

Abstract
Mycobacterium smegmatis MshC catalyzes the ATP-dependent condensation of GlcN-Ins and L-cysteine to form L-Cys-GlcN-Ins, the penultimate step in mycothiol biosynthesis. Attempts to crystallize the native, full-length MshC have been unsuccessful. However, incubation of the enzyme with the cysteinyl adenylate analogue, 5′-O-[N-(L-cysteinyl)-sulfamonyl]adenosine (CSA), followed by a 24-hour limited trypsin proteolysis yielded an enzyme preparation that readily crystallized. The three-dimensional structure of MshC with CSA bound in the active site was solved and refined to 1.6 Å. The refined structure exhibited electron density corresponding to the entire 47 kDalton MshC molecule, with the exception of the KMSKS loop (residues 285–297), a loop previously implicated in the formation of the adenylate in related tRNA synthases. The overall tertiary fold of MshC is similar to that of cysteinyl-tRNA synthetase, with a Rossmann fold catalytic domain. The interaction of the thiolate of CSA with a zinc ion at the base of the active site suggests that the metal ion participates in amino acid binding and discrimination. A number of active site residues were observed to interact with the ligand, suggesting a role in substrate binding and catalysis. Analysis utilizing modeling of the proteolyzed loop and GlcN-Ins docking, as well as the examination of sequence conservation in the active site suggests similarities and differences between cysteinyl-tRNA synthetases and MshC in recognition of the substrates for their respective reactions.
Keywords: Mycobacterium, mycothiol, cysteine ligase, limited proteolysis, three-dimensional structure
Actinomycetes produce mycothiol (MSH, acetyl-cys-GlcN-Ins) as the predominant low molecular weight thiol to minimize oxidative stress and protect against electrophilic toxins (1-4). Among actinomycetes, mycobacteria generate the highest intracellular levels of MSH (5). Mycobacterium smegmatis mutants which are deficient in MSH production become more sensitive towards oxidizing agents, electrophiles, and antibiotics (1-3), indicating the critical role of MSH in the survival and pathogenicity of mycobacteria (1). In contrast, eukaryotes and many eubacteria produce glutathione (GSH). This suggests that the enzymes involved in the mycothiol biosynthetic pathway may be potential targets for selective antimicrobial chemotherapy.
MSH is synthesized via a series of four unique enzyme-catalyzed reactions (6-9), as illustrated in Scheme 1. Mycothiol is reversibly oxidized by cellular oxidants and can react with electrophiles to form the S-conjugates. A detailed biochemical characterization of the cysteine ligase (MshC) from Mycobacterium smegmatis, which is the penultimate enzyme in mycothiol biosynthetic pathway, has been performed (10, 11). MshC catalyzes the ATP-dependent condensation of cysteine and GlcN-Ins via a Ping Pong kinetic mechanism (Scheme 2). It has been proposed based on sequence comparisons that MshC is related to cysteinyl-tRNA synthetase (CysRS), and shares a common evolutionary origin (12). Adenylate-forming enzymes, including class I and II aminoacyl-tRNA synthetases (AaRS) (13), pantothenate synthetase (14), malonyl-CoA synthetase (15), and acyl- and aryl-CoA synthetases/ligases (16-18) catalyze reactions that generally can be divided into two halves. In the first half-reaction, one substrate reacts with ATP to form an adenylate and inorganic pyrophosphate; in the second half-reaction the second substrate reacts with the adenylated substrate to generate the product and AMP. The three-dimensional structures of adenylate-forming enzymes comprise multiple domains that are usually linked by flexible loops to allow conformational changes upon substrate binding or domain alternation. In class I CysRS's, binding of cysteine induces a significant conformational change including the movement of the zinc ion towards the substrate, the movement of the conserved W205 indole ring to interact with the cysteine side chain, as well as the closure of the loop which contains the KMSKS signature motif (19). In the case of 4-chlorobenzoate:CoA ligase, the C-terminal domain rotates by ∼140° to permit alternate catalysis of adenylation and thioester formation reactions (20). Such a large conformational change has been termed “domain alternation”, and was first introduced to describe such structural changes in B12-dependent methionine synthetase (21).
Scheme 1.

