

Abstract
A biphasic gastroretentive floating drug delivery system with multiple-unit mini-tablets based on gas formation technique was developed to maintain constant plasma level of a drug concentration within the therapeutic window. The system consists of loading dose as uncoated core units, and prolonged-release core units are prepared by direct compression process; the latter were coated with three successive layers, one of which is seal coat, an effervescent (sodium bicarbonate) layer, and an outer polymeric layer of polymethacrylates. The formulations were evaluated for quality control tests, and all the parameters evaluated were within the acceptable limits. The system using Eudragit RL30D and combination of them as polymeric layer could float within acceptable time. The drug release was linear with the square root of time. The rapid floating and the controlled release properties were achieved in this present study. When compared with the theoretical release profile, the similarity factor of formulation with coating of RS:RL (1:3)–7.5%, was observed to be 74, which is well fitted into zero-order kinetics confirming that the release from formulation is close to desired release profile. The stability samples showed no significant change in dissolution profiles (p > 0.05). In vivo gastric residence time was examined by radiograms, and it was observed that the units remained in the stomach for about 5 h.
Key words: biphasic release, controlled release, dissolution, polymeric coating
INTRODUCTION
A relatively constant plasma level of a drug is often preferred to maintain the drug concentration within the therapeutic window. However, it is difficult to achieve, especially for once-daily dosage forms, partly because the environment for drug diffusion and/or absorption varies along the gastrointestinal (GI) tract. Normally, drug absorption is slow in the stomach and the large intestine and fast in the small intestine; liquid volume becomes smaller while viscosity of the GI content increases toward the distal segment of the GI tract (1–4). As a result, a constant plasma concentration may not be obtainable even though a dosage form with a zero-order in vitro release is administered. It is conceivable that a delivery system that can provide a release profile with an initial burst of release followed by a relatively steady release or an accelerated release at a late stage may offer a better solution. Such a release profile, namely pseudo-zero-order release with initial burst or bimodal release, may compensate for the lower absorption rate in the stomach and the large intestine.
The production of mini-matrices using a tabletting technique is an attractive alternative to the production of pellets, as the presence of solvents (e.g., water) is avoided, and high production yields like the ones observed in extrusion and spheronization are obtained. Furthermore, due to the manufacturing process, defined size and strengths can easily be produced, with small variability within and between batches (5).
Like other multiple-unit dosage forms (MUDFs), several mini-tablets can be filled into hard capsules that, after disintegration, release these subunits as multiple dosage forms. Because of their size uniformity, regular shape, smooth surface, low porosity, and high attainable strength, mini-tablets can maintain their structure and shape in a more reproducible way than usual pellets or granules, once they have been compressed into a tablet system. This concept can be used to produce a biphasic delivery system combining a fast release together with the slow release component of the drug, This system can produce a rapid rise in the plasma concentrations for some drugs that are requested to promptly exercise the therapeutic effect, followed by a prolonged-release phase in order to avoid repeated administrations (6).
Ranitidine hydrochloride (RH) is a histamine H2-receptor antagonist. It is widely prescribed in active duodenal ulcers, gastric ulcers, Zollinger–Ellison syndrome, gastroesophageal reflux disease, and erosive esophagitis. The recommended adult oral dosage of ranitidine is 150 mg twice daily or 300 mg once daily. The effective treatment of erosive esophagitis requires administration of 150 mg of ranitidine four times a day. A conventional dose of 150 mg can inhibit gastric acid secretion up to 5 h but not up to 10 h. An alternative dose of 300 mg leads to plasma fluctuations; thus, a sustained-release dosage form of RH is desirable (7). The short biological half-life of drug (∼3.5 h) also favors development of a sustained-release formulation. A traditional oral sustained-release formulation releases most of the drug at the colon; thus, the drug should have absorption window either in the colon or throughout the gastrointestinal tract. Ranitidine is absorbed only in the initial part of the small intestine and has 50% absolute bioavailability (8–9). Moreover, colonic metabolism of ranitidine is partly responsible for the poor bioavailability of ranitidine from the colon (10). These properties of RH do not favor the traditional approach to sustained-release delivery. Hence, clinically acceptable sustained-release dosage forms of RH prepared with conventional technology may not be successful. The gastroretentive drug delivery systems can be retained in the stomach and assist in improving the oral sustained delivery of drugs that have an absorption window in a particular region of the gastrointestinal tract. These systems help in continuously releasing the drug before it reaches the absorption window, thus ensuring optimal bioavailability. It is also reported that oral treatment of gastric disorders with an H2-receptor antagonist like ranitidine or famotidine used in combination with antacids promotes local delivery of these drugs to the receptor of the parietal cell wall. Local delivery also increases the stomach wall receptor site bioavailability and increases the efficacy of drugs to reduce acid secretion (11). This principle may be applied for improving systemic as well as local delivery of RH, which would efficiently reduce gastric acid secretion. Several approaches are currently used to prolong gastric retention time. These include floating drug delivery systems, also known as hydrodynamically balanced systems, swelling and expanding systems, polymeric bioadhesive systems, modified-shape systems, high-density systems, and other delayed gastric emptying devices. In context of the above principles, a strong need was recognized for the development of a dosage form to deliver RH in the stomach and to increase the efficiency of the drug, providing sustained action. The present investigation applied a systematic approach to the development of biphasic gastroretentive dosage forms.
