

Abstract
The purpose of this research was to demonstrate the ability of reflectance near-infrared (NIR) spectroscopy for quantitative analysis of an active ingredient in different production steps of a solid formulation. The drug is quantified at two different steps of a pharmaceutical process: after granulation and after tablet coating. Calibration samples were prepared by mixing pure drug, excipients, and batch samples (75–120 mg/g active ingredient) using a simple methodology that can be easily carried out in a laboratory. Partial least squares calibration models were calculated in second-derivative mode using the wavelength range 1,134–1,798 nm. The error of prediction for granulated samples was 1.01% and 1.63% for tablets. The results prove that NIR spectroscopy is a good alternative to other, more time-consuming means of analysis for pharmaceutical process monitoring.
Key words: API determination, NIR spectroscopy, PAT, PLS calibration, tablet content uniformity
INTRODUCTION
Quality control constitutes an essential element of pharmaceutical production processes by virtue of the high safety levels demanded from commercially available formulations. Assuring quality in such formulations entails subjecting the end products to a wide variety of tests; this has an adverse impact on production expeditiousness and commercial competitiveness. In response to this problem, the FDA PAT Guideline (1) established a new working philosophy aimed at replacing end-product controls with monitoring of the whole production process in order to identify any weaknesses with a view to facilitating the correction and assuring quality in the target product by acting throughout the process. One major objective of this initiative was to improve the quality of end products while increasing the efficiency of the production processes; in this way, quality would be built across the process. The best process knowledge, the higher probability to improve the quality of the product.
Fulfilling this objective requires a deep knowledge of the effect of some variables on the process and such knowledge can be acquired by determining the critical parameters and monitoring the changes in order to correct any undesirable deviations.
Implementing the new working philosophy requires the development and use of simple, rapid analytical techniques to expeditiously acquire the information needed (2). Spectroscopic techniques are especially amenable to this use. This is especially so with near-infrared spectroscopy (NIRS). In fact, the NIRS technique has been widely used with success in this context, as reflected by the large number of reported applications in the pharmaceutical industry (3–6), where its flexibility has enabled the obtainment of analytical information for untreated samples of any type with minimal human intervention.
Quantifying the active principle (API) in different steps of a production process and its end product usually helps assess the status of the process and confirm that a given production batch is fit for release. API quantitation usually relies on high performance liquid chromatography (7,8), which uses time-consuming protocols. This has led to its increasing replacement with NIR spectroscopy in recent years, where this technique has proved a useful tool for the simultaneous determination of physical and chemical properties of samples in a clean, expeditious, and non-destructive manner in various steps of the production process (9–11).
Wet granulation is a basic operation intended to improve the flowability of pharmaceutical mixtures because of an increase in the mean particle size. In addition, the granulation process is a common step where the end product is released in form of tablets or granulated mixtures.
Determinations by NIR spectroscopy require the use of multivariate calibration models encompassing all potential sources of variability during routine analysis. Because the end product invariably spans too narrow a concentration range, the models must be constructed from additional, laboratory samples of variable enough concentrations to ensure the required accuracy, precision, and robustness (11). One easy way of expanding the API content range is by using underdosed and overdosed samples prepared by adding small amounts of the excipients and API. Scheiwe et al. (12) determined the API content in tablets by using Transmission mode. This work presented a successful strategy of calibration and routine API content monitoring; however, the experimental approach involved a great effort of sample preparation in a pilot plant installation. The present work shows an easy and novel strategy to prepare the calibration samples in the laboratory for the development of calibration models to analyze intact tablets by NIR spectroscopy. This strategy simplifies the sample preparation if different calibration models need to be calculated for individual manufacturing step. Basically, a few number of each manufacturing step is included in the calibration sample set to include the process variability in the multivariate model.
Altering the amounts of API and excipients in the production process to construct a calibration set with a wide range of API represents a high cost for the company. In the present work, production tablets at nominal contents were milled into powder samples. Once these samples were uniformed milled, API and excipients were added obtaining a calibration set including the chemical variability required to correctly predict production samples. This strategy supposes a great diminution in time and cost.
In this work, two PLS calibration models were redeveloped in order to quantify the API in the pharmaceutical product after two different steps of the production process, namely: wet granulation of the powdered components and tableting and coating this granulate. The first calibration model provides significant information about the conformity parameters of the obtained granulated mixture to be used in the next steps of the process. The second calibration model allows monitoring the API content from the obtained coated tablets.
