

Abstract
The purpose of this research was to evaluate how the presence of oxygen can affect irradiation-induced degradation reactions of PEGd,lPLA and PEG-PLGA multiblock copolymers submitted to gamma irradiation and to investigate the radiolytic behavior of the polymers. PEGd,lPLA, PEG-PLGA, PLA, and PLGA were irradiated by using a 60Co irradiation source in air and under vacuum at 25 kGy total dose. Mw and Mn were evaluated by gel permeation chromatography. The stability study was carried out on three samples sets: (a) polymer samples irradiated and stored in air, (b) polymer samples irradiated and stored under vacuum, and (c) polymer samples irradiated under vacuum and stored in air. The thermal and radiolytic behavior was investigated by differential scanning calorimetry and electron paramagnetic resonance (EPR), respectively. Samples irradiated in air showed remarkable Mw and Mn reduction and Tg value reduction due to radiation-induced chain scission reactions. Higher stability was observed for samples irradiated and stored under vacuum. EPR spectra showed that the presence of PEG units in multiblock copolymer chains leads to: (a) decrease of the radiolytic yield of radicals and (b) decrease of the radical trapping efficiency and faster radical decay rates. It can be concluded that the presence of oxygen during the irradiation process and the storage phase significantly increases the entity of irradiation-induced damage.
Key words: ionizing radiation, multiblock copolymer, polymer stability, radicals, radiolysis mechanism
INTRODUCTION
Gamma irradiation has been widely accepted as an effective technique for sterilizing moisture and heat-sensitive polymers and consequently medical devices based on this kind of polymer (1). However, it has been known that this process can lead to remarkable alterations in the materials during, immediately after, or even days, weeks, or months after irradiations (2). Many fundamental physical and chemical polymers properties can be modified with radiations such as molecular weight, chain length, entanglement, polydispersity, branching, pendant functionality, and chain termination. Physical changes can include embrittlement, discoloration, odor generation, stiffening, softening, enhancement or reduction of chemical resistance, and an increase or decrease in melting (or glass transition) temperature (3).
The extent of these alterations varies from polymer to polymer and it depends on the chemical composition, the structure of polymer, the total radiation dose absorbed, and the rate at which the dose was deposited. The extent is also affected by the environment conditions under which the radiation treatment was carried out, and the post-radiation storage environment (4–9). The environmental conditions under which radiation process is conducted can significantly affect the impact of radiations on the polymer material. The presence of oxygen or air during irradiation produces free radicals that are often rapidly converted to peroxidic radicals. The fate of these radicals depends on the nature of the irradiated polymer, the presence of additives, and other parameters such as temperature, total dose, dose rate, and sample size (2,4,10,11).
In most cases, the polymer radicals generated by gamma irradiation transform into oxidized moieties if oxygen is present in proximity of formed radicals, or remain trapped in the polymer matrix for a certain period of time after irradiation. These trapped radicals may further undergo some reactions during storage time after irradiation, resulting in significant alteration of the physical properties of the irradiated polymer. It is highly probable that post-irradiation effects appear during storage time and they can deeply influence the in vivo polymer performance. Understanding the effects of sterilization method and shelf aging, on the oxidation of polymers is crucial in developing polymeric systems with longer outcomes (12–14).
Even if a number of studies have been performed on the effects of gamma-sterilization on biodegradable polymers for pharmaceutical uses, only a limited number of works investigated the influence of the environmental conditions under which the irradiation process is conducted on the degradation reactions induced by gamma irradiation. Besides, no study has been found in the literature on the effects of gamma irradiation on the multiblock copolymers PEGd,lPLA and PEG-PLGA.
In a previous paper (15), we reported the effects of different gamma irradiation doses (5, 15, 25, and 50 kGy) on PEGd,lPLA and PEG-PLGA polymers and the behavior of these multiblock copolymers during post-irradiation storage time at 4°C, 40% RH. The study showed that gamma irradiation doses significantly affect physical and chemical polymer properties in terms of variations of polymer chemical structure and average molecular weight. The stability study carried out on polymers after irradiation treatment displayed a different behavior related to the polymer composition.
On the basis of results obtained in this previous work (15), the present study was undertaken to evaluate how the presence of oxygen in the samples during irradiation process and during post-irradiation storage time can affect irradiation-induced degradation reactions of PEGd,lPLA and PEG-PLGA multiblock copolymers submitted to gamma irradiation at 25 kGy. On this purpose, a set of samples was irradiated at 25 kGy in the presence of air and a second set under vacuum. The behavior of the multiblock copolymers to irradiation was compared to that of PLA, PLGA polymers. The stability of the polymers irradiated in different environmental conditions was evaluated by gel permeation chromatography (GPC) and differential scanning calorimetry (DSC) immediately after ionizing treatment and during storage in refrigerator (+4°C, 40% RH) for 30, 60, 90, and 120 days.