In this study, mini-tablets filled into capsules (comprising of floating loading dose component and floating prolonged-release component) based on gas formation technique was developed. The drug containing both core mini-tablets was prepared by direct compression method followed by coating of the prolonged-release units with seal coating, effervescent layer, and gas-entrapped polymeric membrane (Eudragit RS30D, RL30D). RH, which is predominantly absorbed in the upper part of GI tract, and a relatively constant plasma level of a drug, which is often preferred to maintain the drug concentration within the therapeutic window, was used as a model compound. The effect of the preparative parameters like amount of the effervescent agent layered onto the seal coated units and type and coating level of the gas-entrapped polymeric membrane on the floating ability and drug release properties of the multiple-unit FDDS were evaluated.
MATERIALS AND METHODS
Materials
Ranitidine HCl was obtained from Dr Reddy’s laboratories Hyderabad, India. Microcrystalline cellulose (MCC; Avicel PH102), hydroxy propyl methyl cellulose (HPMC K100), and HPMC K4M were obtained from ISP, Hyderabad, India. Sodium bicarbonate (Merk, India) was used as an effervescent agent with HPMC (Methocel E15LV) and plasticized with polyethylene glycol 6000 (PEG 6000 SD fines, India) as a binder. Magnesium stearate was purchased from S.D. Fine-Chem Ltd, India, The polymeric layer used was polymethacrylates (Eudragit RL30D and RS30D, Rohm Pharma, Germany) plasticized with acetyl triethyl citrate. All other reagents and solvents used were of analytical grade procured from Merck (Mumbai, India).
Methods
Calculation of the Loading and Sustained Dose (Theoretical Release) Profile of RH
The total dose of RH for biphasic delivery was calculated with available pharmacokinetic data (12–13). As per the zero-order release principle, the rate of delivery must be independent of the amount of drug remaining in the dosage form and constant over time. The release from the dosage form should follow zero-order kinetics, as shown by the equation: 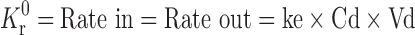 . Where Kr0 is the zero-order rate constant for drug release (amount per time), ke is the first-order rate constant of overall drug elimination (per hour), Cd is the desired drug level in the body (amount per volume), and Vd is the volume in which the drug is distributed. The elimination half-life of ranitidine is 3.5 h (ke = 0.693/3.5 = 0.198 h−1).
. Where Kr0 is the zero-order rate constant for drug release (amount per time), ke is the first-order rate constant of overall drug elimination (per hour), Cd is the desired drug level in the body (amount per volume), and Vd is the volume in which the drug is distributed. The elimination half-life of ranitidine is 3.5 h (ke = 0.693/3.5 = 0.198 h−1).
The loading dose is required to give initial rapid burst of dose so as to attain therapeutic range immediately after dosing.
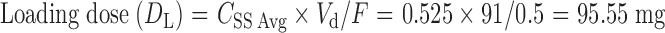 |
DL is the loading dose, CSS Avg is the average steady-state plasma level, Vd is the apparent volume of distribution, and F is the fraction of dose absorbed.
