

Abstract
The type 1 neurotensin receptor (NTS1) belongs to the G-protein coupled receptor (GPCR) family. GPCRs are involved in important physiological processes but for many GPCRs ligand binding sites and other structural features have yet to be elucidated. Comprehensive analyses by mass spectrometry (MS) could address such issues, but they are complicated by the hydrophobic nature of the receptors. Recombinant NTS1 must be purified in the presence of detergents to maintain solubility and functionality of the receptor, to allow testing of ligand or G-protein interaction. However, detergents are detrimental to MS analyses. Hence steps need to be taken to substitute the detergents with MS-compatible polar/organic solvents. Here, we report the characterization of NTS1 by electrospray ionization mass spectrometry (ES-MS) with emphasis on methods to transfer intact NTS1 or its proteolytic peptides into compatible solvents by protein precipitation and liquid chromatography (LC) prior to ES-MS analyses. Molecular mass measurement of intact recombinant NTS1 was performed using a mixture of chloroform/methanol/aqueous TFA as the mobile phase for size exclusion chromatography-ES-MS analysis. In a separate experiment, NTS1 was digested with a combination of CNBr and trypsin and/or chymotrypsin. Subsequent reversed phase LC-ES-MS/MS analysis resulted in greater than 80% sequence coverage of the NTS1 receptor protein including all seven transmembrane domains. This work represents the first comprehensive analysis of recombinant NTS1 receptor using mass spectrometry.
Keywords: Integral membrane protein, intact protein, detergents, electrospray ionization mass spectrometry (ES-MS), G-protein coupled receptor (GPCR), neurotensin receptor
Introduction
The type 1 neurotensin receptor (NTS1) is a G-protein coupled receptor (GPCR) that is predominately expressed in the brain and intestine. Recent studies have indicated that NTS1 is linked to cancer and pain perception [1]. The receptor is activated by the brain-gut tridecapeptide neurotensin. From mutational analyses and computer assisted modeling techniques, it has been proposed that neurotensin interacts with amino acids in TMs IV and VI [2] and extra-cellular loop III [2; 3] of NTS1. The backbone conformation of neurotensin, bound to NTS1, was determined by solid-state nuclear magnetic resonance experiments [4]. Alternative approaches, such as mass spectrometry, would allow further characterization of the receptor-ligand interactions. As a prerequisite for such studies, protocols must be developed to cover the entire sequence of NTS1, which is described below.
Mass spectrometry (MS) has gained wide acceptance for the structural characterization of proteins. Proteins can be analyzed intact or after digestion into peptides. Detection of the intact protein provides molecular mass information and the extent of protein heterogeneity. The analysis of proteolytic peptides allows confirmation of the amino acid sequence (assuming that full sequence coverage is achieved) and a thorough investigation of post-translational modifications i.e.; modification type and location. With continuous improvements in technology and computer software, MS has become not only a key player in protein sequencing but also a tool for the characterization of higher order protein structures, non-covalent complexes and receptor-ligand binding interactions [5; 6].
The under-representation of GPCRs and indeed other integral membrane proteins in MS-based proteomic studies is primarily due to their hydrophobic nature. Membrane proteins aggregate when they are removed from their lipid environment; however, this can be minimized by using suitable detergents. The choice of an appropriate detergent allows efficient extraction from membranes (solubilization), as well as preservation of the receptor’s function and hence 3D structure; a requirement for studying for example receptor-ligand interactions. Detergent composition and concentrations must be optimized experimentally for each receptor in question to maintain its functional integrity. However, detergents are often detrimental to proteolytic digestion and to mass spectrometric analyses [7] and therefore they need to be reduced or completely removed before MS analysis while maintaining receptor solubility in MS-compatible solvents. Despite these challenges, some GPCRs have been successfully studied by MS [8; 9; 10; 11; 12; 13; 14; 15; 16; 17].
To-date, only three GPCRs, rhodopsin [8; 9], the tachykinin NK-1 receptor [17] and recently the cannabinoid 2 (CB2) receptor [14] have had their protein sequences (almost) completely determined using MS. Given its naturally high abundance and availability, the photoreceptor rhodopsin was the first GPCR to be completely sequenced. In initial studies by Barnidge et al. [18], rhodopsin was purified from retinal rod outer segment membranes in the presence of the detergent n-octyl-β-D-glucoside, partially denatured in urea, and then digested with trypsin. The resulting tryptic peptides were separated by reversed phase liquid chromatography (LC) with octyl-glucoside in the mobile phase. Peptides covering the hydrophilic loop regions and hydrophobic peptides from 4 out of 7 TM domains were observed by matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS). The limited coverage of rhodopsin’s hydrophobic regions may have been a consequence of the interfering effects of octyl-glucoside on the MALDI-TOF MS analysis. Following these initial studies, modifications to the rhodopsin preparation protocol for MS analyses were made by several groups [8; 9; 10; 11] improving the sequence coverage of rhodopsin to include all 7 TM domains. While the detergents used to purify the receptor from the rod outer segment membranes varied between protocols, a common feature in all was that the purified receptor was first precipitated, and then digested with cyanogen bromide (CNBr). Precipitation of the receptor prior to digestion helped to reduce the amount of detergent, while high acid concentrations needed for CNBr digestion provided favorable conditions for dissolving the precipitated receptor. The identification of peptides after CNBr treatment was performed either by MALDI-TOF MS [8; 9; 10] or by ES-MS after separation of the CNBr generated peptides by reversed phase LC [11]. Full sequence coverage of rhodopsin by bottom-up MS methods led to further studies on the receptor including the characterization of post-translational modifications [19; 20; 21] and to probing interaction sites with the transducin α-subunit [22]. A different MS sample preparation was employed by Alves et al. for a recombinant tachykinin NK-1 receptor, achieving ~80% sequence coverage [17]. The purified receptor (with FLAG and His-tags) was subjected to one-dimensional sodium dodecylsulfate polyacrylamide gel electrophoresis (1D SDS PAGE). The receptor band was excised and in-gel digestion was performed with a combination of enzymes and/or CNBr. Peptides from the tachykinin receptor were extracted from the gel in the presence of the detergent n-dodecyl-β-D-maltoside at 0.1% and analyzed by MALDI-TOF MS. The addition of dodecyl-maltoside to extraction buffers has been shown to improve the recovery of hydrophobic peptides from in-gel digestions and dodecyl-maltoside at low concentrations seemed tolerable in the MALDI-TOF MS. However, it was noted by the authors that the tachykinin NK-1 receptor peptides appeared as weak signals and most of the intense peaks in the spectrum corresponded to contaminants [17]. Recently, ~90% of the cannabinoid CB2 receptor sequence was observed using MS. The recombinant receptor was digested with trypsin in the presence of the detergent 5-cyclohexyl-1-pentyl-β-D-maltoside (CYMAL5) at low concentration [14] and the resulting peptides were identified by reversed phase LC-ES-MS/MS using formic acid/water/acetonitrile mixtures as the mobile phase. This represented a significant breakthrough as previous attempts reported that 70% of the hydrophobic TM domains remained undetected when an in-gel trypsin digestion was performed [23].
Two GPCRs, rhodopsin and the tachykinin NK-1 receptor, have been analyzed intact by MS. Intact rhodopsin was precipitated with acetone, separated using a mixture of water/formic acid/2-propanol as the mobile phase and analyzed by ES-MS. The recorded mass spectra revealed extensive heterogeneity owing to different glycoforms of rhodopsin and modifications such as phosphorylation and formylation [24]. Recently, the intact tachykinin NK-1 receptor was analyzed using MALDI-TOF MS [17]. The histidine-tagged receptor was expressed in mammalian cells, and bound to either nickel or cobalt chelating magnetic beads for purification. The beads containing the receptor were then directly deposited on the MALDI target. The MADLI mass spectrum indicated the presence of the unmodified receptor as well as a number of glycosylated and truncated forms. These results were confirmed by Western blotting.