Mycothiol biosynthesis. The MshA-D enzymes catalyze a glycosyltransferase, deacetylase, a ligase and an acetytransferase reaction, respectively. The phosphatase is depicted as being unknown at present.
Scheme 2.

The reactions catalyzed by MshC.
MshC from M. smegmatis has been cloned, expressed, purified and kinetically characterized (11). The enzyme is a monomer with a molecular weight of ∼ 47 kDa. A stable analog of the intermediate cysteineadenylate, 5′-O-[N-(L-cysteinyl)sulfamonyl]adenosine (CSA), exhibits competitive inhibition versus ATP, with an inhibition constant of ∼300 nM versus ATP. Previous crystallization efforts of MshC, using a variety of screening conditions and with a number of different molecular constructs failed to produce protein crystals.
Limited proteolysis has been documented as a powerful tool to obtain structural information on stable domains in proteins that have resisted crystallization (22, 23). It has been observed that limited proteolysis occurs exclusively as solvent-accessible and flexible regions of exemplary proteins (24). These flexible regions include disordered N- and C-termini as well as exposed loops or linkers between domains (25), which could be the cause for unsuccessful crystallization. Limited proteolysis generally yields a nicked or cleaved species with the overall fold of the native protein remaining intact. The products of proteolysis often exhibit higher solubility and are often amenable to high-resolution structural analysis (22, 23). We report here the limited proteolysis of the MshC-CSA complex that yields a single cleaved product that readily crystallized, and the three-dimensional structure of the MshC-CSA complex at 1.6 Å resolution.
Materials and Methods
Materials
Trypsin was purchased from Sigma (T-7409). CSA was from TriLink BioTechnologies. Full-length MshC was expressed and purified as described previously (11). Crystal screens were purchased from Hampton Research, and all chemical reagents were purchased from Sigma-Aldrich.
Limited Proteolysis of MshC
The purified full-length MshC protein was incubated with CSA at a molar ratio of 1:4 in 20 mM triethanolamine (TEA), pH 7.9 containing 100 mM NH4SO4. Trypsin was added at a ratio of MshC:Trypsin, 50:1(w/w), at room temperature for 4-24 hours. Aliquots were withdrawn at various time points and quenched with 0.1 mM phenylmethylsulfonylfluoride (PMSF). Samples were then diluted into SDS buffer, boiled and subjected to SDS-PAGE analysis.
Having established conditions to obtain limited proteolysis, approximately 75 mg of purified full-length MshC was concentrated to a volume of 15 ml. The protein solution was incubated with trypsin in the presence of CSA at the ratios described above, with gentle stirring at 4 °C for 24 hours. The proteolysis reaction was stopped at various times by adding 0.1 mM PMSF, the solution was centrifuged to remove precipitant, and then concentrated using a YM 10 Amicon ultracentrifuge membrane.
Purification and assay of the proteolyzed MshC
All purification steps were performed at 4 °C. The concentrated, proteolyzed MshC was loaded onto a HiLoad 16/60 Superdex 75 gel filtration column that was pre-equilibrated with 20 mM TEA, pH 7.9 containing 100 mM NH4SO4. The column was eluted at 0.5 ml/min and fractions containing MshC were pooled and concentrated. The enzyme was then dialyzed against 3 changes of 20 mM TEA, pH 7.9. After removal of precipitated protein by centrifugation, the enzyme solution was loaded onto a Mono Q (1.6/5) column pre-equilibrated with 20 mM TEA, pH 7.9. The protein was eluted with two gradients: a 60 column-volume gradient of NH4SO4 from 0 to 500 mM, followed by a 5 column-volume gradient from 500 to 1,000 mM NH4SO4. The fractions exhibiting A280 were examined by SDS-PAGE, and the relevant fractions were pooled and concentrated. The resulting protein was dialyzed against 3 changes of 20 mM HEPES, pH 7.8, then centrifuged to remove precipitated protein.
The concentration of enzyme was determined using the Bio-Rad protein assay kit with bovine serum albumin as the standard. The native molecular weight and oligomeric state was estimated using gel filtration on a column calibrated with Bio-Rad molecular weight markers. Mass spectrometry was performed on a LTQ electrospray ionization from Thermo Finnigan.