Electron paramagnetic resonance (EPR) spectroscopy is a sensitive and specific technique to study chemical species that have one or more unpaired electrons, such as organic and inorganic free radicals or inorganic complexes possessing a transition metal ion. This technique finds useful applications in the investigation of radiolytic mechanisms and in the analysis of radiolytic intermediates and of their transformation as a function of temperature. In previous works (6,7), EPR has been successfully used to investigate the radiolytic behavior of PLA and PLGA raw polymers and microspheres subjected to gamma irradiation. In this work, EPR technique was applied to evaluate the nature and the concentration of free radicals formed upon exposure of multiblock copolymers to 25 kGy total dose, at 77 K, under high vacuum.
MATERIALS AND METHODS
Materials
The copolymers PEGd,lPLA (Mw 130 kDa), PEG-PLGA (7525 DLG 3C-PEG 6000, Mw 22 kDa), PLA (Mw 78 kDa), and PLGA (7525 DLG 3E, Mw 34 kDa) were purchased from Lakeshore Biomaterials, Birmingham, USA. Tetrahydrofuran (THF) and methylene chloride (CH2Cl2), analytical grade, were from Sigma Aldrich (Milan, Italy). All the reagents were of analytical grade.
Methods
γ-irradiation
Raw polymers were irradiated by using 60Co as irradiation source (Applied Nuclear Energy Laboratory (LENA), University of Pavia) at 1 kGy/h dose rate.
To evaluate how and to what extent oxygen can play a role in the effect of gamma radiation on polymers the samples were irradiated at 25 kGy total dose at room temperature: (a) under vacuum and (b) in the presence of air. It was checked by thermometric control that sample temperature during the irradiation did not appreciably increase above room temperature. Four hundred milligrams of polymer samples were placed in glass tube, closed and irradiated at 25 kGy, room temperature. Samples treated in the absence of oxygen were sealed under vacuum at 10−5 Torr.
Twenty-five kilogray is the minimum absorbed dose considered adequate for the purpose of sterilizing pharmaceutical products without providing any biological validation (1,16). This reason led to the consideration of this irradiation condition that is commonly used by the pharmaceutical industry.
The irradiation pertaining EPR measurements devoted to the identification and characterization of the free radicals was performed at 77 K with a total dose of 25 kGy and dose rate of 1 kGy/h.
Molecular Weight Determination
The molecular weights of polymers were determined by GPC. The GPC system consisted of three Ultrastyragel columns connected in series (7.7 × 250 mm each, with different pore diameters: 104,103, and 500 Å), a pump (Varian 9010 (MI), Italy), a detector Prostar 355 RI (Varian (MI), Italy), and software for computing molecular weight distribution (Galaxie Ws, ver. 1.8 Single-Instrument, Varian (MI), Italy). Sample solutions in THF at a concentration of 20 mg/mL were filtered through a 0.45-μm filter (Millipore, USA) before injection into the GPC system and were eluted with THF at 1 mL/min flow rate. The weight average molecular weight (Mw) of each sample was calculated using monodisperse polystyrene standards, Mw 1,000–150,000 Da. The data were processed as weight average molecular weight (Mw), average molecular number (Mn), and polydispersity index (PI). The analyses have been performed in triplicate on three samples for each type of polymers and irradiation conditions.
Glass Transition Temperature Determination
Glass transition temperature (Tg) was determined by means of a 2910 modulated differential scanning calorimeter (TA Instruments, USA), fitted by a standard DSC cell, and equipped with a liquid nitrogen cooling accessory. Samples of about 10 mg were quantitatively transferred to hermetically sealed aluminum pans and subjected to two cooling and heating cycles from −60°C to 60°C at cooling and heating rates of 5°C/min. The DSC cell was purged with dry nitrogen at 40 mL/min. The baseline correction was performed by recording a run with empty pans. The system was calibrated both in temperature and enthalpy with indium as a standard material. The data were treated with Thermal Solutions software (TA Instruments, USA) and the results were expressed as the mean of three determinations.
Stability Study
The stability study was carried out on three samples sets: (a) polymer samples irradiated at 25 kGy in air and stored at 4°C in open vial, (b) polymer samples irradiated at 25 kGy under vacuum and stored at 4°C in sealed vial, and (c) polymer samples irradiated at 25 kGy under vacuum and stored at 4°C in opened vials. The vials were opened immediately after the irradiation process to permit the oxygen diffusion in the polymer samples. The changes in polymer molecular weight were monitored by GPC for a period of 120 days.