The above examples show that sample preparation prior to MS analysis varies between receptors, possibly reflecting differences in amino acid sequence and purification protocols to obtain purified GPCRs. Therefore, sample preparation methods for MS analysis would need to be refined specifically for each receptor. Factors such as the degree of detergent removal prior to MS, solubility of the intact receptor or receptor peptides in MS-compatible solvents, and efficiency of proteolytic enzymes will influence the ability to achieve full sequence coverage of a particular receptor. With these factors in mind, we present here the analysis of NTS1 by LC-ES-MS. Under optimized conditions, we were able to obtain a mass spectrum of intact NTS1 and achieve 88% coverage of the amino acid sequence from a combination of chemical and proteolytic fragments.
Materials
Water, acetonitrile, cyanogen bromide (5M in acetonitrile) (CNBr), formic acid, ammonium bicarbonate, trifluoroacetic acid (TFA), iodoacetamide (IAM), dithiothreitol (DTT), chloroform, and methanol were purchased from Sigma-Aldrich (St Louis, MO). RapiGest™ SF was purchased from Waters Corporation (Milford, MA). The model TM peptide (IYSKVLVTAIYLALFVVGTVGNSVTAFTLARKKSLQSLQSTVHYHLGSLALSDLLILLLAMPVELY) was synthesized at the Center for Biologics Evaluation and Research (CBER), FDA, Bethesda, MD.
Experimental
Preparation of NTS1
The NTS1 fusion protein MBP-N10-Tev-rT43NTR-CH2-N5G3S-G3S-TrxA-H10 (here referred to as NTS1-1233) consists of the Escherichia coli maltose-binding protein, followed by a linker and a tobacco etch virus (Tev) protease recognition site, the N-terminally truncated rat neurotensin type 1 receptor (NTS1) starting at Thr43, a second Tev protease recognition site at the receptor C-terminus, a linker, the E. coli thioredoxin and a decahistidine tag [25]. The fusion protein was expressed in E. coli and purified as described [25]. Purified NTS1-1223 was incubated overnight at 4°C with Tev protease in the presence of DTT. This generated rT43NTR, with Ser-Gly-Ser at the N-terminus, and with the C-terminus ending in Glu421-Asn-Leu-Tyr-Phe-Gln (calculated average molecular mass of 43,303 Da). We refer to rT43NTR as NTS1. The mixture was resolved by size-exclusion chromatography in detergent buffer (20 mM TrisHCl pH 7.4, 8% glycerol, 1 M NaCl, 0.03% 3-[(3-cholaminidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)/0.006% cholesteryl hemisuccinate (CHS), 0.01% n-dodecyl-β-D-maltoside (LM)) [25] to obtain NTS1.
Intact Protein Analysis by Size-Exclusion Chromatography-Electrospray Ionization Mass Spectrometry (SEC-ES-MS)
Purified NTS1 receptor protein (25 µL at 0.34 mg/mL) in detergent buffer was diluted 1:1 with neat TFA prior to analysis. SEC was performed on a Hewlett-Packard (Palo Alto, CA) binary LC pump using a Super SW2000 size exclusion column (4.6 mm ID × 30 cm, Tosoh Bioscience, Montgomeryville, PA). The mobile phase consisted of chloroform/methanol/0.5% aqueous TFA (1:3:1 v/v/v). The flow rate was set to 200 µl/min and the column temperature to 30°C. The eluent was directly electrosprayed into a single quadrupole mass spectrometer, HP1100 LC-mass selective detector (MSD) (Palo Alto, CA). Data were acquired from m/z 600-2000 every 7 s. Nitrogen was used to assist nebulization and desolvation. The HP ChemStation data system was used for data acquisition and deconvolution. The MS instrument was calibrated with a standard Agilent ES tuning mix.
Analysis of Model Transmembrane Peptides
Purified synthetic transmembrane peptide with sequence IYSKVLVTAIYLALFVVGTVGNSVTAFTLARKKSLQSLQSTVHYHLGSLALSDLLILLLAMPVELY was digested with trypsin in the presence of either 60:40 (v/v) methanol/50 mM ammonium bicarbonate, 80:20 (v/v) acetonitrile/50 mM ammonium bicarbonate or an acid labile detergent (Rapigest) to generate two tryptic peptides comprising TM I (VLVTAIYLALFVVGTVGNSVTAFTLAR) and and TM II (SLQSLQSTVHYHLGSLALSDLLILLLAMPVELY) of NTS1. The digest mixture was analyzed by LC-MS. Briefly, the digest mixture was separated by reversed phased chromatography using a CapLC (Waters) and peptides were mass measured using a Q-ToF II (Micromass/Waters) mass spectrometer.
Protein Precipitation
Purified NTS1 receptor protein in detergent buffer was precipitated with chloroform/methanol/aqueous TFA. Briefly, methanol (150 µL) and chloroform (50 µL) were added to the protein solution (50 µL at 0.34 mg/mL) and mixed. A solution of 10% trifluoroacetic acid (aqueous) (100 µL) was added, followed by brief mixing. The two phases were separated by centrifugation (8,944 × g at 10°C, for 20 min), and the precipitated protein accumulated between the two solvent phases. The bulk of the upper and lower phases was removed without disrupting the precipitated protein at the interface. Methanol (150 µL) was added to the precipitate and mixed. The protein pellet was recovered by centrifugation. The majority of the methanol was removed with a pipette tip and the protein pellet was then allowed to dry.
Digestion of NTS1 Receptor
For CNBr cleavage, the dried protein was dissolved in 90% aqueous formic acid (100 µL). CNBr (5 µL) was added to the dissolved receptor and mixed for 2 hours at room temperature in the dark, after which CNBr was removed using a vacuum centrifuge. Peptides were acidified with 0.5% TFA (500 µL) and left to stand overnight at room temperature. TFA was removed using a vacuum centrifuge and peptides were washed with water (2×100 µL) with drying in between each wash step. The peptides generated by the CNBr treatment were dissolved in 25 µL 0.2% RapiGest solution before performing reduction and alkylation of cysteines. To reduce the disulfide bonds, the sample solution was adjusted to 4 mM DTT and incubated at 55°C for 30 minutes. For the subsequent alkylation of free cysteine residues, the solution was adjusted to 8.3 mM iodoacetamide and incubated at room temperature in the dark for 30 minutes. The CNBr generated, cysteine alkylated peptides were further digested with trypsin and/or chymotrypsin in the presence of 0.2% RapiGest. A 1:5 enzyme-to-protein ratio was used. Digestion was carried out at 37°C overnight. RapiGest was then hydrolyzed by the addition of formic acid at a final concentration of 30%. The peptide mixture was incubated at 37°C for 30 minutes and then centrifuged for 5 minutes. The supernatant was transferred to a clean vial and stored at −20°C until required for analysis.
