability to overcome barriers (enzymatic degradation and low permeability) and to develop safe and effective therapies. Potential approaches to enhance sCT absorption include the use of formulation additives (absorption enhancers, enzyme inhibitors etc.) to limit the activity of intestinal enzymes or increase its intestinal transport and the preparation of mucoadhesive polymeric carriers to protect sCT from proteolytic degradation in the intestine (2,12,13,33,35). These studies reported that improving the intestinal absorption of sCT and significant reduction in enzymatic degradation. However, the toxicity of mucoadhesive polymeric carriers over a long period is still unclear and human data are required to regulate their potential use for clinical applications and also the use of enzyme inhibitors may also affect the absorption of other peptides or proteins that would normally be degraded (35). A major drawback of these inhibitors is their high toxicity, especially during chronic drug therapy (35). Furthermore, the use of absorption enhancers is limited by the fact that once cell membranes are permeabilized or tight junctions opened, transport is enhanced not only for peptide and protein drugs but also for undesirable molecules present in the gastrointestinal tract (35). Another approach is site-specific bioconjugation with PEG of sCT to help overcome enzymatic barrier in intestine or colon (9,10). This conjugation technology has been used to improve the therapeutic efficacy of sCT. Youn et al. (9) and Mansoor et al. (10) reported that the PEGylation of sCT profoundly improves its enzymatic stability while retaining its biological activities and thus enhances its therapeutic potential. In this current study, we used a new strategy for producing orally active sCT prodrug using site-specific biotinylation. We prepared salmon calcitonin–biotin conjugates and characterized the transport properties and hypocalcemic effects of conjugates. The present results suggest the ability of macromolecule conjugates with targeting moieties such as biotin to be transported efficiently across the intestinal membranes.
A further component of this study is under way to determine the molecular mechanism(s) of carrier-mediated uptake of the salmon calcitonin–biotin conjugates via SMVT and the stability of biotinylated-sCT derivatives against intestinal enzymes. The transport of the conjugates in the Caco-2 cell monolayer expressing SMVT will be investigated through inhibition studies that were performed by co-incubating competitive substrates of SMVT (biotin, pantothenic acid, desthiobiotin, etc.). In addition, the stabilities of mono/di-bio sCTs in gastrointestinal systems might be increased to prepare biotinylated-sCT derivatives-loaded drug delivery systems or bio-PEGylated-sCT derivatives.
Conclusion
In this study, Lys11-mono-bio-sCT and Lys11,18-di-bio-sCT were prepared and characterized. The biotinylation of sCT (especially di-bio-sCT) increased its intestinal absorption and obtained a significantly higher and prolonged hypocalcemic effect compared to sCT. These findings show that biotinylated-sCT might be a promising drug candidate for oral osteoporosis therapy.
References
- 1.Caliceti P., Veronese F. M. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Drug Deliv. Rev. 2003;55:1261–1277. doi: 10.1016/S0169-409X(03)00108-X. [DOI] [PubMed] [Google Scholar]
- 2.Song K. H., Chung S. J., Shim C. K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control Release. 2005;106:298–308. doi: 10.1016/j.jconrel.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Na D. H., Youn Y. S., Lee S. D., Son M. W., Kim W. B., DeLuca P. P., Lee K. C. Monitoring of peptide acylation inside degrading PLGA microspheres by capillary electrophoresis and MALDI-TOF mass spectrometry. J. Control Release. 2003;92:291–299. doi: 10.1016/S0168-3659(03)00366-3. [DOI] [PubMed] [Google Scholar]
- 4.Azria M. The calcitonins, Physiology and pharmacology. Basel: Karger; 1989. [Google Scholar]