EPR Study
EPR measurements were carried out on polymer samples by using an X Band Brucker EMX/12. Two sets of experiments were performed starting with samples irradiated at the liquid N2 temperature (77 K). The EPR spectra were first recorded at 77 K, in order to identify the primary species, and subsequently after stepwise increases of the temperature above 77 K up to room temperature. This procedure was aimed to obtain suitable conditions for the reactions of the primary species and for the identification of the secondary radicals. The EPR spectra were analyzed by computer simulation using Hamiltonian including the Zeeman electronic and nuclear terms and the hyperfine terms with isotropic and anisotropic components for the g and hyperfine tensors:
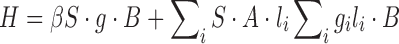 |
The microwave power level was set below 0.1 mW to avoid power saturation.
RESULTS AND DISCUSSION
Effect of Oxygen
Standard conditions for sterilization of polymeric drug delivery systems using ionizing radiation is still a matter of debate and they should be defined considering the physical–chemical properties of the polymer submitted to gamma radiation treatment and how the raw polymer responds to the irradiation process (3,10,17). The environment conditions under which radiation processing is conducted can significantly affect polymer properties. For example, the presence of oxygen and air during irradiation treatment produces free radicals that are often rapidly converted to peroxidic radicals. The fate of these peroxidic radicals depends on the nature of the irradiated polymer, the presence of additives, and other parameters such as temperature, total dose, dose rate, and sample size.
These preliminary remarks make allowance for the need to evaluate the effect of oxygen on the physical and chemical properties of the polymeric materials that are submitted to sterilization treatment with gamma irradiation.
In Table I, we collected the GPC results obtained on polymer samples (PEGd,lPLA, PEG-PLGA, PLA, and PLGA) non-irradiated and irradiated in the presence of oxygen and under vacuum.
Table I.
GPC Results of PEGd,lPLA, PEG-PLGA Multiblock Copolymers and PLA, PLGA Polymers During Irradiation at 25 kGy in Presence of Air and Under Vacuum
| Nonirradiated | Irradiated 25 kGy | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In presence of oxygen | Under vacuum | ||||||||||||||
| Polymer | Mw | Mn | PI | Mw | Mn | PI | ΔMw (%) | ΔMn (%) | G(s)/G(x) | Mw | Mn | PI | ΔMw (%) | ΔMn (%) | G(s)/G(x) |
| PEGd,lPLA | 105,500 | 65,300 | 1.61 | 59,000 | 34,000 | 1.73 | 44 | 47.9 | 7.56 | 60,600 | 35,800 | 1.7 | 42.5 | 45.17 | 6.60 |
| PLA | 77,900 | 44,300 | 1.75 | 48,000 | 31,500 | 1.52 | 38.4 | 28.9 | 10.45 | 48,400 | 26,300 | 1.8 | 37.8 | 40.6 | 8.56 |
| PEG-PLGA | 21,750 | 18,150 | 1.19 | 17,000 | 10,500 | 1.62 | 21.8 | 42.1 | 12.87 | 19,000 | 14,600 | 1.3 | 15.5 | 19.5 | 12.64 |
| PLGA | 36,000 | 22,500 | 1.6 | 26,700 | 18,500 | 1.44 | 25.8 | 17.7 | 9.46 | 27,300 | 17,700 | 1.54 | 24.1 | 21.3 | 10.08 |
The effect of oxygen on Mw, Mn, and PI were not very significant for PEGd,lPLA, PLA, and PLGA polymers. A remarkable reduction of Mw and Mn was observed between PEG-PLGA samples irradiated under vacuum (absence of oxygen) and the samples treated in the presence of oxygen.
The evacuation of the PEG-PLGA samples reduces oxygen diffusion in the polymer matrix and leads to a lower extent of Mw and Mn reduction. The Mw and Mn reduction (%) of PEG-PLGA was 15.5% and 19.5% after irradiation under vacuum, against the 21.8% and 42.1% detected on samples submitted to gamma irradiation in the presence of air. The great sensitivity of this multiblock copolymer to oxidative degradation may be due to the high percentage of PEG in its structure. PEG as plasticizer was expected to induce important intrinsic modification in the polymer such as: (1) significant changes in local structure/microstructure; (2) enhancement in fraction of amorphous phase; (3) increasing free volume and the flexibility of the polymer chains (18–20).
Irradiated PEG-PLGA polymer segments in the amorphous region are mobile due to the low glass transition temperature (<33°C). It is also possible that the radicals trapped in the crystalline regions of the polymer can migrate along the polymer main chain and finally appear in the amorphous region.
When oxygen molecules enter into the amorphous region of polymer, they react with free hydrogen atoms (free radicals), increasing amorphous regions and creating creeks which could lead to a reduction of Mw and Mn.