Crystallization & Data Collection
The proteolyzed, inhibitor-bound MshC, (5 mg/ml in 20 mM HEPES, pH 7.5) was screened for crystallization in a 1:1 ratio using Hampton 1&2 screens along with other in-house sparse-matrix screens by vapor diffusion under silicon oil. Crystals formed in 2 M NH4SO4, 0.1 M HEPES, pH 7.9, and 2% (w/v) PEG400 within 7 days at 18 °C. The hexagonal crystals (∼0.1 × 0.1 × 0.1 mm) were briefly (∼30 s) exposed to cryo-conditions consisting of the crystallization buffer plus 20% (v/v) glycerol and immediately flash frozen. Initial data were collected on an RaxisIV to 2.2 Å resolution and were indexed using Mosflm (26) into space group H3 with unit cell dimensions a =123.5 Å, b = 123.5 Å, c =186.0 Å.
Native and heavy metal-soaked diffraction data were collected at the NSLS at Brookhaven National Laboratory on beamline X12C. A single crystal was soaked for 30 seconds in cryo-buffer containing 50 mM trimethylleadacetate, and data were collected at a wavelength of 0.9486 Å to a resolution of 2.0 Å. The unit cell dimensions were isomorphous to our previous native data set, and revealed clear peaks in isomorphous difference Patterson maps. Initial heavy atom phases were obtained using the PHENIX (27) software by applying the SAD technique. An overall figure of merit of 0.40 was sufficient to phase the SAD data. This yielded density maps in which the autobuilding program ARPWARP (28) was able to fit about 75% of the initial backbone and some side chains. The partially completed model was used within the molecular replacement software, MOLREP (29), to phase a final 1.6 Å resolution native data collected at beamline X12C. Further model building in Coot (30) and structural refinement were performed in REFMAC (31). Due to the high resolution of the data, non-crystallographic constraints were not applied, and the two monomers in the asymmetric unit were refined independently. The data collection and refinement statistics are shown in Table 1.
Table 1.
| Data Collection | Native | Heavy Atom |
|---|---|---|
| Resolution (Å) | 92.9-1.6 (1.66-1.60) | 50.0-2.0 (2.13-2.00) |
| Completeness | 99.7% (98.3%) | 100.0% (100%) |
| Redundancy | 4.0 (3.7) | 11.3 (11.4) |
| I/sigma(I) | 37.0 (3.1) | 19.7 (6.77) |
| Rmerge | 0.062 (0.349) | 0.086 (0.295) |
| FOM | 0.41 (0.26) | |
| Wavelength (Å) | 1.1000 | 0.9486 |
| Space Group | H 3 | H 3 |
| Unit cell (Å) £ | a=123.5, b=123.5, c=186.0, α=β=90.0°, γ=120.0° | |
| Unique Reflections | 139,087 | 97,521 (8,954) |
| Refinement Statistics | ||
| Rwork (%) | 0.185 (0.259) | |
| Rfree (%) | 0.207 (0.307) | |
| Resolution Range (Å) | 37.1-1.6 (1.64-1.60) | |
| Refined Reflections | 131,736 (10,031) | |
| Reflections in Rfree | 6,973 | |
| Number of Atoms | ||
| Protein | 6,227 | |
| CSA Inhibitor | 58 | |
| Solvent | 868 | |
| Zinc Ions | 2 | |
| Average B-factors (Å2) | ||
| Protein | 22.51 | |
| Inhibitor | 16.06 | |
| Solvent | 37.06 | |
| Zinc Ions | 14.83 | |
| All Atoms | 22.51 | |
| RMS Deviations | ||
| bonds (Å) | 0.011 | |
| angles (°) | 1.656 | |
| Ramachandra Plot | 94.7% core, 5.2% allowed, 0.1% generously, 0.0 % disallowed | |
| PDB accession code | 3C8Z | |
statistics for the highest resolution shell are shown in parenthesis,
no Sigma(F) data cutoff was applied,
unit cells were isomorphous.