LC-ES-MS/MS Analysis of NTS1 Fragments
An aliquot of the NTS1 digest mixture (10 µL) was loaded onto a Waters Symmetry 300 C18 NanoEase™ trap column (5µm) (Waters, Milford, MA) using 0.2% aqueous formic acid (mobile phase A) for desalting. Peptides were subsequently separated on a C18 column (VC-10-C18WS-150, Micro-Tech Scientific, Vista, CA) using a gradient with mobile phase B containing 0.2% formic acid in acetonitrile. The eluent from the column was directed into the nano-electrospray source of the mass spectrometer fitted with a 360 µm OD, 75 µm ID, 10 µm tip Picotip emitter (New Objective, Woburn, MA). Reversed phase LC was performed using a CapLC system (Waters) and tandem mass spectrometric analysis was performed using a 7-Tesla linear ion trap Fourier transform ion cyclotron resonance (LTQ-FTICR) mass spectrometer (ThermoFisher, San Jose, CA). Data were acquired in data-dependent mode using Xcalibur software. In each cycle, the five most intense ions detected in FT-ICR mode were automatically isolated and fragmented by CID in the linear ion trap. Bioworks (ThermoFisher) was used to generate dta files and a perl script was used to generate a Mascot Generic Format (MGF) file that was then used to search the Swissprot protein database using the MASCOT (Matrix Science, London, UK) search engine. Typical search parameters include: 10ppm error for MS mode and 0.5 Da for MS/MS mode. The identified peptides were verified by manual interpretation of MS/MS spectra.
Results and Discussion
A combination of the detergents CHAPS and LM in the presence CHS and glycerol is essential to maintain NTS1 in soluble and functional form during solubilization and purification[26]. Although critical for purification, these reagents have adverse effects on proteolytic digestion and LC-ES-MS analyses. Therefore, the NTS1 sample was treated in a manner to substitute the detergents with LC-ES-MS compatible solvents whilst maintaining receptor solubility throughout. The amino acid sequence of recombinant NTS1 is shown in figure 1.
Figure 1.
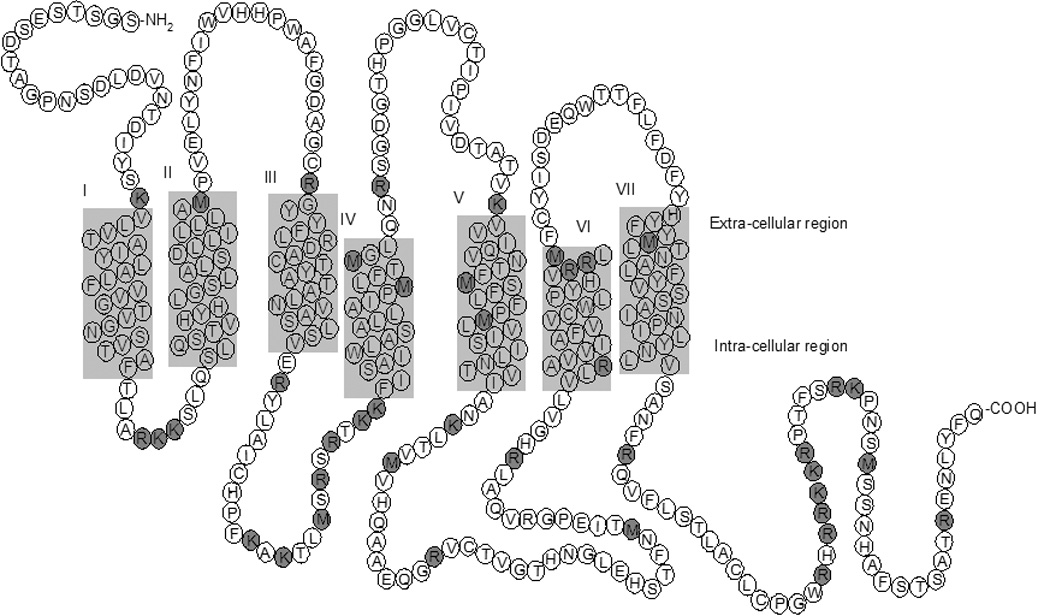
Schematic diagram of the recombinant type 1 rat neurotensin receptor (NTS1). The proposed seven transmembrane domains are labeled I-VII [39]. Predicted CNBr and tryptic cleavage sites are highlighted with dark grey circles.
Analysis of Intact NTS1 Receptor
To analyze the intact NTS1 receptor by ES-MS, it was necessary to dissolve the receptor in MS-compatible solvents. Typical electrospray solvents such as acetonitrile/aqueous acid or methanol/aqueous acid mixtures do not adequately dissolve hydrophobic proteins. A number of research groups showed that the addition of chloroform, which is MS-compatible, to these solvent mixtures improved the solubility of membrane proteins, and hence enabled acquisition of mass spectra of intact seven transmembrane proteins such as bacteriorhodopsin [27; 28] and bacterioopsin [29; 30]. In subsequent studies, the use of chloroform/methanol/aqueous acid was extended further by Whitelegge and co-workers by employing this mixture (4:4:1 v/v/v) as the mobile phase for the separation of membrane proteins by size-exclusion chromatography (SEC) coupled to ES-MS [31; 32; 33; 34]. The method has been successfully applied to the characterization of intact thylakoid membrane proteins [34] and cytochrome b6f complexes [33]. The chloroform/methanol/aqueous acid mixture (4:4:1 v/v/v) employed by Whitelegge and co-workers served as starting point to find suitable mobile phases for the analysis of recombinant NTS1 by SEC-MS. NTS1 precipitated upon addition of the 4:4:1 chloroform/methanol/aqueous formic acid mixture to receptor-containing detergent buffer in preparation for SEC. Therefore, alternative solvent combinations were tested. Adding TFA to receptor-containing detergent buffer, and using a mobile phase of chloroform/methanol/0.5% aqueous TFA (1:3:1 v/v/v) for SEC was found to keep NTS1 soluble and therefore amenable to SEC-MS.
Figure 2a shows the total ion chromatogram (TIC) from SEC-MS analysis of the intact NTS1 receptor using chloroform/methanol/0.5% aqueous TFA (1:3:1 v/v/v) as the mobile phase. The peak eluting at 11−12 minutes corresponded to monomeric NTS1. The summed mass spectrum is shown in figure 2b and deconvolution of the multiply charged spectrum gave a single component with a molecular mass of 43,321 Da. The mass discrepancy between the experimental molecular mass of NTS1 and of that calculated from amino acid sequence shown in figure 1 (43,303 Da) can be accounted for by an oxidation and/or sodium adduct. High acid concentrations employed to solubilize the receptor is the likely cause of amino acid oxidation and sodium was present from the sample preparation. The MSD mass spectrometer used here for intact protein analysis was unable to resolve these products but their presence can be confirmed. The calculated errors between the experimental and theoretical molecular mass of these products are within the error range of the mass spectrometer. The summed mass spectrum of the peak eluting at 13–14 minutes in the TIC showed ion signals from multiple components. Deconvolution of these signals resulted in multiple components with molecular masses approximately twice that of NTS1, suggesting that the second peak (13–14 minutes) corresponded to NTS1 dimeric aggregates. The detergent CHAPS elutes after 20 minutes (figure 2a) and was thus sufficiently separated from NTS1 to allow the molecular mass determination of the receptor.
Figure 2.