PEGd,lPLA did not respond in the same manner, the Mw and Mn reductions evaluated after irradiation under vacuum did not present significant differences compared to the variations observed on polymer samples irradiated in air. This could be due either to the low percentage of hydrophilic polymer (5% mol) in the multiblock polymer with respect to PEG-PLGA (60% mol) or the high molecular weight (105 kDa) of PEGd,lPLA polymer.
PLA and PLGA polymer samples present hydrophobic features and the chains have a lower mobility compared to PEG-PLGA; consequently, the oxygen molecules meet more difficulties to permeate through the polymer chains. Oxygen diffusion rate in PLA and PLGA samples could be so slow that its presence during the irradiation process did not affect in a remarkable way the extent of Mw and Mn reduction induced by gamma-rays.
The G(s)/G(x) calculated by mathematical equation (14) values collected in Table I illustrates the dominance of chain scission over cross-linking reactions either in the polymer samples irradiated in air or under vacuum.
It is reasonable to predict that such a change in Mw and Mn after irradiation would affect thermal properties like the glass transition temperature of the amorphous polymers. Table II resumes the Tg values of PEGd,lPLA, PLA, PEG-PLGA, and PLGA polymers determined by DSC. DSC analyses were performed on polymer samples before and after irradiation at 25 kGy, under vacuum and in presence of oxygen.
Table II.
DSC Results of PEGd,lPLA, PEG-PLGA Multiblock Copolymers and PLA, PLGA Polymers Nonirradiated and Irradiated at 25 kGy in the Presence of Air and Under Vacuum
| Polymer | Nonirradiated, Tg (°C)a | Irradiated at 25 kGy, Tg (°C)a | |
|---|---|---|---|
| In presence of oxygen | Under vacuum | ||
| PEGd,lPLA | 43.2 | 35.8 | 37.8 |
| PLA | 47.3 | 43.3 | 46.2 |
| PEG-PLGA | 35.2 | 32.6 | 32.5 |
| PLGA | 42.4 | 39.9 | 42.0 |
aMaximum standard error value ±0.5
PEGd,lPLA and PEG-PLGA Tg values (43.23°C and 35.24°C), detected before radiation treatment were always lower than Tg values of PLA and PLGA (47.3°C and 42.4°C). They are consistent with the plasticizing effect of PEG chains present in the multiblock copolymers.
The irradiation of polymers resulted in a reduction of Tg value due to radiation-induced chain scission reactions which led to a remarkable decrement of molecular weight of polymers and consequently an increase of free volume and chain mobility.
The absence of oxygen during the irradiation of PEGd,lPLA, PLA, and PLGA led up to a lower reduction of Tg compared to polymers irradiated in air.
A different behavior was recorded for PEG-PLGA multiblock copolymer since the Tg value of samples irradiated under vacuum (32.51°C) was pretty the same as that of sample treated by gamma-rays in the presence of air (32.61°C). This feature can be explained by admitting the formation of intermolecular hydrogen bonding interactions between end terminal groups (–COOH, –OH) of polymer chains, as a consequence of chain scission reactions which take place during the sterilization process. The presence of oxygen molecules likely promotes the formation of the hydrogen bonding interactions, which leads to an alteration of the polymer chain mobility. The reduction of the chain mobility involves an increase of the transition temperature with respect to what is expected.
Stability Study
It is well recognized that gamma-irradiation treatment causes property changes in poly-α-orthoesters, and generally this kind of effects last for long periods of time because of latent free radicals. Combination reactions between trapped radicals and/or oxidation reactions can occur during storage time, resulting in cross-linking or chain scission events. The combined action of ionizing radiation and oxygen on polymeric materials may rapidly lead to severe alterations of the polymer properties. The resulting effects are strongly dependent on the chemical structure of the polymer (11,14,21).
The stability study was carried out on samples irradiated at 25 kGy and stored in different conditions to evaluate the role of oxygen in the degradation reactions induced by gamma irradiation during the storage time. The stability study was performed on three sample sets: (a) polymer samples irradiated in air and stored at 4°C in an open vial, (b) polymer samples irradiated under vacuum and stored at 4°C in a sealed vial, and (c) polymer samples irradiated under vacuum and stored at 4°C in opened vials. In the last case, the vials were opened immediately after the irradiation process to permit oxygen diffusion in the polymer matrix. The changes in molecular weight, as a function of storage time, were detected by GPC on irradiated samples.