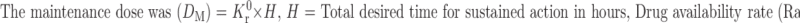 |
Ke is the overall elimination rate constant (per hour), Kr0 = 0.198 × 95.55 = 18.92 mg/h. Thus Kr0 (Rate of drug input) is 18.92 mg/h, which should also have been equal to the elimination constant so as to maintain the steady-state condition. For a system in which the maintenance dose releases drug by a zero-order process for a specified period of time, the total dose is as follows:
 |
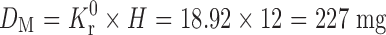 |
If the maintenance dose begins the release of the drug at the time of dosing, it will add to that which is provided by the initial dose, thus increasing the initial drug level. In this case, a correction factor is needed (Kr0 × Tp) to account for the added drug from the maintenance dose. This correction factor is the amount of drug provided during the period from t = 0 to the time of the peak drug level, Tp. If DL is 95.55 mg, Kr0 is 18.92 mg/h, and Tp is 2 h, then the corrected loading dose (DL*) = DL − (Kr0 × Tp) = 57.73 mg.
Thus, the total dose required (DT) = DL* + DM = 57.73 + 227 = 284.73 mg.
The total dose of ranitidine rounded off to 300 mg (which is equivalent to 336 mg of ranitidine hydrochloride, with 67 mg loading dose and 269 mg maintenance dose). Thus, the biphasic GRDDS which has loading dose for initial burst release and floating multiple units for maintenance dose was formulated.
Preparation of Biphasic Gastroretentive Drug Delivery System
The biphasic GRDDS consists of floating loading dose component (FLDC) and floating prolonged-release component (FPRC).
Preparation of FLDC
FLDC was prepared by direct compression method. All formulation ingredients (RH—33.5%, MCC—49.5%, croscaramellose sodium—5%, and sodium bicarbonate—10%) were sifted through # 40 mesh and homogeneously blended in blender for 5 min, then magnesium stearate—2% was (sifted through # 60 mesh) added to the above blend and mixed for 3 min in double cone blender (VJ Instruments, Maharashtra, India). The final blend was compressed into mini-tablets using 6-mm-size concave punches and corresponding dies on rotary compression machine (Riddhi, India).
Preparation of FPRC
The core mini-tablets contained RH—44.8%, MCC—16.5%, HPMC K100—25%, and HPMC K4M—12% as release rate controlling agents were blended same as method mentioned above. Then, magnesium stearate—2% was (sifted through # 60 mesh) added to above blend and mixed for 3 min in double cone blender. The final blend was compressed into core mini-tablets using 6-mm-size concave punches and corresponding dies on rotary compression machine (Riddhi, India).
Physical Properties of the Final Blend and Core Units
Physical properties such as bulk density, tapped density, compressibility index, Hausner ratio, and the angle of repose of final blend were determined; tapped density was determined by using a tapped density tester (Campbell, India). Percent compressibility and Hausner ratio were calculated using Eqs. 1 and 2:
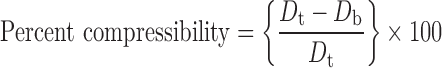 |
1 |
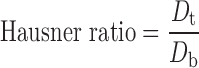 |
2 |
where Dt and Db are tapped and bulk densities.
Compressed mini-tablets were characterized for weight variation and thickness (n = 20) using analytical balance and digital micrometer (Mitutoyo, Japan). Crushing strength (n = 6) was measured with Monsanto tester, friability (n = 6), with (Roche type friabilator, at 25 rpm for 100 revolutions) and drug content.
Coating of the Core Prolonged-Release Component
The core units were coated with three successive layers; first with seal coat, effervescent layer (sodium bicarbonate), and then with polymethacrylate (Eudragit RS30D, RL30D, and RS30D:RL30D) as an outer polymeric coating layer. For coating of seal coat HPMC solution plasticized with PEG 6000 (10%, w/w based on the solids content) and then layered onto the core units. An effervescent agent was incorporated into HPMC solution plasticized with PEG 6000 (10%, w/w based on the solids content) and then layered onto the seal coated units. On a dry solid basis, the ratio of HPMC to sodium bicarbonate was 2:6 w/w. The coating level of effervescent layer was 12% (optimized) weight gain, and the solids content of coating solution was kept constant at 8% (w/w).