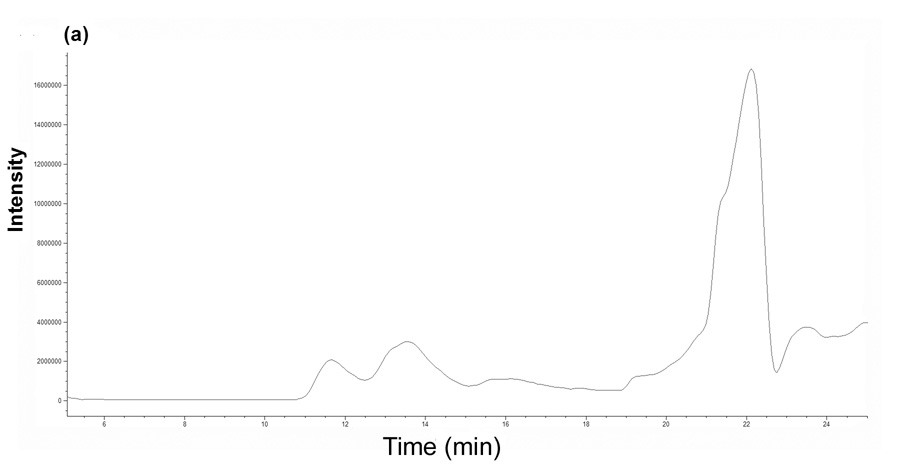

Intact protein analysis of NTS1 using SEC-MS. (a) Total ion chromatogram and (b) ES mass spectrum of the NTS1 receptor (summed spectra between 12–13 minutes from the total ion chromatogram). The charge states of NTS1 are shown in bold. Deconvolution of the ES spectrum resulted in a molecular mass of 43,321 Da (theoretical value is 43,303 Da based on amino acid sequence shown in figure 1).
Digestion and LC-ES-MS/MS Analysis of NTS1 Fragments
As an initial experiment, we performed digestion of purified NTS1 in detergent buffer with trypsin. The resulting peptides were identified by reversed phase LC-MS/MS using acetonitrile/water/0.2% formic acid mixtures as the mobile phase. With this approach, less than 20% of the receptor sequence was covered, with the majority of the observed peptides corresponding to regions from the hydrophilic intra-and extra-cellular loops (data not shown). Several factors may account for these results. First, low sequence coverage may have been caused by the presence of high concentrations of detergents which can affect the ionization efficiency of peptides. Extracted ion chromatograms of CHAPS (m/z 615.40) and LM (m/z 511.31) showed the monomers and their multimers eluting as broad peaks overshadowing coeluting NTS1 peptides (data not shown). The strategy to remove detergent by SEC, as was done with the intact receptor, could not be applied to peptide separation because the molecular masses of CHAPS and LM (monomer and multimers) are too similar to the NTS1 peptides. Second, NTS1 may have stayed tightly folded in the presence of the detergents CHAPS/LM/CHS such that trypsin could not access all cleavage sites in NTS1 leading to long hydrophobic peptide due to trypsin missed cleavages. Third, some of the tryptic peptides are predicted to contain one or more TM domain due to lack of tryptic cleavage sites within these regions. These long hydrophobic tryptic peptides may not be amenable to separation by reversed phase LC, have poor ionization efficiency or fragmentation by collision induced dissociation (CID) and thus could not be observed.
To test whether digestion efficiency, reversed phase LC separation, electrospray ionization and fragmentation by CID are influenced by the length and amino acid composition of TM domains, a portion of the rat NTS1 receptor sequence was synthesized and used as a model TM peptide. The amino acid sequence of this synthetic peptide (IYSKVLVTAIYLALFVVGTVGNSVTAFTLARKKSLQSLQSTVHYHLGSLALSDLLILLLAMPVELY) corresponded to TM I, loop I and TM II of NTS1 (figure 1). We recognize that the synthetic peptide represents only a small portion of NTS1 and that trypsin digest conditions optimal for this peptide may not necessarily present conditions suitable for the entire protein. Nonetheless, we used this peptide as representative of the typical length and hydrophobicity of peptides potentially produced from digestion of NTS1 and therefore as a useful model to investigate appropriate experimental conditions for their retention and separation by reversed phase LC and also ES ionization and fragmentation by CID. This particular portion of the NTS1 receptor was chosen because in previous digestions no peptides corresponding to the TM I domain were observed, but peptides corresponding to the TM II domain were found frequently. The synthetic peptide was digested with trypsin in the presence of either 60:40 (v/v) methanol/50 mM ammonium bicarbonate, 80:20 (v/v) acetonitrile/50 mM ammonium bicarbonate or an acid labile detergent (Rapigest) to generate two tryptic peptides comprising TM I and TM II of NTS1. These digestion buffers were chosen because the synthetic NTS1 peptide dissolved in them and they have been shown to enhance the number of peptides in tryptic digests of integral membrane proteins[35; 36; 37]. Figure 3 shows the extracted ion chromatograms of the triply charged peptides, VLVTAIYLALFVVGTVGNSVTAFTLAR (TM I) and SLQSLQSTVHYHLGSLALSDLLILLLAMPVELY (TM II), from a LC-ES-MS analysis of a trypsin digestion of the synthetic NTS1 peptide in the presence of 0.2% Rapigest in ammonium bicarbonate. These two peptides could be separated by reversed phase LC using acetonitrile/water/0.2% formic acid mixtures and were successfully analyzed by ES-MS and ES-MS/ MS. Therefore, it is probable that the observation of low sequence coverage in previous experiments with the NTS1 receptor was due to inefficient digestion rather than to the properties of the TM peptides.
Figure 3.
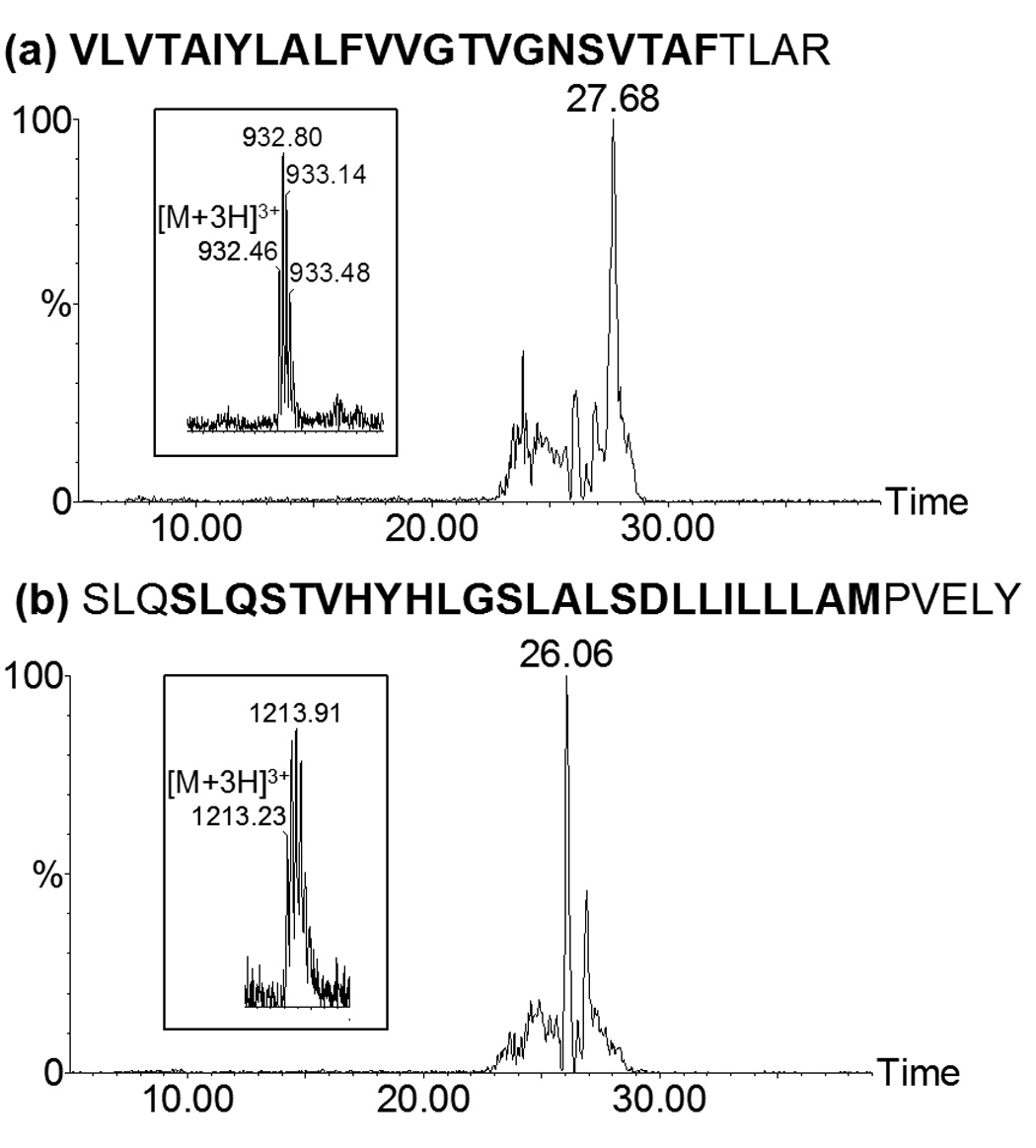
LC-MS analysis of a trypsin digestion mixture of the synthetic NTS1 peptide IYSKVLVTAIYLALFVVGTVGNSVTAFTLARKKSLQSLQSTVHYHLGSLALSDLLILLLAMPVEY. Extracted ion chromatograms and ES mass spectrum of the triply charged tryptic peptides (a) VLVTAIYLALFVVGTVGNSVTAFTLAR which eluted at 27.68 min, and (b) SLQSLQSTVHYHLGSLALSDLLILLLAMPVELY which eluted at 26.06 min. Mass spectra of the parent ion for each peptide are shown in the inserts. Bold text indicates amino acids in the TM domains. Mass spectra were acquired using an electrospray Q-ToF II mass spectrometer.