Figure 1a plots the GPC results obtained on PEGd,lPLA polymer samples in terms of Mw variations. Polymer samples irradiated in air and stored in the presence of oxygen showed at the 120th day Mw reduction higher than the samples irradiated under vacuum. The Mw reduction of PEGd,lPLA samples irradiated in the presence of air amounted to 43.53% after 120 days. For the samples irradiated under vacuum, a significant lower Mw reduction between 24.17% and 29.94% was observed. The sample irradiated under vacuum and stored in the presence of oxygen showed a significative reduction of Mw between the 30th and 60th day of storage due to the oxidative degradation reactions induced by oxygen diffused in the polymeric network. PEGd,lPLA samples submitted to gamma-irradiation under vacuum and kept in these conditions (absence of oxygen) for all storage time are more stable: a percentage reduction from 18.97% to 24.17% between the 30th and 120th day was observed.
Fig. 1.

Stability study of a PEGd,lPLA and b PLA polymers treated in different conditions: irradiated and stored in air (diamonds); irradiated and stored under vacuum (squares); irradiated under vacuum and then open to air (circles)
PLA polymer irradiated under vacuum and kept in sealed vials for all storage time presented a percentage of Mw reduction of 8.57% after 120 days; the polymer samples irradiated under vacuum and stored in open vial (in presence of oxygen) did not show a significant difference in comparison with the sample stored under vacuum (Fig. 1b). These results could be attributed to the high hydrophobicity of polymer, the Tg value, and consequently the low mobility of polymer chains that limit the migration of oxygen molecules along the polymer main chains. A gradual increase of molecular weight reduction was observed for PLA polymer sample treated in air; the percentage of Mw reduction calculated at the end of incubation time (120th day) was 29.65%. The slow reduction detected in between the 30th and 60th day could be due to the time needed for oxygen molecules to diffuse through polymeric matrix.
The high Mw decay obtained for PEGd,lPLA with respect to PLA can be a consequence of the presence of PEG segments in the polymer network that improve the penetration of oxygen.
The results obtained from the stability study of multiblock copolymer PEG-PLGA are plotted in Fig. 2a. Polymer sample treated under vacuum are stable during storage time: an Mw reduction of 4% was detect after 120 days of storage. The sample irradiated in air and stored in air showed a gradual Mw reduction; a significative increase was detected at the 30th day of storage until it reached a reduction of 26.06%. The sample irradiated under vacuum and stored in the presence of air showed a remarkable decrease of Mw between the 30th and 90th day; the percent reduction ranged between 12.85% and 28.45%. No more variations were observed up to the 120th day. After 120 days, the Mw reduction was 27% equal to the values detected for the samples treated in air. These results can be justified considering the composition of polymer PEG-PLGA: 60 mol% of the polymer is based on polyethylene glycol and only 40 mol% is PLGA. The high percentage of PEG in the matrix improves in a significative way the diffusion of oxygen in the polymer; when the vial is opened to air, a massive amount of oxygen can permeate through the polymeric chains and react with the radicals trapped in the polymer matrix.
Fig. 2.

Stability study of a PEG-PLGA and b PLGA polymers treated in different conditions: irradiated and stored in air (diamonds); irradiated and stored under vacuum (squares); irradiated under vacuum and then stored in open air (circles)
PLGA polymer showed a slight Mw reduction either for the samples treated under vacuum or for the PLGA polymer irradiated under vacuum and opened to air during storage time. The Mw decay ranged between 2% and 12% (Fig. 2b).The Mw reduction observed for the samples treated in air were higher than those observed for the samples irradiated under vacuum; after 120 days of storage, the percent reduction measured was 18.5%.
Comparatively, PEGd,lPLA Mw reduction resulted to be more sensitive to irradiation than PEG-PLGA as shown by the Mw reduction observed under vacuum. This behavior confirms that higher Mw polymers undergo higher degradation upon irradiation, as explained in a previous work (15). On the contrary, PEG-PLGA resulted to being more sensitive than PEGd,lPLA to oxygen effects.
EPR Study
Free Radical Products of the PLA and PLGA Radiolysis
The EPR measurements have dealt with samples of neat PLGA and PLA and samples of PLA-PEG (5 mol% PEG) and PLGA-PEG (60 mol% PEG) copolymers which were analyzed on a comparative basis. The EPR spectra from PLA and PLGA samples recorded at 120 K after irradiation at 77 K and after warming at 290 K are shown in Fig. 3. The superimposition makes it fairly evident that PLA and PLGA afforded almost identical spectra with really minor differences in line shape, splitting, and intensity distributions. This implies that a prominent mechanism of radiolysis exists, driving the free radical radiation damage at the tertiary carbon sites of the lactide units. According to previous observations (6,7), the dominant component in the low-temperature patterns was the chain scission radical −CH•(CH3). Two different structures for this radical can be predicted: –OCH•(CH3) (A) and CH•(CH3)C(=O)O– (B) depending on weather the bond rupture takes place at the C–C or the O–C site.
Fig. 3.