Thus, the calibration models obtained were applied to various steps of the production process and the two ensuing quantitation methods were subsequently validated in accordance with the ICH and EMEA guidelines (13,14).
MATERIALS AND METHODS
Pharmaceutical Formulation
The formulation studied consisted of tablets containing 135 mg/g dexketoprofen trometanol as API; PH101 microcrystalline cellulose (500 mg/g) as major excipient; and maize starch, carboxymethyl starch, and glycerol distearate as minor excipients. The tablets were coated with a thin film by spraying a solution consisting of OPADRY lacquer (85%) and propylene glycol (15%) diluted in purified water, and finally dried.
Although dexketoprofen (DKP) was present as its trometamol salt, the reference values were expressed as DKP (1 g of DKP in acid form is equivalent to 1.476 g of DKP trometamol). The nominal content in DKP of the production samples was 93 mg/g.
All studied samples were supplied by Laboratorios Menarini, S.A. (Badalona, Spain).
Production of the Pharmaceutical
The production process of the tablets involves several steps including joint granulation of the API and some excipients, addition of the other ingredients, blending, pressing, and coating. The tablets are cylindrical in shape and weigh 270 mg on average.
In the first granulation step, a mixture of DKP, cellulose, and starch is granulated in a fluid bed reactor that is supplied with a binder solution and then dried; the average duration of this step is 270 min. The resulting granulate is then homogeneously blended with the other excipients (carboxymethyl starch and glycerol distearate) in the second step. Next, the granulated product is pressed and homogenized to obtain cores (non-coated tablets). In the fourth, final step, the tablets are coated with a film of lacquer.
Two calibration models to quantify the API after blending of the granulate and excipients, and also after coating of the tablets (i.e., after the second and fourth steps), were constructed.
Samples
Calibration models were constructed by using sets of samples obtained from different batches depending on the particular step of the production process.
Production Samples
The production samples were coated tablets from 20 different production batches obtained over a period of 6 months. The calibration set for tablets was constructed from intact coated tablets. On the other hand, the model for the granules (end of the second step) was constructed from tablets that were previously milled and underdosed or overdosed to expand the concentration range.
All samples were additionally analyzed by using a UV–Vis reference method developed and validated by Laboratorios Menarini, S.A.
Underdosed and Overdosed Samples
Commercial samples invariably span a narrow API concentration range (around ±5% of the label claim), so these samples are unsuitable for constructing calibration models. Therefore, the API content range must be expanded by using additional laboratory samples prepared by underdosing and overdosing milled production tablets.
The samples used to construct the two calibration models were milled tablets from production batches and doped (underdosed and overdosed) samples spanning the DKP concentration range 75–120 mg/g.
Regardless of introducing physical variability in the calibration set (particle size), milling production tablets in the laboratory allows expanding the API range. One alternative strategy could be realized in the granulation process in the laboratory, but the time consumption is higher and, moreover, this manual granulation can provide some variability not included in the production process.
Overdosed samples were prepared from powdered production tablets that were supplied with accurately weighed amounts of API. Underdosed samples were also prepared from powdered production tablets which, however, were supplied with known amounts of a mixture of the excipients in the same proportions as in the pharmaceutical preparation.
Both types of samples were mixed in a Turbula shaker until the NIR spectra exhibited no appreciable changes between consecutive measurements.
Recording of NIR Spectra
Spectra were recorded on a model 5000 spectrophotometer from Foss NIRSystems (Silver Spring, MD, USA) equipped with a rapid content analyzer module and governed via the software Vision v. 2.51. A ceramic plate was used to obtain a reference spectrum prior to measuring each sample. All spectra were the averages of 32 scans performed at 2-nm intervals over the range 1,100–2,498 nm.
Aliquots of production and doped granulated samples were placed in a quartz cell and the reflectance spectra were recorded in triplicate with turnover between recordings. The resulting average spectra were used to construct and validate the models, and also to quantify the API in the pharmaceutical preparation.
The spectra for coated production tablets were obtained for both sides by placing each tablet on the quartz window of the recording module. The spectra for the two sides were averaged for subsequent processing.