Methods to improve the efficiency of proteolytic enzymes are to reduce or completely remove detergents and salts and/or to denature NTS1 prior to digestion. We therefore chose to precipitate the receptor using a mixture of chloroform/methanol/aqueous TFA instead of separating the protein by 1D SDS PAGE prior to digestion. Our previous efforts with in-gel digestion of the receptor resulted in low coverage of NTS1 peptides; this may have been due to poor digestion efficiency and recovery of hydrophobic peptides from the gel matrix. Various buffer compositions were tested to dissolve the precipitated NTS1 receptor, including ammonium bicarbonate buffer with varying percentages of methanol, acetonitrile, Rapigest, or urea at various concentrations, but these attempts were all unsuccessful. It was found that precipitated NTS1 receptor required more than 50% of either formic or trifluoroacetic acid to dissolve. Under these conditions, trypsin would be inactive, however cleavage with CNBr at methionine residues would be feasible since this reaction requires acidic conditions. No peptides were observed after CNBr treatment of NTS1. It is possible that the CNBr reaction was inefficient, resulting in peptides, which were too large. These large hydrophobic peptides may have precipitated before or during their LC separation and/or were bound irreversibly to the reversed phase chromatographic material and therefore were not observed by ES-MS. To generate peptides of lengths suitable for LC-ES-MS/MS analysis, trypsin and/or chymotrypsin digestions were performed in the presence of the acid-labile surfactant Rapigest after treatment with CNBr. The concentration of formic acid (30% final) used to hydrolyze Rapigest after digestion was not reduced prior to LC-ES-MS/MS analysis, as a high acid concentration was found to help maintain the hydrophobic peptides soluble. LC-ES-MS/MS analysis of the peptide mixture generated from CNBr treatment followed by trypsin digestion of the NTS1 receptor resulted in 84% sequence coverage. Table 1 shows the list of peptides that were observed; peptides that overlap in sequence due to missed cleavages are not shown. Peptide identifications were confirmed using accurate peptide mass (less than 10 ppm) and MS/MS data. Since the acquisition m/z range was 300–2000, multiply charged peptides outside this range (e.g.; AK, TLM) were not detected. All seven TM domains were either partially or completely covered by a single peptide, for example VLVTAIYLALFVVGTVGNSVTAFTLAR (TM I), or by several peptides. The observation of peptides generated from CNBr and/or tryptic cleavages within TM domains, for example (R)GYYFLR and (R)DACTYATALNVASLSVER, both from TM V, indicated that NTS1 was sufficiently denatured to expose CNBr and tryptic cleavage sites. The CID MS/MS spectra for these peptides are shown in figure 4. As expected, the high formic acid concentrations required for maintaining solubility of NTS1 receptor and its peptide fragments caused oxidation and formylation of amino acid residues in several peptides. The peptide derived from TM I, VLVTAIYLALFVVGTVGNSVTAFTLAR, was present both as the unmodified and formylated form. An MS/MS spectrum was acquired for the formylated peptide only (figure 4), but the presence of the unmodified form could be confirmed by the observation of the triply charged ion in the FT-ICR mass spectrum with high mass accuracy (<10 ppm).
Table 1.
Peptides identified from LC-ES-MS/MS analysis of CNBr cleavage followed by trypsin digestion of the precipitated NTS1 receptor. Letters in bold correspond to TM domains. Overlapping peptides are not shown. Formyl = formylation, Hse_lact = homoserine lactone, ox = oxidation and * = carbamidomethylation.
| Peptide sequence | Residue number | Experimental mass (m/z) | Theoretical mass (m/z) | Mass Accuracy (ppm) | Charge state |
|---|---|---|---|---|---|
| SGSTSESDTAGPNSDLDVNTDIYSK | 1–25 | 1280.568 | 1280.562 | 4.3 | 2+ |
| VLVTAIYLALFVVGTVGNSformylVTAFTLAR | 26–52 | 941.877 | 941.872 | 4.7 | 3+ |
| SLQ SLQSTVHY | 55–65 | 631.824 | 631.822 | 2.0 | 2+ |
| LLILLLAMHse_lact | 75–82 | 851.600 | 851.597 | 3.9 | 1+ |
| PVELYNFIWVHHPWAFGDAGC*R | 83–104 | 668.574 | 668.572 | 2.8 | 4+ |
| GYYFLR | 105–110 | 409.713 | 409.713 | 0 | 2+ |
| DAC*TYATALNVASLSVER | 111–128 | 970.977 | 970.972 | 4.3 | 2+ |
| DAC*TYATALNVASLSVERYLAIC*HPFK | 111–137 | 1024.181 | 1024.176 | 5.0 | 3+ |
| YLAIC*HPFK | 129–137 | 574.800 | 574.800 | 0 | 2+ |
| LASALLAIPMoxLFTMoxGLQNR | 156–174 | 1046.572 | 1046.568 | 3.8 | 2+ |
| LFTMoxGLQNR | 166–174 | 548.285 | 548.285 | 0 | 2+ |
| SGDGTHPGGLVC*TPIVDTATVK | 175–196 | 728.033 | 728.032 | 2.1 | 3+ |
| VVIQVNTFMHse_lact | 197–205 | 1002.563 | 1002.562 | 1.3 | 1+ |
| SFLFPMHse_lact | 206–211 | 693.362 | 693.361 | 2.5 | 1+ |
| LVISILNTVIANK | 212–224 | 699.442 | 699.440 | 3.0 | 2+ |
| LTVMVHQAAEQGR | 225–237 | 720.376 | 720.375 | 1.4 | 2+ |
| VC*TVGTHNGLEHSTFNMHse_lact | 238–254 | 928.425 | 928.423 | 2.4 | 2+ |
| VC*TVGTHNGLEHSTFNMoxTIEPGR | 238–260 | 858.402 | 858.401 | 1.8 | 3+ |
| HGVLVLR | 266–272 | 397.256 | 397.256 | 0 | 2+ |
| AVVIAFVVC*WoxLPYHVR | 273–288 | 973.034 | 973.029 | 4.5 | 2+ |
| FYHYFYMHse_lact | 307–313 | 511.725 | 511.724 | 1.9 | 2+ |
| LTNALFYVSSAINPILYNLVSANFR | 314–338 | 1400.763 | 1400.755 | 5.3 | 2+ |
| QVFLSTLAC*LC*PGWR | 339–353 | 904.457 | 904.453 | 4.5 | 2+ |
| KformylRPTFSR | 358–364 | 307.175 | 307.175 | 0 | 3+ |
| KPNSMoxSSNHAFSTSATR | 365–381 | 613.620 | 613.620 | 0 | 3+ |
| SformylSNHAFSTSATR | 370–381 | 647.295 | 647.295 | 0 | 2+ |
| ENLYFQ | 382–387 | 813.378 | 813.378 | 0 | 1+ |
Figure 4.