Results and Discussion
Limited Proteolysis of MshC
Initial crystallization experiments utilizing full-length MshC were unsuccessful, so limited proteolysis was attempted to delineate rigid domains, leaving a core which might be more amenable to crystallization. In other systems, the addition of substrates or tight-binding inhibitors has been shown to significantly alter the susceptibility of certain loops to limited proteolysis (22, 23). In this case, the bisubstrate analog CSA was chosen as it binds to MshC with nanomolar affinity (11). As shown in Figure 1 (lanes 2-5), the nearly complete proteolysis of MshC occurred by trypsin in the absence of CSA, generating fragments at 32, 25, 21 and 13 kDa after four hours, which were further degraded after 24 hours of proteolysis. In contrast, in the presence of CSA, the 32, 21 and 13 bands are observed at 4 hours and after 24 hours these bands remained intact (lanes 6-10). The 24-hour proteolized MshC solution was purified initially on a Superdex 75 gel filtration column, which separated smaller fragments from the 32, 21 and 13 kDa fragments. Elution from a Mono Q column yielded a protein that could be resolved into 13 and 32 kDa fragments. Typically, from 50 mg of purified full-length MshC we recovered ∼ 5 mg of the purified and proteolyzed protein. The molecular mass of the 32 kDa core domain observed on SDS-PAGE, was more precisely determined to be 32,434.5 Da by electrospray ionization mass spectrometry, which is in agreement with a domain composed of residues 1-284 (data not shown). In addition the size of the remaining C-terminal residues 298-412 is in agreement with the 13 kDa fragment observed on SDS-PAGE. While the presence of additional nonspecific nicks in the 13 kDa domain are supported by the SDS-PAGE and mass spectrometry analysis (data not shown), they are not clearly observed in the crystal structure. Such non-specific cuts in the C-terminal 13 kDa domain during the extended 24 hr proteolysis would not be visualized in the crystal structure, as over the sum of the population, these random nicks would give rise to overall occupied density. While a few weak spots in the C-terminal peptide chain density were observed, none could be obviously attributed to proteolysis. In this manner, nonspecific cuts in the C-terminal domain loops could result in degradation of the 13 kDa band on SDS while not being observed in the crystal structure. Alternatively, the unnicked protein could have been selectively crystallized.
Figure 1.

SDS-PAGE visualization of MshC proteolysis. Lanes 1 and 12, molecular weight markers; Lanes 2-5, proteolysis of MshC with 2% (w/w) trypsin digestion at 0, 1, 4, and 24 h; Lanes 6-9, proteolysis of MshC-CSA complex with 2% (w/w) trypsin digestion at 0, 1, 4, and 24 h; lane 10, proteolyzed MshC after purification using gel filtration and anion exchange chromatographies (as detailed in the Materials and Methods); lane 11, proteolyzed MshC-CSA crystals dissolved in water.
MshC Crystal Structure
The crystal structure of MshC revealed two nearly identical (RMSD=0.61 Å) nearly full-length 47 kDa monomers in the asymmetric unit. All residues of the full-length protein were observed, with the exception of residues 285-297, which was likely the loop removed during proteolysis. The overall structure and fold of the MshC monomer is shown in Figures 2 & S2. The three-dimensional structure of MshC is similar to that of CysRS (19) and other class I AaRS's (32-42). MshC shares 36.1% primary sequence identity to class I CysRS, suggesting a common evolutionary heritage (12). A superposition of MshC and CysRS (PDB 1U0B) yielded an RMSD of 2.70 Å for overlapping Cα-atoms, excluding the additional extended anticodon-binding domain of the CysRS. All members of this enzyme superfamily contain a Rossmann nucleotide binding fold along with two highly conserved signature motifs: HIGH and KMSKS (43, 44). Both motifs participate in the activation of an amino acid with ATP. In MshC, the isoleucine residue in the conserved HIGH motif is replaced with a structurally conserved leucine residue. The KMSKS motif is located on a flexible loop directly following the final C-terminal β-strand of the Rossman fold in both CysRS and the structurally-related methionyl-tRNA synthetases (MetRS) (19). In the MshC structure, the KMSKS motif and loop (residues 289-293) are not observed, as they were proteolyzed. This allowed MshC to form a crystal lattice dimerization interface within the asymmetric unit. An alpha helix forms the ‘lower lip’ of the active site and self-associates in the proteolyzed crystal dimer, whereas this interaction may be sterically hindered in the presence of the loop (Figure S3). Gel filtration results of the native MshC indicate that the monomer is the biologically relevant form. The dimer observed in the asymmetric unit is a non-physiological artifact of the proteolysis.
Figure 2.