Processing of NIR Spectra
PLS1 models were based on various sample sets to determine the API in two steps of the production process. To this end, the software Unscrambler v. 9.2 from CAMO Process AS (Oslo, Norway) has been used.
The spectral pretreatments used to construct the calibration models included standard normal variate and first and second derivatives. Derivative spectra were obtained by using the Savitzky–Golay algorithm with an 11-point moving window and a second order polynomial. The models were obtained by cross-validation and the optimum number of factors was taken to be that resulting in the minimum PRESS value (Eq. 1) and the quality of the resulting models was assessed in terms of the relative standard errors of calibration (% RSEC) and prediction (% RSEP) (Eq. 2):
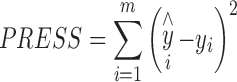 |
1 |
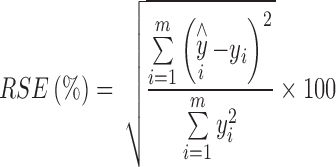 |
2 |
where 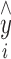 and yi correspond to the drug concentration of each sample predicted by the NIR method and reference method, respectively.
and yi correspond to the drug concentration of each sample predicted by the NIR method and reference method, respectively.
UV Reference Method
The API (dexketoprofen) contents of the production samples were determined by ultrasonicating an amount of ca. 0.25 g of milled sample in 90 ml of Milli-Q water at 37°C for 15 min. Then, an aliquot of 2 ml was diluted to 50 ml of a 3:1 MeOH/H2O mixture and its UV spectrum recorded. The dexketoprofen content was obtained by multiple linear regression of the 210–400 nm region of the spectrum, using that for pure API in MeOH/H2O as reference.
RESULTS AND DISCUSSION
We aimed at establishing simple calibration models with an adequate predictive ability for use in various steps of the production process with a view to facilitating the straightforward, expeditious determination of the dexketoprofen content of the product. To this end, two calibration models were constructed to be used after the granulation step (model 1) and for quality control of the end product (coated tablets) prior to its release (model 2). Both models were constructed from samples in two different forms, namely: milled coated tablets and intact coated tablets. Following validation, each calibration model was used to determine the API content in the specific step of the production process in order to check its appropriateness and allow the process to continue. Obtaining an accurate, precise, and robust calibration model requires using a calibration set consisting of samples encompassing all potential sources of physical and chemical variability in the samples to be subsequently determined.
Figure 1 shows the absorbance spectra for the API and the two types of samples studied (milled tablets, non-coated and coated tablets). As can be seen, the three spectra exhibited an identical profile (i.e., no difference in chemical composition between samples was apparent); however, the spectra were shifted by effect of differences in physical properties between samples. Thus, the tablets exhibited higher absorbance values by effect of the pressing; the higher the pressure used was, the higher was the absorbance (3). Because the sole difference between the two types of tablets was the presence of the lacquer coating—which was in a very low proportion and absorbed very slightly—in one, the spectra were essentially identical.
Fig. 1.

NIR spectra for coated and non-coated tables, granulated samples and the pure API
Analysis of Granulated Samples
Model 1 was constructed from milled coated tablets in addition to milled tablets underdosed and overdosed intended to expand the API content range in order to encompass the whole chemical variability of the production process. These samples were used in combination with various spectral ranges and treatments. Figure 2a shows the scatter plot of calibration samples (production samples with nominal content of API and over/subdosed samples covering the API range). PC2 was the factor responsible to the API variation. Production samples are grouped while dosed samples are expanded through the PC2. Table Ia shows the most salient parameters of the model samples and the predictions obtained for granulated production samples. Second-derivative spectra were those providing the best results. The model was constructed from 20 samples of which 13 were of the doped type and seven contained the nominal API level. These three PLS factors provided acceptable calibration statistics (99.1% Y-explained variance) and predictive ability (RSEP = 1.01%) with granulated production samples. As can be seen from Table I, the model exhibited a good predictive ability with granulated samples. Once this model was capable of correctly predicting samples at the end of the granulation process, coated and non-coated tablets were evaluated with this model to test the chance of using the same model at the end of the process.
Fig. 2.

a PCA scatter plot from absorbance spectra over 1,134–1,798 nm for dosed and production samples. b PCA scatter plot from absorbance spectra over 1,134–1,798 nm for coated and non-coated tablets, granulated samples, and dosed samples
Table I.