EPR spectra of PLGA vs PLA after irradiation under vacuum at 77 K, recorded at 120 K and after temperature annealing at 290 K
The species B was likely to be of prominent importance on the basis of the analogy of its hf and g tensor with those of the analog radical CH3CH•C(O=O)O− resulting from the radiation-induced deamination of alanine (22). A second component in the low-temperature spectrum of PLGA and PLA was the quartet signal with an average splitting of 22 G due to the chain radical –C(=O)O–C•(CH3)–O– formally arising from the splitting of the tertiary C–H bond at the lactide sites. The relative abundance of this latter species increased on warming towards room temperature because of the contribution by the H abstraction reactions by the chain scission radicals (Fig. 3).
The selectivity of the radiolysis mechanism towards the formation of the –O(=O)C–CH•(CH3) radical in both the PLA homopolymer and the PLGA 75/25 random copolymer could be explained with the greater lability of the tertiary ester anion-radicals toward the β-scission at the C–O bond leading to a carboxylate anion (precursor of carboxylic acids, a major radiolysis product) and the tertiary alkyl radical, according to the reaction:
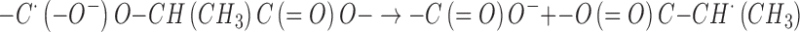 |
1 |
The strong similarity of the PLA and PLGA EPR spectra implied that in PLGA the decomposition reaction of the analog glycolide anion radicals –C•(–O−)O–CH2C(=O)O– was of minor importance presumably because of their greater stability and the occurrence of charge migration leading to a final localization of the spins at the carboxyls adjacent to tertiary units.
The presence of the H abstraction radicals –C•(CH3)– among the species trapped at 77 K called for a mechanism involving excited species or reactions with low or negligible activation energy. Within the latter hypothesis, H abstractions by the ester cation radicals at tertiary C–H sites could be invoked:
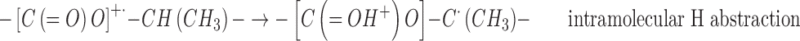 |
2 |
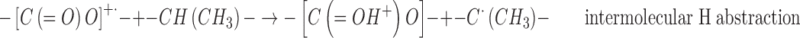 |
3 |
Support for this hypothesis come from Sevilla experiments (23) which have demonstrated that ester cation-radicals are capable of undergoing H abstraction at 77 K giving neutral radicals.
Free Radical Products of PEG Radiolysis
The free radical products of the solid-state radiolysis of PEG resembled closely those identified in the irradiated high-molecular weight analog. The low temperature pattern was reckoned with C–H and C–C bond scissions with formation of the radicals –OCH2• and –CH•–O– (Fig. 4); the post-irradiation annealing at room temperature caused the gradual decay of the initial species and their replacement with a doublet of 15 G centered at g = 2.005; this latter signal was characteristic of the aldehydic radical (5,24) –CH•–CHO.
Fig. 4.

EPR spectra of PEG homopolymer after irradiation under vacuum at 77 K, recorded at increasing of temperature from 120 to 298 K
The low-field shift of the g value in the aldehydic radical was a distinctive feature which was a consequence of the delocalization of the spin on the carbonyl oxygen. The formation of this species was reckoned with the cage reaction between neighboring aldehyde-radical couples according to the mechanism:
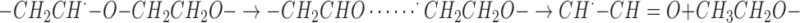 |
The aldehydic radicals were stable for months at room temperature under vacuum.
Effect of PEG on the Radical Products of the PLA and PLGA Radiolysis
The presence of PEG units in the PEGd,lPLA copolymer chains (5% of PEG) and in the PLGA-PEG copolymer chains (60% of PEG) had two major consequences:
-
A decrease of the radiolytic yield of radicals –C(=O)O–C•(CH3)–O– at 77 K. This species was practically absent in the low-temperature spectrum of PEG-PLGA (60% of PEO; Fig. 5) but it showed up with increasing intensity on warming toward 290 K as a consequence of the activation of H abstraction reactions (Fig. 6).
The same effect by PEG was observed when dealing with the PEGd,lPLA copolymer (Fig. 6). In this case, however, due to the lower concentration of PEG, the obliteration of the C(=O)O–C•(CH3)–O– signal at 77 K, it was not complete but it appeared with a lower intensity as compared to neat PLA or PLGA (Fig. 6—spectrum recorded at 125 kGy).
The increase of the segmental chain mobility in the polymer matrix resulting in a decrease of the radical trapping efficiency and ultimately in faster radical decay rates at 290 K. Indeed, a 20 min storage at 290 K was sufficient to cause a 98% decrease of the overall initial radical concentration (Fig. 7b) while in PLA and PLGA the residual radical concentration after the same storage time at 290 K was approximately 28% (Fig. 3). This effect is consistent with the observed Tg decrease and the enhanced radiolytic effects on Mn and Mw in the presence of air.
Fig. 5.