The coating solution was sprayed onto the core units in a coating pan (Allegro, India). The conditions for coating were shown as follows: tablets charge 100 g, preheating temperature 40 ± 5°C, preheating time 20 min, inlet air temperature 50 ± 5°C, spray rate 8–10 ml/min. Seal-coated and then effervescent-layered units were dried in the coating chamber for 30 and 60 min, respectively, at 40 ± 5°C. The prepared units were then removed from the coating pan and stored in a closed container for further processing. The effervescent-layered units were subsequently coated with polymethacrylates dispersions (Eudragit RS30D, RL30D, or RS30D:RL30D) to achieve a weight gain of 5% to 10% (w/w) to obtain the complete multiple-unit FDDS. A plasticizer (acetyl triethyl citrate 20% w/w based on polymer solids content) was added into the polymer dispersion, then the whole dispersion was stirred throughout the coating process. The solids content of the coating dispersions was 10% (w/w). The coating conditions were as follows: tablets charge 100 g, preheating temperature 40 ± 5°C, preheating time 20 min, inlet air temperature 45 ± 5°C, spray rate 3–5 ml/min. The units were further dried in the coating chamber for 60 min after the coating was finished to evaporate the residual moisture. The prepared units were then removed from the coating chamber and stored in a closed container for further experiments.
The biphasic GRDDS comprising of FLDC and FPRC equivalent to 336 mg of RH were placed in hard gelatin capsules and evaluated.
Scanning Electron Microscopy
The core, effervescent-layered and final coated mini-tabs were mounted onto the stages after coating with gold under vacuum. The surface morphology for checking the uniform coating of the units was observed under scanning electron microscopy (SEM).
Floating Behavior
The floating abilities of the effervescent-layered units and the final coated effervescent-layered units (complete multiple-unit FDDS) were determined using USP II apparatus (50 rpm, 37 ± 0.5°C, 900 ml, pH 1.2, enzyme free). Units were placed in the medium; the time required to float was measured by visual observation.
In Vitro Dissolution
The release rate of drug from mini-tablets filled into capsules was determined (n = 3) using USP Apparatus 2 (paddle method). The dissolution test was performed using 900 ml of 0.1 N HCl at 37 ± 0.5°C and 50 rpm. A sample (5 ml) of the solution was withdrawn from the dissolution apparatus hourly for 12 h, and the samples were replaced with fresh dissolution medium. The samples were filtered through a 0.45-μm membrane filter and diluted to a suitable concentration with 0.1 N HCl. Absorbance of these solutions was measured at 317 nm using a UV/Vis double-beam spectrophotometer (Elico India). Cumulative percentage drug release was calculated using an equation obtained from a standard curve.
Kinetic and Mechanism of Drug Release
Dissolution data of the optimized formulation were fitted to various mathematical models (14) (Zero-order, First-order, Higuchi, and Korsmeyer–Peppas model).
Stability Studies
The formulation was kept in the humidity chamber (LabTop, India) and maintained at 25°C/60% relative humidity and 40°C/75% relative humidity for 3 months. At the end of the studies, samples were analyzed for physicochemical parameters. For the comparison of release profiles of initial and stability samples, “difference factor” f1 and “similarity factor” f2, were calculated (15). The difference factor (f1) measures the percent error between the two curves over all time points and was calculated as follows
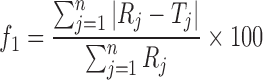 |
3 |
where n is the number of sampling points, and Rj and Tj are the percent dissolved of the reference and test products at each time point j. The two release profiles are considered to be similar if f1 value is lower than 15 (between 0 and 15). The similarity factor (f2) is a logarithmic transformation of the sum of squared error of differences between the test Tj and the reference products Rj over all time points. It was calculated using the following equation
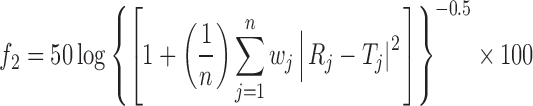 |
4 |
where wj is an optional weight factor and other terms are as defined earlier. The two dissolution profiles are considered to be similar if f2 value is more than 50 (between 50 and 100).