- 5.Lee K. C., Park M. O., Na D. H., Youn Y. S., Lee S. D., Yoo S. D., Lee H. S., DeLuca P. P. Intranasal delivery of PEGylated salmon calcitonins: hypocalcemic effects in rats. Calcif Tissue Int. 2003;73:545–549. doi: 10.1007/s00223-002-0034-9. [DOI] [PubMed] [Google Scholar]
- 6.Lee Y. H., Leesman G. D., Makhey V., Yu H., Hu P., Perry B., Sutyak J. P., Wagner E. J., Falzone L. M., Stern W., Sinko P. J. Regional oral absorption, hepatic first-pass effect, and non-linear disposition of salmon calcitonin in beagle dogs. Eur. J. Pharm. Biopharm. 2000;50:205–211. doi: 10.1016/S0939-6411(00)00102-8. [DOI] [PubMed] [Google Scholar]
- 7.Sinko P. J., Smith C. L., McWhorter L. T., Stern W., Wagner E. J., Gilligan J. P. Utility of pharmacodynamic measures for assessing the oral bioavailability of peptides. 1. Administration of recombinant salmon calcitonin in rats. J. Pharm. Sci. 1995;84:1374–1378. doi: 10.1002/jps.2600841120. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y. H., Sinko P. J. Oral delivery of salmon calcitonin. Adv. Drug Deliv. Rev. 2000;42:225–238. doi: 10.1016/S0169-409X(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 9.Youn Y. S., Jung J. Y., Oh S. H., Yoo S. D., Lee K. C. Improved intestinal delivery of salmon calcitonin by Lys18-amine specific PEGylation: stability, permeability, pharmacokinetic behavior and in vivo hypocalcemic efficacy. J. Control Release. 2006;114:334–342. doi: 10.1016/j.jconrel.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Mansoor S., Youn Y. S., Lee K. C. Oral delivery of mono-PEGylated sCT (Lys18) in rats: regional difference in stability and hypocalcemic effect. Pharm. Dev. Technol. 2005;10:389–396. doi: 10.1081/pdt-65686. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Chow D., Heiati H., Shen W. C. Reversible lipidization for the oral delivery of salmon calcitonin. J. Control Release. 2003;88:369–380. doi: 10.1016/S0168-3659(03)00008-7. [DOI] [PubMed] [Google Scholar]
- 12.Prego C., Torres D., Alonso M. J. Chitosan nanocapsules as carriers for oral peptide delivery: effect of chitosan molecular weight and type of salt on the in vitro behaviour and in vivo effectiveness. J. Nanosci. Nanotechnol. 2006;6:2921–2928. doi: 10.1166/jnn.2006.429. [DOI] [PubMed] [Google Scholar]
- 13.Prego C., Torres D., Fernandez-Megia E., Novoa-Carballal R., Quiñoá E., Alonso M. J. Chitosan-PEG nanocapsules as new carriers for oral peptide delivery. Effect of chitosan pegylation degree. J. Control Release. 2006;111:299–308. doi: 10.1016/j.jconrel.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Fuentes M., Torres D., Alonso M. J. New surface-modified lipid nanoparticles as delivery vehicles for salmon calcitonin. Int. J. Pharm. 2005;296:122–132. doi: 10.1016/j.ijpharm.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Guggi D., Kast C. E., Bernkop-Schnürch A. In vivo evaluation of an oral salmon calcitonin-delivery system based on a thiolated chitosan carrier matrix. Pharm. Res. 2003;20:1989–1994. doi: 10.1023/B:PHAM.0000008047.82334.7d. [DOI] [PubMed] [Google Scholar]
- 16.Sang Y. H., Gwan P. T. Biodegradable nanoparticles containing protein-fatty acid complexes for oral delivery of salmon calcitonin. J. Pharm. Sci. 2004;93(2):488–495. doi: 10.1002/jps.10573. [DOI] [PubMed] [Google Scholar]
- 17.Yamabe K., Kato Y., Onishi H., Machida Y. Potentiality of double liposomes containing salmon calcitonin as an oral dosage form. J. Control Release. 2003;89:429–436. doi: 10.1016/S0168-3659(03)00160-3. [DOI] [PubMed] [Google Scholar]
- 18.Said H. M., Redha R., Nylander W. A carrier-mediated, Na+ gradient dependent transport for biotin in human intestinal brush border membrane vesicles. Am. J. Physiol. 1987;253:G631–G636. doi: 10.1152/ajpgi.1987.253.5.G631. [DOI] [PubMed] [Google Scholar]
- 19.Prasad P. D., Wang H., Huang W., Fei Y. J., Leibach F. H., Devoe L. D., Ganapathy V. Molecular and functional characterization of the intestinal Na+ dependent multivitamin tranporter. Arch. Biochem. Biophys. 1999;366:95–106. doi: 10.1006/abbi.1999.1213. [DOI] [PubMed] [Google Scholar]
- 20.Zempleni J. Uptake, localization, and noncarboxylase roles of biotin. Annu. Rev. Nutr. 2005;25:175–196. doi: 10.1146/annurev.nutr.25.121304.131724. [DOI] [PubMed] [Google Scholar]