a EPR spectra of PLGA vs PEG-PLGA (60 mol%) multiblock copolymer recorded at 120 K following irradiation at 77 K. Spectrum b shows the difference between PLGA and PEG-PLGA
Fig. 6.

EPR spectra of PEGd,lPLA (5 mol% of PEG) recorded as function of temperature after irradiation at 77 K
Fig. 7.

EPR spectra of PEG-PLGA (60mol% of PEG) recorded as function of temperature after irradiation at 77 K
On the basis of the mechanism proposed for the in source low-temperature generation of the –CH•(CH3)– radicals in the radiolysis of PLGA and PLA, the effect of PEG can be rationalized by assuming that it interferes with the intermolecular H abstraction reaction (3) by the ester cation-radicals and that the intramolecular H abstraction mechanism (2) is of minor importance. The cation-radicals will therefore be driven toward alternative reaction paths, like the C–O scission, thus affording an additional contribution to the formation of the tertiary chain scission radicals –C•(CH3)–. The H abstraction at the tertiary C–H sites leading to –CH•(CH3)– radicals was not inhibited above the Tg because of the great enhancement of segmental mobility in the mixed PLGA-PEG matrix.
The EPR spectral sequence recorded from the PLGA-PEG copolymer (60% of PEO) during the storage at 290 K, beside showing a faster decay rate, was diagnostic of the buildup of novel signal with a peak to peak splitting of approximately 12 (Fig. 7b). This spectrum, which account for less than 2% of the initial radical concentration, was tentatively suggested to arise from the superimposition of the quartet signal due to the PLGA radical –C•(CH3)– with the doublet shifted toward the low field of the aldehyde radical –CH•C(=O)H radical from PEG radiolysis (Fig. 7).
In this temperature range, the radical decay process involved almost exclusively the radical –C•(CH3)– as observed by the difference spectrum C, Fig. 7.
CONCLUSIONS
The presence of oxygen during the irradiation process and the storage phase influences the entity of the radiation damage and the stability of polymers after sterilization by ionizing radiation. Polymer radicals remaining trapped in the matrix of PEGd,lPLA and PEG-PLGA multiblock copolymers after irradiation by gamma rays can undergo some reactions long after irradiation time. The findings obtained in this study are expected to contribute both to minimize the damages induced by gamma irradiation on multiblock copolymers based on PLA, PLGA, and PEG and to improve the stability of PEGd,lPLA and PEG-PLGA polymers after ionizing treatment.
References
- 1.European Medicines Agency. The use of ionizing radiation in the manufacture of medicinal products European Guidelines 3AQ4a. (1991).
- 2.Loo J. S. C., Ooi C. P., C Boey F. Y. Degradation of poly(lactide-co-glycolide) (PLGA) and poly(L-lactide) (PLLA) by electron beam radiation. Biomaterials. 2005;26:1359–1367. doi: 10.1016/j.biomaterials.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 3.K. J. Hemmerich. Radiation sterilization, Polymer materials selection for radiation-sterilized products. Medical Device and Diagnostic Industry MDDI Feb. 2000, p. 78.
- 4.Bushell J. A., Claybourn M., Williams H. E., Murphy D. M. An EPR and ENDOR study of gamma and beta-radiation sterilization in poly(lactide-co-glycolide) polymers and microspheres. J. Control. Release. 2005;110(1):49–57. doi: 10.1016/j.jconrel.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Dorati R., Genta I., Montanari L., Buttafava A., Faucitano A., Conti B. The effect of γ-irradiation on PLGA/PEG microspheres containing ovalbumin. J. Control. Release. 2005;107:78–90. doi: 10.1016/j.jconrel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 6.Faucitano A., Buttafava A., Montanari L., Cilurzo F., Conti B., Genta I., Valvo L. Radiation induced free radical reactions in polymer/drug systems for controlled release: an EPR investigation. Radiat. Phys. Chem. 2003;67:61–72. doi: 10.1016/S0969-806X(02)00404-8. [DOI] [Google Scholar]
- 7.Montanari L., Costantini M., Signoretti E.C., Valvo L., Santucci M., Bortolomei M., Fattibene P., Onori S., Faucitano A., Conti B., Genta I. Gamma-irradiations effects on poly(d,l-lactie-co-glycolide) microspheres. J. Control. Release. 1998;56(2):219–229. doi: 10.1016/S0168-3659(98)00082-0. [DOI] [PubMed] [Google Scholar]
- 8.Haugen H. J., Brunner M., Pellkofer F., Aigner J., Will J., Wintermantel E. Effect of different γ-irradiation doses on cytotoxicity and material properties of porous polyether–urethane polymer. J. Biomed. Mater. Res. B. Appl. Biomate. 2006;80B(2):415–423. doi: 10.1002/jbm.b.30612. [DOI] [PubMed] [Google Scholar]
- 9.Claybourn M., Gray H., Murphy D. M., Purnell I. J., Rowlands C. C. Electron magnetic resonance study of gamma-irradiated poly(lactide-co-glycolide) microspheres. J. Control. Release. 2003;91(3):431–438. doi: 10.1016/S0168-3659(03)00269-4. [DOI] [PubMed] [Google Scholar]
- 10.Sintzel M. M. B., Merkli A., Tabatabay C., Gurny R. Influence of irradiation sterilization on polymers used as drug carriers—a review. Drug Dev. Ind. Pharm. 1997;23(9):857–878. doi: 10.3109/03639049709148693. [DOI] [Google Scholar]
- 11.Martini L., Collett J. H., Attwood D. The influence of gamma irradiation on the physicochemical properties of a novel triblock copolymer of ε-caprolactone and ethylene oxide. J. Pharm. Pharmacol. 1997;49(6):601–605. doi: 10.1111/j.2042-7158.1997.tb06852.x. [DOI] [PubMed] [Google Scholar]
- 12.R. Garcia, B. Howard, R. LaRue, G. Parton, and J. Walke. Strategies for Gamma Sterilization of Pharmaceuticals. Pharmaceutical & Medical Packaging News. PMPN May 2004.