In Vivo X-ray Studies
The in vivo tests discussed below were performed on six healthy male volunteers whose ages were between 25 and 32 years and weighed between 60 and 71 kg (approval for the study was taken from University Ethical Committee, UCPSc, Kakatiya University, Warangal, AP, India); 18% of BaSO4 was added to the part of the final formulation (the amount of BaSO4 that allows visibility by X-ray, but does not preclude the floating of tablets, was experimentally determined). Labeled Floating MUDF (Placebo) was given to subjects with 250 ml of water after a light, 308 kcal breakfast. Following ingestion, gastric radiography was undertaken at 0.5, 2, 3, and 5 h, and the duration of the mini-tablets stayed in the stomach was observed.
RESULTS AND DISCUSSION
To maintain constant plasma concentration of RH makes it a candidate for incorporation in a sustained-release form. In order to develop an optimized sustained-release RH dosage form, we tested floating MUDF systems comprising different uncoated and coated mini-tablets in a gelatin capsule.
In FLDC, microcrystalline cellulose was used because of its good compaction properties. Croscarmellose sodium was used as a super disintegrant to obtain an immediate release of the drug and sodium bicarbonate to liberate carbon dioxide upon contact with acidic medium.
In order to delay the drug release, corresponding to the prolonged-release component of the biphasic system, HPMC was used as matricial agents to control release of the drug from the mini-tablets. In matricial systems, the characteristics of the matrix forming agent play an important role in the release mechanism(s) of the drug. Among the hydrophilic polymers, HPMC is one of the most commonly used carriers for the preparation of oral controlled drug delivery systems due to its ability to swell upon jellification once in contact with medium. The gel becomes a viscous layer acting as a protective barrier to both the influx of water and the efflux of the drug in solution (16,17).
The final blend of optimized batch showed good flowability (angle of repose 28°) and compressibility 17%, Hausner ratio 1.2, and friability (percent) of core mini-tablets was 0.5%.
Figure 1 shows the design of biphasic Multiple-Unit GRDDS. The (FPRC) system consisted of drug-containing core mini-tabs coated with seal coat (to prevent direct contact of core with effervescent layer), effervescent layer, and polymeric membrane, respectively. Since sodium bicarbonate itself could not adhere to the units, HPMC was used as a binder in the effervescent layer. An ideal coating material for a floating system should be highly water permeable in order to initiate the effervescent reaction (upon contact with acidic medium, sodium bicarbonate present in the second layer will react with acid and liberate CO2 by neutralization reaction) and the floating process rapidly. However, the wet or hydrated coatings should also be impermeable to the generated CO2 so as to promote and maintain floatation (18). Regarding their mechanical properties, the polymeric coatings should be sufficiently flexible in wet state to be able to withstand the pressure of the generated gas and to avoid rupturing. Krogel and Bodmeier reported that the cellulosic polymers were not suitable candidates for FDDS. Cellulose acetate was too rigid and did not expand sufficiently when in contact with dissolution media, while ethyl cellulose was not flexible and ruptured easily upon CO2 formation. Gas bubbles were released rapidly after the burst of coating. According to these reasons, the higher flexibility polymer, polymethacrylates (Eudragit RS30D, RL30D, and RS30D:RL30D) were chosen and investigated as a polymeric layer in this study. Upon contact with the gastric fluid, the fluid permeated into the effervescent layer through the outer polymeric coating layer. Carbon dioxide was liberated via neutralization reaction and was entrapped in the polymeric layer. After that, the swollen mini-tabs with a density less than 1.0 g/ml floated and maintained the buoyancy; therefore, the drug was released from the system for a long time. To develop the multi-unit FDDS based on gas formation technique, several studies were necessary to identify the formulation variables providing the desired system properties, rapid expansion, and formation of low-density system within minutes after contact with gastric fluids and maintaining the buoyancy in stomach with controlled release. The effect of the preparative parameters such as amount of the effervescent agent layered onto the seal coated mini-tabs, type and coating level of the polymeric layer on the floating ability, and drug release of the multiple-unit FDDS were evaluated.
Fig. 1.

Design of biphasic floating GRDDS
Figure 2a shows the appearance of the external morphology of the core units under SEM. The core units were with a slightly rough surface. The surface of the effervescent-layered units was slightly smoother (Fig. 2b), and the smoothest was the surface of effervescent-layered units coated with polymeric membrane (Eudragit RL30D/RS30D; Fig. 2c).
Fig. 2.