The overall tertiary structure of MshC displayed as a ribbon colored according to fold domains: The CP domain is green, the Rossmann Fold domain is blue, the anticodon binding domain is red, and the SC fold is light blue. The zinc metal is displayed as a pink sphere. The CSA inhibitor (carbons yellow, oxygen red, sulfur orange, and nitrogen blue) is bound in the active site. An N-terminal coil and ß-hairpin are colored yellow, and the sites of proteolysis are marked with asterisks.
Similar to class I AaRS's, the biologically relevant monomeric structure of MshC can be divided into two primary domains and two connecting domains: i) the active site Rossman fold; ii) an inter-domain connective polypeptide, designated as the CP domain; iii) a C-terminal antiparallel α-helix domain (termed the anticodon binding domain in CysRS); and iv) a domain that contains the KMSKS signature motif and connects the C-terminal domain and Rossman fold, known as the stem-contact (SC) fold (Figure 2) (19).
The Rossman fold of MshC is composed of a five-stranded parallel β-sheet, surrounded by α-helices. As observed in other class I AaRS structures, the Rossmann fold of MshC is divided into two halves comprising residues 37–146 and 230–280, which are interrupted by the CP domain (19). As shown in Figure 3, the adenosine portion of the CSA inhibitor, as well as the zinc ion, are tightly associated with the nucleotide-binding motif. The CSA ligand is bound by residues (T46, T58, T83, G249, D251, and I283), which reside on multiple small loops between the core set of β-sheets and α-helices. The CP domain of MshC is folded into a four-stranded antiparallel β-sheet motif, similar to all class I AaRS structures (19). The C-terminal domain of MshC consists of a series of α-helices composed of residues 332–412. In the case of CysRS, this C-terminal domain is responsible for the recognition and specific binding of the anticodon region of tRNAcys (19, 45). The MshC SC-fold, like that found in CysRS and other class I tRNA synthetases, is composed of a β-α-α-β-α motif and contains the KMSKS motif (19, 32-42). In the case of CysRS this sequence is important for the binding and positioning of the L-shaped tRNA molecule as well as amino acid activation. In the case of tyrosyl-tRNA synthase, this loop domain was observed in both an open, flexible conformation in the unliganded enzyme and in a rigid ATP-bound state (46). Based on these structural results, we propose that this loop in MshC participates in the formation of the adenylate in the first half reaction, and may also participate in the binding of the GlcN-Ins substrate in the second phase of the reaction.
Figure 3.

Binding of CSA and zinc in the active site of MshC. A) The inhibitor density is displayed as a Fo-Fc omit map contoured at 3σ. B) Schematic illustration of the interactions of CSA with residues in the active site of MshC. C) Tetrahedral geometry of the active site zinc, in a 2Fo-Fc map contoured at 1σ. All bond distances are in Å.
The refined structure reveals that a zinc ion is coordinated to the terminal thiol group of CSA, as well as the side chain thiol groups of C43 and C231, and the NE2 atom of H256. The zinc atom is coordinated to these residues at distances of 2.1-2.4 Å, and forms a tetrahedral ligand field geometry (Figure 3C). The ligand field geometry and thiol-metal distances are most suggestive of an active site zinc ion, however, we cannot rigorously rule out a nickel ion being present after Ni-NTA affinity chromatography. These zinc coordination residues are conserved with those in CysRS, i.e., C28, C209, and H234 (19), and attempts to generate zinc-free CysRS have been unsuccessful (47). Mutagenesis of the zinc coordinating residues generated mutant forms of the enzyme with native structure but impaired activity, suggesting a catalytic rather than structural role for the zinc atom (47, 48).