Examples of CID MS/MS spectra of NTS1 peptides that were observed in the LC-ES-MS/ MS analysis of CNBr followed by trypsin digestion of precipitated NTS1.
(* = carbamidomethylation and formyl = formylation ). Mass spectra of the parent ion for each peptide are shown in the inserts.
Figure 5 shows the base peak ion (BPI) chromatogram for a typical LC-ES-MS/MS analysis of a CNBr and trypsin digestion mixture of NTS1 and extracted chromatograms for peptide SGSTSESDTAGPNSDLDVNTDIYSK and VLVTAIYLALFVVGTVGNSformylVTAFTLAR, which, based on their Bull and Breese values [38] represent the most hydrophilic and hydrophobic peptides in the digestion mixture, respectively. Peptide retention times were observed to increase with increasing hydrophobicity, a trend generally observed with reversed phase LC separations. Peptide ion intensities of hydrophobic peptides were observed to be lower when compared to hydrophilic peptides. One explanation for this difference could be poor recovery of hydrophobic peptides from the reversed phase column and electrospray ionization efficiency.
Figure 5.
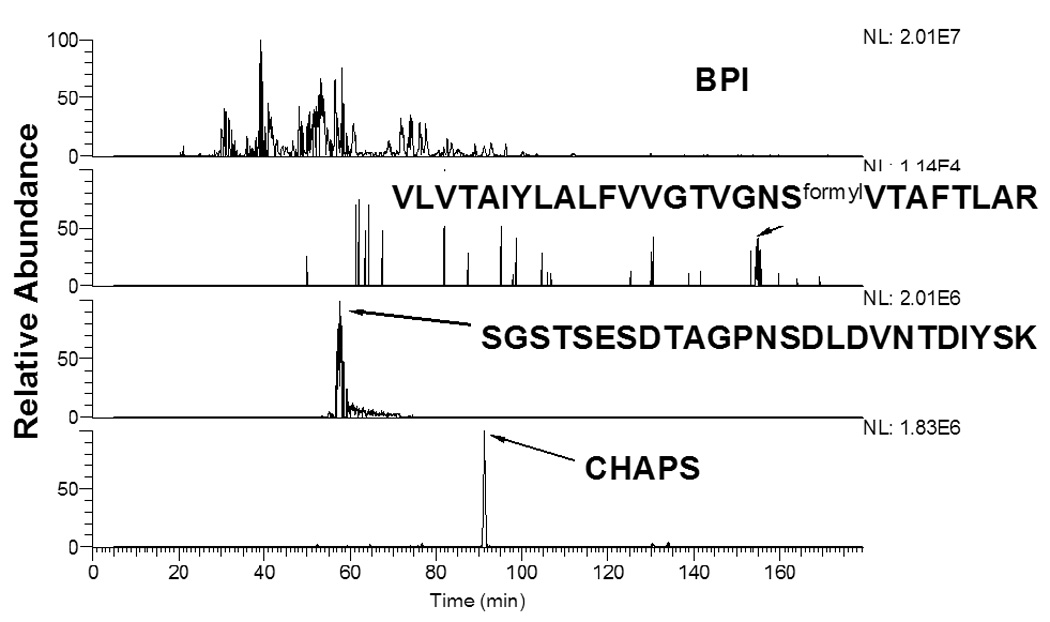
Base peak ion (BPI) chromatogram of LC-MS/MS analysis of CNBr followed by trypsin digestion of the precipitated NTS1 receptor. Extracted ion chromatograms of the most hydrophilic (SGSTSESDTAGPNSDLDVNTDIYSK, [M+2H]2+) and hydrophobic (VLVTAIYLALFVVGTVGNSformylVTAFLAR, [M+3H]3+) peptides based on their Bull and Breese values and the detergent CHAPS.
To further improve sequence coverage of the NTS1 receptor, a combined trypsin and chymotrypsin digestion was performed after CNBr treatment of the precipitated protein (table 2). This resulted in 83% coverage of the NTS1 sequence with the observation of extra-cellular loop III fragments, which were not obtained from CNBr and trypsin digestion alone. But peptides from TM IV were now absent. Overall 88% sequence coverage was achieved when the observed peptides from independent digestion methods (CNBr+trypsin and CNBr+trypsin+chymotrypsin) were combined (figure 6).
Table 2.
Peptides identified from LC-ES-MS/MS analysis of CNBr cleavage followed by trypsin and chymotrypsin digestion of the precipitated NTS1 receptor. Letters in bold correspond to TM domains. Overlapping peptides are not shown. Formyl = formylation, Hse_lact = homoserine lactone, ox = oxidation, Hse = homoserine and * = carbamidomethylation.
| Peptide sequence | Residue number | Experimental mass (m/z) | Theoretical mass (m/z) | Mass Accuracy (ppm) | Charge state |
|---|---|---|---|---|---|
| SGSTSESDT AGPNSDLDVNTDIYSK | 1–25 | 1280.571 | 1280.562 | 7.0 | 2+ |
| VLVTAIYLALFVVGTVGNSVTAF | 26–49 | 785.454 | 785.451 | 3.0 | 3+ |
| SLQSLQSTVHY | 55–65 | 631.824 | 631.822 | 1.7 | 2+ |
| HLGSLALSformylDLL | 66–76 | 583.827 | 583.825 | 4.5 | 2+ |
| ILLLAMoxPVELY | 77–87 | 645.876 | 645.873 | 4.5 | 2+ |
| *NH2-PVELYNFIWVHHPWAFGDAGC*R | 83–104 | 682.831 | 682.827 | 5.1 | 4+ |
| DAC*TYATALNVASLSVER | 111–128 | 970.980 | 970.973 | 7.0 | 2+ |
| YLAIC*HPFK | 129–137 | 383.537 | 383.536 | 2.7 | 3+ |
| KFISAIWLASALL | 149–161 | 716.935 | 716.932 | 5.0 | 2+ |
| LAIPMHse_lact | 161–165 | 496.314 | 496.313 | 2.8 | 1+ |
| LFTMoxGLQNR | 166–174 | 548.286 | 548.285 | 2.8 | 2+ |
| SGDGTHPGGLVC*Tformyl/PIVDTATVK | 175–196 | 1105.550 | 1105.542 | 7.6 | 2+ |
| VVIQVNTFMHse_lact | 197–205 | 501.787 | 501.785 | 4.0 | 2+ |
| *NH2-SFLFPMoxLVISILNTVIANK | 206–224 | 731.753 | 731.750 | 4.4 | 3+ |
| LTVMoxVHQAAEQGR | 225–237 | 728.375 | 728.372 | 3.6 | 2+ |
| VC*TVGTHNGLEHSTF | 238–252 | 829.887 | 829.883 | 4.1 | 2+ |
| NMoxTIEPGR | 253–260 | 933.450 | 933.446 | 4.5 | 1+ |
| C*YISDEQWTTF | 293–303 | 725.307 | 725.303 | 5.0 | 2+ |
| LFDFYHYF | 304–311 | 576.266 | 576.264 | 4.3 | 2+ |
| LFDFYHYFYMHse | 304–313 | 708.323 | 708.319 | 5.5 | 2+ |
| LTNALFYVSSAINPILY | 314–330 | 950.022 | 950.017 | 5.7 | 2+ |
| YVSSAINPILYNLVSANFR | 320–338 | 1071.079 | 1071.073 | 5.6 | 2+ |
| QVFLSTLAC*LC*PGW | 339–352 | 826.407 | 826.402 | 5.5 | 2+ |
| Kformyl/PNSMoxSSNHAF | 365–375 | 632.277 | 632.275 | 2.7 | 2+ |
| KPNSMoxSSNHAFSTSATR | 361–381 | 919.928 | 919.926 | 1.5 | 2+ |
| ENLYFQ | 382–387 | 813.380 | 813.378 | 2.8 | 1+ |
Figure 6.