SEM of a core units, b effervescent-layered units, and c complete final coated units
The floating ability of the effervescent-layered units and the effervescent-layered units coated with polymeric membrane (complete multiple-unit FDDS) were investigated with respect to the amount of the effervescent agent coated and type and level of the polymeric coating (Fig. 3). The system should float within a few minutes after contact with gastric fluid to prevent the dosage form from transiting into the small intestine together with food (19). The percent coating level of effervescent layer was evaluated and found that about 12% of effervescent layer is required for floating the units within minutes. The effervescent-layered units floated within 10 s after being placed in acidic media. The floating time of the effervescent-layered units was quite short (less than 3 h) because HPMC dissolved and there was no polymeric layer which could entrap the generated CO2 gas. Therefore, the complete multiple-unit FDDS (effervescent-layered units coated with polymeric membrane) was prepared and evaluated for floating ability. Eudragit RL30D, RS30D, and in combination were used as polymeric layer. The multiple-unit FDDS using Eudragit RL30D and Eudragit RS:RL30D as polymeric layer floated completely within 4 min. The floating lag time was found to be in the order of the following: effervescent-layered < RL—5% < RS:RL (1:3)—5% < RS:RL (1:3)—7.5% < RS:RL(1:3)—10%. The time to float of the systems decreased with increasing amount of effervescent agent and increased with increasing level of polymeric coating layer. The higher amount of effervescent agent caused faster and higher CO2 generation (18). With increasing level of coating, the floating started later due to the delayed water penetration through the thicker coating. The duration of floating was longer than 12 h. It was indicated that Eudragit RL30D and RS:RL combination of polymeric layer was impermeable to the generated CO2 and could maintain the floatation. The multiple-unit FDDS coated with Eudragit RS30D as polymeric layer did not float within 20 min even used high effervescent coating level (15% w/w weight gain). Eudragit RS30D might not be permeable enough for dissolution medium to induce the effervescent reaction and generate sufficient amount of CO2 to make the units float. Eudragit RL30D is a highly water-permeable polymer according to its hydrophilic content, quaternary ammonium groups in the structure (20–21). It has twice as many quaternary ammonium groups and is more hydrophilic than Eudragit RS. A faster and higher CO2 generation caused by increasing the level of effervescent resulted in higher swelling of polymeric layer and subsequent floating. It is therefore hydrated faster and resulted in a shorter time to float (18). Based on these results, Eudragit RL30D and combination of RS:RL30D were the polymers of choice as outer polymeric coating layer in this multiple-unit FDDS.
Fig. 3.

Effect of polymer coatings on floating lag time at 12% of effervescent coating level
The release of RH from the biphasic floating MUDF coated with Eudragit RL30D and RS:RL30D as polymeric layer was shown in Fig. 4. The effects of polymer type and coating level on drug release were investigated. Since only the multiple-unit FDDS using Eudragit RL30D and combination of RS:RL as an outer polymeric layer coated mini-tablets could float, the drug release of this system was investigated for further study. The drug release slightly decreased with increasing level of polymeric coating from 5% to 10%. The higher membrane thickness retarded water penetration, resulting in decreasing drug release (18,22). The drug release from the system using Eudragit RL30D and RS:RL combinations as an outer polymeric coating layer was found to be linear with time.
Fig. 4.

Cumulative percentage of RH released from formulations versus time (n = 3)
The correlation coefficient (r2) was used as an indicator of the best fitting for the models considered. Some release mechanisms can be better elucidated indirectly, on basis of exponent n. The results (Table I) reveal that all formulations were best fitted in the Higuchi model. The mechanism of drug release from these mini-tablets was found to be diffusion-controlled as seen from r2 values of Higuchi model. The n values for these systems were in the range of 0.57–0.63, which can be regarded as indicators of both phenomena (transport corresponding to coupled drug diffusion in the hydrated matrix and polymer relaxation) commonly called anomalous non-Fickian transport.
Table I.