- 21.Ramanathan S., Pooyan S., Stein S., Prasad P. D., Wang J., Leibowitz M. J., Ganapathy V., Sinko P. J. Targeting the sodium-dependent multivitamin transporter (SMVT) for improving the oral absorption properties of a retro-inverso Tat nonapeptide. Pharm. Res. 2001;18:950–956. doi: 10.1023/A:1010932126662. [DOI] [PubMed] [Google Scholar]
- 22.Ramanathan S., Qui B., Pooyan S., Zhang G., Stein S., Prasad P. D., Wang J., Leibowitz M. J., Sinko P. J. Targeted PEG-based bioconjugates enhance the cellular uptake and transport of a HIV-1 TAT nonapeptide. J. Control Release. 2001;77:199–212. doi: 10.1016/S0168-3659(01)00474-6. [DOI] [PubMed] [Google Scholar]
- 23.Koda Y., Shiotani K., Toth I., Tsuda Y., Okada Y., Blanchfield J. T. Comparison of the in vitro apparent permeability and stability of opioid mimetic compounds with that of the native peptide. Bioorg. Med. Chem. Lett. 2007;17:2043–2046. doi: 10.1016/j.bmcl.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee N. S., Kumar C. K., Ortiz A., Rubin S. A., Said H. M. Molecular mechanism of the intestinal biotin transport process. Am. J. Physiol. 1999;277:C605–C613. doi: 10.1152/ajpcell.1999.277.4.C605. [DOI] [PubMed] [Google Scholar]
- 25.Lee S. H., Lee S., Youn Y. S., Na D. H., Chae S. Y., Byun Y., Lee K. C. Synthesis, characterization, and pharmacokinetic studies of PEGylated glucagons-like peptide-1. Bioconjug. Chem. 2005;16:377–382. doi: 10.1021/bc049735+. [DOI] [PubMed] [Google Scholar]
- 26.Youn Y. S., Na D. H., Yoo S. D., Song S. C., Lee K. C. Chromatographic separation and mass spectrometric identification of positional isomers of polyethylene glycol-modified growth hormone-releasing factor (1–29) J. Chromatogr. A. 2004;1061:45–49. doi: 10.1016/j.chroma.2004.10.062. [DOI] [PubMed] [Google Scholar]
- 27.Cholewinski M., Lückel B., Horn H. Degradation pathways, analytical characterization and formulation strategies of a peptide and a protein. Calcitonine and human growth hormone in comparison. Pharm. Acta Helv. 1996;71:405–419. doi: 10.1016/S0031-6865(96)00049-0. [DOI] [PubMed] [Google Scholar]
- 28.Yoo S. D., Jun H., Shin B. S., Lee H. S., Park M. O., Deluca P. P., Lee K. C. Pharmacokinetic disposition of polyethylene glycol-modified salmon calcitonins in rats. Chem. Pharm. Bull. (Tokyo) 2000;48:1921–1924. doi: 10.1248/cpb.48.1921. [DOI] [PubMed] [Google Scholar]
- 29.Hirai S., Yasbiki T., Mima H. Effect of surfactants to nasal absorption of insulin in rats. Int. J. Pharm. 1981;9:165–172. doi: 10.1016/0378-5173(81)90009-0. [DOI] [Google Scholar]
- 30.Calceti P., Salmaso S., Walker G., Bernkop-Schnürch A. Development and in vivo evaluation of an oral insulin–PEG delivery system. Eur. J. Pharm. Sci. 2004;22:315–323. doi: 10.1016/j.ejps.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Mahato R. I., Narang A. S., Thoma L., Miller D. D. Emerging trends in oral delivery of peptide and protein drugs. Crit. Rev. Ther. Drug Carr. Syst. 2003;20:153–214. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i23.30. [DOI] [PubMed] [Google Scholar]
- 32.Shah R. B., Khan M. A. Protection of salmon calcitonin breakdown with serine proteases by various ovomucoid species for oral drug delivery. J. Pharm. Sci. 2004;93:392–406. doi: 10.1002/jps.10557. [DOI] [PubMed] [Google Scholar]
- 33.Shah R. B., Khan M. A. Regional permeability of salmon calcitonin in isolated rat gastrointestinal tracts: transport mechanism using Caco-2 Cell monolayer. AAPS J. 2004;6:1–5. doi: 10.1208/aapsj060431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chae S. Y., Jin C. H., Shin H. J., Youn Y. S., Lee S., Lee K. C. Preparation, characterization, and application of biotinylated and biotin-PEGylated glucagon-like peptide-1 analogues for enhanced oral delivery. Bioconjug. Chem. 2008;19:334–341. doi: 10.1021/bc700292v. [DOI] [PubMed] [Google Scholar]
- 35.el-Khafagy S., Morishita M., Onuki Y., Takayama K. Current challenges in non-invasive insulin delivery systems: a comparative review. Adv. Drug Deliv. Rev. 2007;59(15):1521–1546. doi: 10.1016/j.addr.2007.08.019. [DOI] [PubMed] [Google Scholar]