Sequence coverage of the NTS1 receptor. The proposed TM domains [39] are highlighted in bold. Peptides that were observed in the LC-MS/MS analysis of a CNBr+trypsin and CNBr+trypsin+chymotrypsin digestion of precipitated NTS1 are underlined. The peptides observed cover 88% of the protein sequence shown.
CHAPS and LM were not completely removed after performing the chloroform/methanol/TFA precipitation, but their ion abundances were reduced approximately 10-fold. Both CHAPS (extracted ion chromatogram in figure 5) and LM were now observed as sharp peaks and eluted at positions distant from the peptides. This suggests that interactions between the hydrophobic protein core and detergent molecules were largely disrupted by the chloroform-methanol precipitation, so that released detergent molecules could be observed as distinct entities. Reducing detergent concentrations by performing a protein precipitation unfolded the protein and thus allowed efficient digestion of NTS1 and the LC-ES-MS/MS analysis of both hydrophobic and hydrophilic peptides, thus increasing the overall protein sequence coverage.
Conclusion
A mixture of detergents and salts are essential to maintain solubility and functionality of GPCRs, whether they originate from natural sources or are expressed in a recombinant system. In order to perform comprehensive analysis of GPCRs by MS, experimental methodology is required to minimize detergents and salts in the receptor sample. With an appropriate, MS-compatible sample preparation we were able to perform MS analyses both with the intact NTS1 and receptor peptides. To determine the molecular mass of the intact monomeric NTS1 receptor, detergents were substituted with a chloroform/methanol/aqueous TFA mixture by SEC. This solvent mixture was ES-MS compatible and a mass spectrum of multiply charged NTS1 was obtained. For receptor sequence analysis, NTS1 was precipitated to reduce detergent interference and then digested with either CNBr+trypsin or CNBr+trypsin+chymotrypsin. Peptides observed in the LC-ES-MS/MS analysis of the digestion mixtures covered more than 80% of the protein sequence, including all seven TM domains. The work presented here provides an important step towards the detailed characterization of NTS1 by MS. It is anticipated that these MS methods will allow the characterization of post-translational modifications and ligand binding sites on NTS1, and provide complementary structural information to NMR and X-ray crystallography studies. Together, these techniques will enhance our understanding of the structure-function relationship of the neurotensin receptor.
Acknowledgements
We thank G. Abdoulaeva and N. Y. Nguyen from the Center for Biologics Evaluation and Research, (CBER), FDA, Bethesda, MD for the synthesis of the model TM peptide. This work was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Neurological Disorders and Stroke, and the Betty and Gordon Moore Foundation. Experiments by J.T.C.H. and S.H. were conducted at the Proteomics and Mass Spectrometry Facility of NIDDK, part of the experiments by J.F.W. and R.G. were conducted at the Laboratory of Molecular Biology, NIDDK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kitabgi P. Targeting neurotensin receptors with agonists and antagonists for therapeutic purposes. Curr. Opin. Drug Discov. Devel. 2002;5:764–776. [PubMed] [Google Scholar]
- 2.Barroso S, Richard F, Nicolas-Etheve D, Reversat JL, Bernassau JM, Kitabgi P, Labbe-Jullie C. Identification of residues involved in neurotensin binding and modeling of the agonist binding site in neurotensin receptor 1. J. Biol. Chem. 2000;275:328–336. doi: 10.1074/jbc.275.1.328. [DOI] [PubMed] [Google Scholar]
- 3.Pang YP, Cusack B, Groshan K, Richelson E. Proposed ligand binding site of the transmembrane receptor for neurotensin(8–13) J. Biol. Chem. 1996;271:15060–15068. doi: 10.1074/jbc.271.25.15060. [DOI] [PubMed] [Google Scholar]
- 4.Luca S, White JF, Sohal AK, Filippov DV, van Boom JH, Grisshammer R, Baldus M. The conformation of neurotensin bound to its G protein-coupled receptor. PNAS. 2003;100:10706–10711. doi: 10.1073/pnas.1834523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sinz A. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J. Mass Spectrom. 2003;38:1225–1237. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
- 6.Sinz A. Investigation of protein-ligand interactions by mass spectrometry. Chemmedchem. 2007;2:425–431. doi: 10.1002/cmdc.200600298. [DOI] [PubMed] [Google Scholar]
- 7.Ogorzalek Loo RR, Dales N, Andrews PC. The Effect of Detergents on Proteins Analyzed by Electropsray Ionization. In: Chapman J, editor. Protein and Peptide Analysis by Mass Spectrometry. Humana Press Inc.; 1996. pp. 141–160. [DOI] [PubMed] [Google Scholar]
- 8.Ball LE, Oatis JE, Dharmasiri K, Busman M, Wang JY, Cowden LB, Galijatovic A, Chen N, Crouch RK, Knapp DR. Mass spectrometric analysis of integral membrane proteins: Application to complete mapping of bacteriorhodopsins and rhodopsin. Protein Sci. 1998;7:758–764. doi: 10.1002/pro.5560070325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kraft P, Mills J, Dratz E. Mass spectrometric analysis of cyanogen bromide fragments of integral membrane proteins at the picomole level: Application to rhodopsin. Anal. Biochem. 2001;292:76–86. doi: 10.1006/abio.2001.5072. [DOI] [PubMed] [Google Scholar]
- 10.Ablonczy Z, Kono M, Crouch RK, Knapp DR. Mass spectrometric analysis of integral membrane proteins at the subnanomolar level: Application to recombinant photopigments. Anal. Chem. 2001;73:4774–4779. doi: 10.1021/ac015563n. [DOI] [PubMed] [Google Scholar]
- 11.Ablonczy Z, Crouch RK, Knapp DR. Mass spectrometric analysis of integral membrane proteins at the subpicomolar level: Application to rhodopsin. J. Chromatogr. B-Analyt. Technol. Biomed. Life Sci. 2005;825:169–175. doi: 10.1016/j.jchromb.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Trester-Zedlitz M, Burlingame A, Kobilka B, von Zastrow M. Mass spectrometric analysis of agonist effects on posttranslational modifications of the beta-2 adrenoceptor in mammalian cells. Biochemistry. 2005;44:6133–6143. doi: 10.1021/bi0475469. [DOI] [PubMed] [Google Scholar]
- 13.Roos M, Soskic V, Poznanovic S, Grodovac-Zimmermann J. Post-translational modifications of endothelin receptor B from bovine lungs analyzed by mass spectrometry. J. Biol. Chem. 1998;273:924–931. doi: 10.1074/jbc.273.2.924. [DOI] [PubMed] [Google Scholar]