The Correlation Coefficient (r 2) Values for Different Formulations
| Release models | r 2 | |||
|---|---|---|---|---|
| RL 5% | RS:RL (1:3) 5% | RS:RL (1:3) 7.5% | RS:RL (1:3) 10% | |
| First order | 0.9425 | 0.9383 | 0.9618 | 0.9456 |
| Zero order | 0.9861 | 0.9867 | 0.994 | 0.9803 |
| Higuchi | 0.9914 | 0.9883 | 0.9893 | 0.9717 |
The analysis of the parameter dissolution data, after storage at 40°C/75% RH and 25°C/60% RH for 3 months was shown in Fig. 5. There is no statistically significant difference between the initial and stability samples (p > 0.05), indicating that the two dissolution profiles are considered to be similar (f2 value is 75, 83 and f1 value is 5, 3, respectively).
Fig. 5.

Dissolution profiles of a initial and after storage at b 40°C and 75% RH, c 25°C and 60% RH for 3 months
Following the ingestion of the final formulation prepared by the addition of BaSO4 to the release layer, the gastric residence time of mini-tablets was examined by radiogram, and it was observed that the units remained in the stomach for about 5 h (Fig. 6).
Fig. 6.

In vivo gastric residence time of floating MUDF by X-ray studies a 0.5, b 2, c 3, and d 5 h
CONCLUSION
Ideally, a sustained-release formulation should release the required quantity of drug with predetermined kinetics in order to maintain an effective drug plasma concentration. To achieve this, the delivery system should be formulated so that it releases the drug in a predetermined and reproducible manner. By considering the drug’s biopharmaceutical and pharmacokinetic profile, one can determine the required release from the formulation. The biphasic GRDDS remains buoyant in drug release media with disintegration of FLDC and prolonged release of FPRC. There was an initial burst release of RH from the gastroretentive system, and release of RH from FPRC was sustained over 12 h compared with conventional commercial tablets, which released complete drug within 30 min. It was found that the initial burst release of 20% during the first hour is in accordance with loading dose requirement. The results for fitting of the kinetic models for RH release reveal that all formulations of GRDDS best fits in the zero-order model. The mechanism of drug release from these tablets was found to be diffusion as seen from r2 values of Higuchi model.
In the selection of optimized GRDDS formulation along with buoyancy characteristics, the dissolution profiles of GRDDS were compared with the theoretical release profile as there was no standard reference sustained-release product of RH available in the market. The similarity factor (f2) of all GRDDS ranged from 63 to 74.
When compared with the theoretical release profile, the similarity factor (f2) of formulation with coating of RS:RL (1:3)—7.5% was observed to be 74, which is well fitted into zero-order kinetics, confirming that the release from formulation is close to desired release. The in vivo gastric residence time was examined by radiograms, and it was observed that the units remained in the stomach for about 5 h. Further in vivo study has to be carried out in healthy human volunteers to access the bioavailability of drug.
Acknowledgments
The authors would like to thank Dr Reddy’s Laboratories (Hyderabad, India) for providing the gift sample of ranitidine hydrochloride, and one of the authors (Meka Lingam) is thankful to the UGC India for providing Junior Research Fellowship.
References
- 1.Shah A. C. Design of oral sustained release drug delivery systems: in vitro/in vivo Considerations. In: Yacobi A., Halperin-Walega E., editors. Oral Sustained Release Formulation Design and Evaluation. New York: Pergamon; 1988. pp. 35–56. [Google Scholar]
- 2.Rubinstein A., Li V. H. K., Gruber P., Robinson J. R. Gastrointestinal-physiological variables affecting the performance of oral sustained release dosage forms. In: Yacobi A., Halperin-Walega E., editors. Oral Sustained Release Formulation Design and Evaluation. New York: Pergamon; 1988. pp. 125–156. [Google Scholar]