Figures of Merit of the Models for Tablets and Granulated Samples
| Calibration model | ||
|---|---|---|
| (a) Granulate | (b) Tablets | |
| Spectral pretreatment | 2nd der. S–G | 2nd der. S–G |
| Spectral range (nm) | 1,134–1,798 | 1,134–1,798 |
| No. of PLS factors | 3 | 3 |
| Y-explained variance % | 99.12 | 99.31 |
| Calibration set | ||
| Dosed and milled samples | 20 | 13 |
| Tablets | – | 5 |
| Concentration range (mg/g) | 75.6–120.6 | 75.6–120.6 |
| RSEC | 1.04 | 0.97 |
| Prediction set | ||
| Granulated samples | 20 | – |
| RSEP | 1.01 | – |
| Tablets | 28 | 28 |
| RSEP | 5.03 | 1.63 |
The model for granulated samples was used to analyze tablets, both cores and coated. As can be seen from Fig. 3a, the ensuing predictions were invariably overestimated and subject to an RSEP greater than 5%; this suggests that differences in compactness between samples resulted in substantial errors, so the model was unsuitable for tablets.
Fig. 3.

Residuals obtained in the prediction of tablets and granulated samples with models 1 a and 2 b
Analysis of Tablets
The NIR spectra of granulated samples used in the previous model and the spectra of coated and non-coated tablets were subjected to principal component analysis (PCA). Figure 2b shows the scatter plot for a PCA of second-derivative spectra obtained with the Savitzky–Golay algorithm. As can be seen, the first factor (PC1) essentially contained the physical differences between sample types exposed by Fig. 1. Based on the results of Fig. 2, PC4 was the factor that more clearly showed the chemical variability of the samples; thus, while the coated tablets—which contained virtually the same amount of API—lay in the center of the PC4 axis, the milled samples—which included underdosed and overdosed samples—lay in different positions along the axis depending on the particular API content.
Constructing a model capable of accurately predicting the API contents of intact tablets required using samples spanning a wide content range. The inability to alter coated tablets to this end led us to use the set of samples previously employed to expand the range for model 1. The model was constructed from 13 granulated samples spanning the API content and five intact coated tablets containing the nominal API level. This strategy allowed including both chemical variability and physical variability—a result of pressing the granulated samples, mainly—to be incorporated by using coated production tablets. Although the incorporation of coated tablets into the model introduced chemical variability in the coating due to the small amount of film coating on the tablets, its effect on the resulting spectra was minimal (see Fig. 1). In this figure, differences in the spectra of coated and non-coated tablets cannot be observed.
Table Ib shows the figures of merit of the PLS1 model as used to predict coated tablets at the end of the production process (model 2). The model was constructed by using various spectral ranges and treatments, and also different types of samples. The best choice was that based on the same parameters as the previous model, namely: three PLS factors, which sufficed to accurately predict production tablets.
The predictions for coated tablets had an RSEP of 1.63%, which is substantially lower than that provided by model 1; also, the residuals were randomly distributed around 0, so the model can be deemed accurate for predicting samples at the end of the process.
Using model 2 to analyze non-coated tablets (cores) provided predictions with an RSEP of 2.30%. The predictions were systematically overestimated (see Fig. 3b) for all batches, which can be ascribed to the absence of the lacquer coating.
Finally, model 2 was used to predict granulated samples, with which it gave an RSEP of 3.43%. Contrary to non-coated samples, all samples were underestimated (Fig. 3b).
Validation of the NIR Method for Determining the API
The two NIR models were validated prior to use in routine analyses of the intermediate granulate obtained in the production process and also of individual coated tablets—which were assessed for content uniformity. To this end, the validation guidelines of the International Conference on Harmonisation (ICH) and the European Agency for the Evaluation of Medicinal products (EMEA) were followed to assess the selectivity, linearity, accuracy, precision (as repeatability and intermediate precision), and robustness of the proposed method.
The selectivity was established by identifying the sample prior to quantifying the analyte. Identification was based on a previously built library of second-derivative spectra for the pharmaceutical and its individual ingredients recorded over the whole spectra range (1,100–2,498 nm); identification was deemed positive if the correlation coefficient exceeded the threshold (0.98). All production samples studied were positively identified and all pure components in the formulation were accurately discriminated, with correlation coefficients lower than the threshold.