- 14.Suma NYZvonok, John Williams, Shujia Dai, Keling Dong, Thomas Rejtar, Barry KargerL, Makriyannis, Alexandros Comprehensive Proteomic Mass Spectrometeric Characterization of Human Cannabinoid CB2 Receptor. J. Proteome Res. 2007;6:2068–2079. doi: 10.1021/pr060671h. [DOI] [PubMed] [Google Scholar]
- 15.Christoffers KH, Li H, Keenan SA, Howells RD. Purification and mass spectrometric analysis of the mu opioid receptor. Mol. Brain Res. 2003;118:119–131. doi: 10.1016/j.molbrainres.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Christoffers KH, Li H, Howells RD. Purification and mass spectrometric analysis of the delta opioid receptor. Mol. Brain Res. 2005;136:54–64. doi: 10.1016/j.molbrainres.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Alves ID, Sachon E, Bolbach G, Millstine L, Lavielle S, Sagan S. Analysis of an intact G-protein coupled receptor by MALDI-TOF mass spectrometry: Molecular heterogeneity of the tachykinin NK-1 receptor. Anal. Chem. 2007;79:2189–2198. doi: 10.1021/ac062415u. [DOI] [PubMed] [Google Scholar]
- 18.Barnidge DR, Dratz EA, Sunner J, Jesaitis AJ. Identification of transmembrane tryptic peptides of rhodopsin using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Protein Sci. 1997;6:816–824. doi: 10.1002/pro.5560060408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KA, Craven KB, Niemi GA, Hurley JB. Mass spectrometric analysis of the kinetics of in vivo rhodopsin phosphorylation. Protein Sci. 2002;11:862–874. doi: 10.1110/ps.3870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papac DI, Oatis JE, Crouch RK, Knapp DR. Mass-Spectrometric Identification of Phosphorylation Sites in Bleached Bovine Rhodopsin. Biochemistry. 1993;32:5930–5934. doi: 10.1021/bi00074a002. [DOI] [PubMed] [Google Scholar]
- 21.Papac DI, Thornburg KR, Bullesbach EE, Crouch RK, Knapp DR. Palmitylation of a G-Protein Coupled Receptor - Direct Analysis by Tandem Mass-Spectrometry. J. Biol. Chem. 1992;267:16889–16894. [PubMed] [Google Scholar]
- 22.Wang X, Kim SH, Ablonczy Z, Crouch RK, Knapp DR. Probing rhodopsin-transducin interactions by surface modification and mass spectrometry. Biochemistry. 2004;43:11153–11162. doi: 10.1021/bi049642f. [DOI] [PubMed] [Google Scholar]
- 23.Filppula S, Yaddanapudi S, Mercier R, Xu W, Pavlopoulos S, Cai J, Pierce WM, Makriyannis A. Purification and mass spectroscopic analysis of human CB2 cannabinoid receptor expressed in the baculovirus system. J. Pept. Res. 2004;64:225–236. doi: 10.1111/j.1399-3011.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 24.Whitelegge JP, Gundersen CB, Faull KF. Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci. 1998;7:1423–1430. doi: 10.1002/pro.5560070619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White JF, Trinh LB, Shiloach J, Grisshammer R. Automated large-scale purification of a G protein-coupled receptor for neurotensin. FEBS Lett. 2004;564:289–293. doi: 10.1016/S0014-5793(04)00195-4. [DOI] [PubMed] [Google Scholar]
- 26.Grisshammer R, White JF, Trinh LB, Shiloach J. Large-scale Expression and Purification of a G-protein-coupled Receptor for structure Determination - an overview. J. Struct. and Funct. Genomics. 2005;6:159–163. doi: 10.1007/s10969-005-1917-6. [DOI] [PubMed] [Google Scholar]
- 27.Hufnagel P, Schweiger U, Eckerskorn C, Oesterhelt D. Electrospray ionization mass spectrometry of genetically and chemically modified bacteriorhodopsins. Anal. Biochem. 1996;243:46–54. doi: 10.1006/abio.1996.0480. [DOI] [PubMed] [Google Scholar]
- 28.Barnidge DR, Dratz EA, Jesaitis AJ, Sunner J. Extraction method for analysis of detergent-solubilized bacteriorhodopsin and hydrophobic peptides by electrospray ionization mass spectrometry. Anal. Biochem. 1999;269:1–9. doi: 10.1006/abio.1999.4012. [DOI] [PubMed] [Google Scholar]
- 29.Schaller J, Pellascio BC, Schlunegger UP. Analysis of hydrophobic proteins and peptides by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1997;11:418–426. [Google Scholar]
- 30.Schindler PA, Vandorsselaer A, Falick AM. Analysis of Hydrophobic Proteins and Peptides by Electrospray-Ionization Mass-Spectrometry. Anal. Biochem. 1993;213:256–263. doi: 10.1006/abio.1993.1418. [DOI] [PubMed] [Google Scholar]
- 31.Whitelegge JP, le Coutre J, Lee JC, Engel CK, Prive GG, Faull KF, Kaback HR. Toward the bilayer proteome, electrospray ionization-mass spectrometry of large, intact transmembrane proteins. PNAS. 1999;96:10695–10698. doi: 10.1073/pnas.96.19.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.le Coutre J, Whitelegge JP, Gross A, Turk E, Wright EM, Kaback HR, Faull KF. Proteomics on full-length membrane proteins using mass spectrometry. Biochemistry. 2000;39:4237–4242. doi: 10.1021/bi000150m. [DOI] [PubMed] [Google Scholar]
- 33.Whitelegge JP, Zhang HM, Aguilera R, Taylor RM, Cramer WA. Full subunit coverage liquid chromatography electrospray ionization mass spectrometry (LCMS+) of an oligomeric membrane protein - Cytochrome b6f complex from spinach and the cyanobacterium Mastigocladus laminosus. Mol. Cell. Proteomics. 2002;1:816–827. doi: 10.1074/mcp.m200045-mcp200. [DOI] [PubMed] [Google Scholar]
- 34.Gomez SM, Nishio JN, Faull KF, Whitelegge JP. The chloroplast grana proteome defined by intact mass measurements from liquid chromatography mass spectrometry. Mol. Cell. Proteomics. 2002;1:46–59. doi: 10.1074/mcp.m100007-mcp200. [DOI] [PubMed] [Google Scholar]
- 35.Blonder J, Conrads TP, Yu LR, Terunuma A, Janini GM, Issaq HJ, Vogel JC, Veenstra TD. A detergent- and cyanogen bromide-free method for integral membrane proteomics: Application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics. 2004;4:31–45. doi: 10.1002/pmic.200300543. [DOI] [PubMed] [Google Scholar]
- 36.Fischer F, Wolters D, Rogner M, Poetsch A. Toward the complete membrane proteome - High coverage of integral membrane proteins through transmembrane peptide detection. Mol. Cell. Proteomics. 2006;5:444–453. doi: 10.1074/mcp.M500234-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Yu YQ, Gilar M, Gebler JC. A complete peptide mapping of membrane proteins: a novel surfactant aiding the enzymatic digestion of bacteriorhodopsin. Rapid Commun. Mass Spectrom. 2004;18:711–715. doi: 10.1002/rcm.1374. [DOI] [PubMed] [Google Scholar]
- 38.Bull HB, Breese K. Surface-Tension of Amino-Acid Solutions - Hydrophobicity Scale of Amino-Acid Residues. Arch. Biochem. Biophys. 1974;161:665–670. doi: 10.1016/0003-9861(74)90352-x. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka K, Masu M, Nakanishi S. Structure and Functional Expression of the Cloned Rat Neurotensin Receptor. Neuron. 1990;4:847–854. doi: 10.1016/0896-6273(90)90137-5. [DOI] [PubMed] [Google Scholar]