- 3.Mayersohn M. Principles of drug absorption. In: Banker G. C., Rhodes C. T., editors. Modern Pharmaceutics. New York: Marcel Dekker; 1979. pp. 23–89. [Google Scholar]
- 4.Gupta P. K., Robinson J. R. Oral controlled-release delivery. In: Kydonieus A., editor. Treatise on Controlled Drug Delivery. New York: Marcel Dekker; 1992. pp. 255–313. [Google Scholar]
- 5.Rouge N., Cole E. T., Doelker E., Buri P. Screening of potentially floating excipients for Minitablets. S.T.P. Pharm. Sci. 1997;7:386–392. [Google Scholar]
- 6.Maggi L., Machiste E. O., Torre M. L., Conte U. Formulation of biphasic release tablets containing slightly soluble drugs. Eur. J. Pharm. Biopharm. 1999;48:37–42. doi: 10.1016/S0939-6411(99)00019-3. [DOI] [PubMed] [Google Scholar]
- 7.Somade S., Singh K. Comparative evaluation of wet granulation and direct compression methods for preparation of controlled release ranitidine HCL tablets. Indian J. Pharm. Sci. 2002;64:285. [Google Scholar]
- 8.Lauritsen K. Clinical pharmacokinetics of drugs used in the treatment of gastrointestinal diseases. Clin. Pharmacokinet. 1990;19(11–31):94–125. doi: 10.2165/00003088-199019020-00002. [DOI] [PubMed] [Google Scholar]
- 9.Grant S. Ranitidine: an updated review of its pharmacodynamics and pharmacokinetic properties and therapeutic use in peptic ulcer and other allied diseases. Drugs. 1989;37:801–870. doi: 10.2165/00003495-198937060-00003. [DOI] [PubMed] [Google Scholar]
- 10.Basit A., Lacey L. Colonic metabolism of ranitidine: implications for its delivery and absorption. Int. J. Pharm. 2001;227:157–165. doi: 10.1016/S0378-5173(01)00794-3. [DOI] [PubMed] [Google Scholar]
- 11.M. Coffin, and A. Parr, inventors. Glaxo Inc. Ranitidine solid dosage form. US patent 5 407 687. April 18, 1995.
- 12.J. G. Wagner. Biopharmaceutics and pharmacokinetics. 1st ed. Org. Intelligence publications, 22, 1971, pp. 148–157.
- 13.A. M. Hamid, M. S. Harris, T. Jaweria, and I. Y. Rabia. Once-daily tablet formulation and in vitro release evaluation of cefpodoxime using hydroxypropyl methylcellulose: a technical note. AAPS PharmSciTech.7:(3) Article 78 (2006). [DOI] [PMC free article] [PubMed]
- 14.Costa P., Lobo J. M. S. Modeling and comparison dissolution profiles. Int. J. Pharm. 2001;13:123–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 15.Moore J. W., Flanner H. H. Mathematical comparison of curves with an emphasis on in- vitro dissolution profiles. Pharm. Tech. 1996;20:64–74. [Google Scholar]
- 16.Colombo P., Bettini R., Santi P., Peppas N. A. Swellable matrices for controlled drug delivery: gel-layer behaviour, mechanisms and optimal performance. PSTT. 2000;3:198–204. doi: 10.1016/s1461-5347(00)00269-8. [DOI] [PubMed] [Google Scholar]
- 17.Kiil S., Dam-Johansen K. Controlled drug delivery from swellable Hydroxyl propyl methylcellulose matrices: model-based analysis of observed radial front movements. J. Contr. Release. 2003;90:1–21. doi: 10.1016/S0168-3659(03)00122-6. [DOI] [PubMed] [Google Scholar]
- 18.Krögel I., Bodmeier R. Floating or pulsatile drug delivery systems based on coated effervescent cores. Int. J. Pharm. 1999;187:175–184. doi: 10.1016/S0378-5173(99)00189-1. [DOI] [PubMed] [Google Scholar]
- 19.Iannuccelli V., Coppi G., Bernabei M. T., Cameroni R. Air compartment multipleunit system for prolonged gastric residence. Part I. Formulation study. Int. J. Pharm. 1998;174:47–54. doi: 10.1016/S0378-5173(98)00229-4. [DOI] [Google Scholar]
- 20.I. Ghebre-Sellassie, R. U. Nesbitt, and J. Wang. Eudragit aqueous dispersions as pharmaceutical controlled release coatings. In J. W. McGinity (ed.), Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms, 2nd ed., Marcel Dekker, New York, pp. 267–286.
- 21.Bauer K. H., Lehmann K., Osterwald H. P., Rothgang G. Coated Pharmaceutical Dosage Forms. Stuttgart: Medpharm Scientific; 1998. pp. 63–119. [Google Scholar]
- 22.Ichigawa M., Watanabe S., Miyake Y. A new multiple-unit oral floating dosage system. I. Preparation and in vitro evaluation of floating and sustained release characteristics. J. Pharm. Sci. 1991;80:1062–1066. doi: 10.1002/jps.2600801113. [DOI] [PubMed] [Google Scholar]