By contrast, the model for tablets required distinguishing between coated and non-coated tablets; this was done by using a sub-library of second-derivative spectra obtained over the spectral range 1,100–2,188 nm. The discriminating criterion used was the Euclidian distance in the space of three PCs, with a threshold of 0.46. This sub-library allowed coated tablets to be accurately distinguished from non-coated tablets (cores).
Table II shows the most salient results for the other validation parameters of the granulated product and tablets as determined in accordance with the ICH and EMEA guidelines for NIR methods (12). The proposed NIR method met all requirements in both guidelines, which testifies to its usefulness as a routine analytical method for the pharmaceutical industry.
Table II.
Validation Parameters for the API Quantitation with the Models for Tablets and Granulated Samples
| Model | |||
|---|---|---|---|
| Granulate | Tablets | ||
| Compound | Correlation coefficient | ||
| Selectivity | Enantyum granulated | >0.99 | – |
| Coated tablets | – | 1 | |
| Uncoated tables | – | 0.99 | |
| Maize starch | 0.75 | 0.76 | |
| Microcrystalline cellulose | 0.90 | 0.86 | |
| Carboxymethyl starch | 0.79 | 0.70 | |
| Dexketoprofen | 0.31 | 0.30 | |
| Glycerol palmitoestearate | 0.24 | 0.33 | |
| Parameter | Results | ||
| Linearity | n | 20 | 8 |
| Concentration range (mg/g) | 75.6–120.6 | 74.1 – 112.5 | |
| Intercept | −0.02 ± 4.54 | 3.9 ± 10.6 | |
| Slope | 1.00 ± 0.05 | 0.96 ± 0.12 | |
| R | 0.995 | 0.993 | |
| Accuracy | n | 10 | 10 |
| Avg. difference (mg/g) | −0.33 | 0.17 | |
| SD | 1.87 | 1.00 | |
| t exp | 1.11 | 0.54 | |
| t crit | 2.23 | 2.26 | |
| Repeatability | Replicates | 6 | 6 |
| Mean NIR (mg/g) | 94.64 | 91.22 | |
| CV (%) | 0.39 | 0.21 | |
| Intermediate | Days (3) | F exp 13.7 | F exp 0.66 |
| F tab 19.00 | F tab 19.00 | ||
| Precision | Analysts (2) | F exp 0.07 | F exp 0.09 |
| F tab 18.51 | F tab 18.51 | ||
| CV (%) | 0.72 | 1.17 | |
| Robustness | n | 20 | 14 |
| t exp | 0.56 | 0.41 | |
| t crit | 2.09 | 2.16 | |
Once model 2 was validated, the determination of API concentration in coated tablets was assessed. The US Pharmacopoeia recommends individual analyses of ten tablets from the same batch and using the average value for the ten determinations in order to determine content uniformity in production batches.
While residuals of up to 15% in individual analyses are deemed acceptable, the average residual should not exceed 5% of the nominal value. Table III shows the results of API concentration in ten coated tablets in the same batch with the validated model 2.
Table III.
API Concentration in Coated Tablets as Determined with Model 2
| Reference value | Prediction (mg/g) | Residual | Residual % |
|---|---|---|---|
| 91.480 mg/g | 92.112 | −0.632 | −0.69 |
| 91.217 | 0.263 | 0.29 | |
| 92.154 | −0.674 | −0.74 | |
| 91.833 | −0.353 | −0.39 | |
| 89.906 | 1.574 | 1.72 | |
| 89.809 | 1.671 | 1.83 | |
| 89.897 | 1.583 | 1.73 | |
| 93.281 | −1.801 | −1.97 | |
| 92.129 | −0.649 | −0.71 | |
| 93.244 | −1.764 | −1.93 | |
| Mean value | 91.558 | −0.078 | −0.09 |
CONCLUSIONS
The use of NIR reflectance spectroscopy allows the API content of a pharmaceutical preparation to be controlled throughout the production process and batches to be checked for acceptability in this respect prior to commercial release. The two models were constructed by using a low number of samples prepared in the laboratory and provided accurate predictions (errors lower than 1.7%). The methodology used to construct the models can be extended to more complex processes involving a greater number of steps with little additional effort.