- 13.M. Bernkopf. Sterilisation of Bioresorbable Polymer Implants. Medical Device Technology MDT May/June 2007. [PubMed]
- 14.Al-Ma’adeed M. A., Al-Qaradawi I. Y., Madi N., Al-Thani N. J. The effect of gamma irradiation and shelf aging in air on the oxidation of ultra-high molecular weight polyethylene. Appl. Surf. Sci. 2006;252(9):292. [Google Scholar]
- 15.Dorati R., Colonna C., Serra M., Genta I., Modena T., Pavanetto F., Perugini P., Conti B. γ-irradiation of PEGd,lPLA and PEG-PLGA multiblock copolymers: I Effect of irradiation doses. AAPS PharmSciTech. 2008;9(2):718–725. doi: 10.1208/s12249-008-9103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gèze A., Venier-Julienne M. C., Cottin J., Faisant N., Benoit J. P. PLGA microsphere bioburden evaluation for radiosterilization dose selection. J. Microencapsul. 2001;18(5):627–636. doi: 10.1080/02652040010019424. [DOI] [PubMed] [Google Scholar]
- 17.Woo L., Sandford C. L. Comparison of electron beam irradiation with gamma processing for medical packaging materials. Radiat. Phys. Chem. 2002;63:845–850. doi: 10.1016/S0969-806X(01)00664-8. [DOI] [Google Scholar]
- 18.Ahlneck C., Zografi G. The molecular basis of moisture effects on the physical and chemical stability of drugs in the solid state. Int. J. Pharm. 1990;62(2–3):87–95. doi: 10.1016/0378-5173(90)90221-O. [DOI] [Google Scholar]
- 19.Oksanen C. A., Zografi G. Molecular mobility in mixtures of absorbed water and solid poly(vinylpyrrolidone) Pharm. Res. 1993;10(6):791–799. doi: 10.1023/A:1018988506336. [DOI] [PubMed] [Google Scholar]
- 20.Hancock B. C., Zografi G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm. Res. 1994;11(4):471–477. doi: 10.1023/A:1018941810744. [DOI] [PubMed] [Google Scholar]
- 21.Buchanan F. J., White J. R., Sim B., Downes S. The influence of gamma irradiation and aging on degradation mechanisms of ultra-high molecular weight polyethylene. J. Mater. Sci: Mater. Med. 2001;12(1):29–37. doi: 10.1023/A:1026796817483. [DOI] [PubMed] [Google Scholar]
- 22.Heydari M. Z., Malinen E., Hole E. O., Sagstuen E. Alanine radicals 2. The composite polycrystalline alanine EPR spectrum studied by ENDOR, thermal annealing, and spectrum simulation. J. Phys. Chem. A. 2002;106:8971–8977. doi: 10.1021/jp026023c. [DOI] [Google Scholar]
- 23.Becker D., Swart S., Sevilla M. D. Electron spin resonance investigation of intramolecular hydrogen transfer and alkyl attack in ester cation radicals. J. Phys. Chem. 1985;89(12):2638–2646. doi: 10.1021/j100258a041. [DOI] [Google Scholar]
- 24.Faucitano A., Buttafava A., Martinotti F., Ferloni P., Magistris A. The mechanism of gamma-radiolysis of polymethylene, polypropylene and poly-n-butylene oxides: an ESR investigation. Radiat. Phys. Chem. 1992;40(5):347–355. [Google Scholar]