Acknowledgements
The authors are grateful to Spain’s MCyT for funding this research within the framework of Project CTQ2006-12923. Also, we wish to thank Laboratorios Menarini, S.A. (Badalona, Spain) for kindly supplying the studied samples.
M. Alcalà thanks the National Science Foundation Engineering Research Center for Structured Organic Particulate Systems for the postdoctoral grant funding.
References
- 1.Guidance for Industry. PAT—A framework for innovative pharmaceutical development, manufacturing, and quality Assurance. US Department of Health and Human Services, Food and Drug Administration, Center of Drug Evaluation (CDER), and Research, Center for Veterinary Medicine (CVM), Office of Regulatory Affairs (ORA), 2004.
- 2.Kueppers S., Haider M. Process analytical chemistry—future trends in industry. Anal. Bioanal. Chem. 2003;376:313–315. doi: 10.1007/s00216-003-1907-0. [DOI] [PubMed] [Google Scholar]
- 3.Chalus P., Roggoa Y., Walter S., Ulmschneider M. Near-infrared determination of active substance content in intact low-dosage tablets. Talanta. 2005;66:1294–1302. doi: 10.1016/j.talanta.2005.01.051. [DOI] [PubMed] [Google Scholar]
- 4.Blanco M., Alcala M. Content uniformity and tablet hardness testing of intact pharmaceutical tablets by near infrared spectroscopy A contribution to process analytical technologies. Anal. Chim. Acta. 2006;557:353–359. doi: 10.1016/j.aca.2005.09.070. [DOI] [Google Scholar]
- 5.Blanco M., Alcalá M., González J. M., Torras E. A process analytical technology approach based on near infrared spectroscopy: tablet hardness, content uniformity, and dissolution test measurements of intact tablets. J. Pharm. Sci. 2006;95:2137–2144. doi: 10.1002/jps.20653. [DOI] [PubMed] [Google Scholar]
- 6.C. Peroza, M. A. Santos, and R. J. Romañach. Quantitation of drug content in a low dosage formulation by transmission near infrared spectroscopy. AAPS Pharm. Sci, Tech. 7(1) (2006), Article 29. [DOI] [PMC free article] [PubMed]
- 7.Sajeev C., Jadhav P. R., RaviShankar D., Saha R. N. Determination of flurbiprofen in pharmaceutical formulations by UV spectrophotometry and liquid chromatography. Anal. Chim. Acta. 2002;463:207–217. doi: 10.1016/S0003-2670(02)00426-9. [DOI] [PubMed] [Google Scholar]
- 8.Abourashed A., Khan A. High-performance liquid chromatography determination of hydrastine and berberine in dietary supplements containing goldenseal. J. Pharm. Sci. 2001;90:7817–822. doi: 10.1002/jps.1035. [DOI] [PubMed] [Google Scholar]
- 9.Donoso M., Kildsig D. O., Ghaly E. S. Prediction of tablet hardness and porosity using near-infrared diffuse reflectance spectroscopy as a nondestructive method. Pharm. Dev. Tech. 2003;8:357–366. doi: 10.1081/PDT-120024689. [DOI] [PubMed] [Google Scholar]
- 10.Peinado A. C., van den Berg F., Blanco M., Bro R. Temperature-induced variation for NIR tensor-based calibration. Chemom. Intel. Lab. Sys. 2006;83:75–82. doi: 10.1016/j.chemolab.2006.01.006. [DOI] [Google Scholar]
- 11.Blanco M., Alcala M. Use of near-infrared spectroscopy for off-line measurements in the pharmaceutical industry. In: Bakeev K., editor. Process Analytical Technology. Oxford: Blackwell; 2005. pp. 375–376. [Google Scholar]
- 12.Scheiwe M. W., Schilling D., Aebi P. Near Infrared spectroscopy analysis of intact diclofenac coated tablets in transmission. Pharmazeutische Industrie. 1999;61:179–183. [Google Scholar]
- 13.ICH Q2B: Validation of Analytical procedures: Methodology, Consensus Guideline, International Conference on Harmonisation (ICH), 1998. http://www.bcg-usa.com/docs/ich/ICHQ2B.pdf
- 14.Note for guidance on the use of near infrared spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations, 2003. http://www.fda.gov/Cber/gdlns/cmprprot.pdf