There is clear electron density for the bi-substrate analogue, CSA, with an average B-factor of 16.06 Å2, which is similar to that of neighboring protein residues (Figure 3A). The CSA molecule is stabilized in the MshC active site in four positions: interactions with the adenine group, the 2′-oxygen on the ribose ring, the α-amino group of the cysteinyl moiety, and the Zn2+-thiolate interaction. The adenine ring binds in a narrow pocket flanked on either side by the planer backbone atoms of G54 (part of the HIGH signature sequence) and the sidechain of M282. The side chain oxygen of T58 hydrogen bonds to the N3 position, while I283 makes hydrogen bonding interactions with N1 and N6 through amide and carbonyl backbone atoms, respectively. The 2′-ribose hydroxyl is hydrogen bonded to the sidechain carboxyl of D251 and to the amide nitrogen of G249. In the third set of interactions, hydrogen bonds are observed between the inhibitor α-amino nitrogen and the sidechain hydroxyls of T46 and T83, along with an interaction to the mainchain carbonyl of G44. Additionally, H55, although 3.5 Å away from the ribose ring oxygen of CSA, is highly conserved in MshC and CysRS from different species, and might play an important role in cysteine adenylation (19). In the structure of MetRS crystallized with its bi-substrate analogue (MSA) (42), the ligand is stabilized not only by the interactions discussed above for MshC, but also at the non-bridging oxygen moiety of the adenylate mimic. The sulfamoyl non-bridging oxygen atom of MSA is hydrogen bonded to MetRS H24, corresponding to H55 in MshC, which is part of the HIGH motif. Although in the structure of MshC, H55 is 3.5 Å away from ribose oxygen atom, it is possible that such a hydrogen bond would form with a bona fide cysteinyladenylate intermediate. Figure 4 illustrates the similarity of the binding of the CSA to MshC to the binding of MSA to MetRS.
Figure 4.
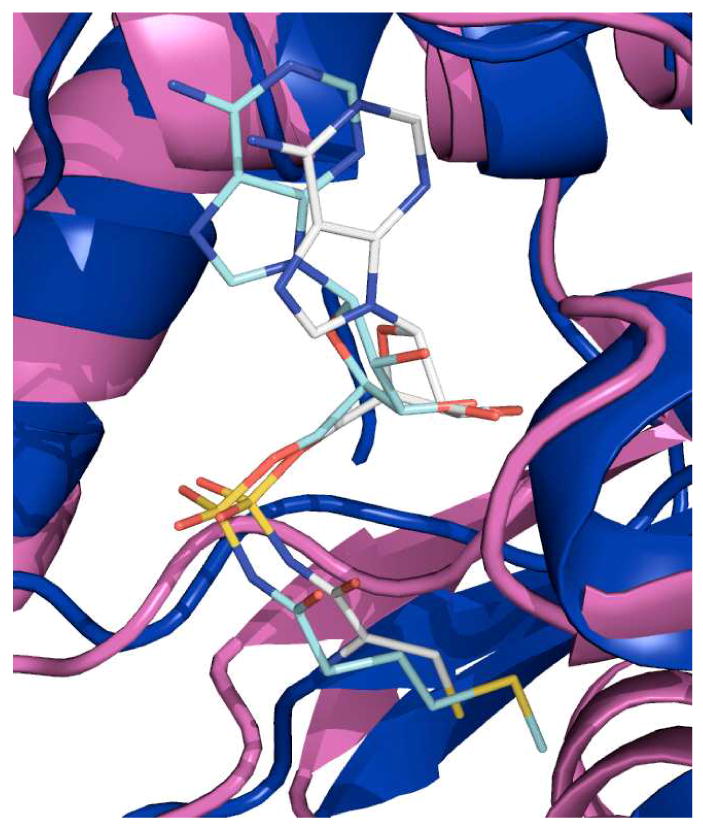
Structural superposition of MshC-CSA complex with MetRS-MSA complex. The CSA molecule (gray carbons) and the MSA molecule (light blue carbons) show similar binding positions within their respective enzyme active sites.
The cysteine portion of CSA binds at the base of the active site cleft in a position similar to the amino acid binding sites of other class I AaRs (19, 32-42), and interacts with residues in the first half of the Rossmann fold (i.e., T46 and T83). The cysteine thiol forms a direct contact with the zinc atom at a distance of 2.4 Å. This zinc-thiolate interaction was suggested to be responsible for amino acid discrimination in CysRS (49). Another study showed that zinc binds the serine hydroxyl group with significantly lower affinity than the thiolate moiety of cysteine (50). The inherently strong and specific zinc-thiolate interaction presumably allows MshC to select cysteine versus serine without the need for any editing step (47). The class II threonine tRNA synthetase (ThrRS) also contains an active site zinc atom that is used to coordinate the side chain hydroxyl group of the substrate threonine (51, 52). Since this is not an interaction that can easily discriminate between threonine, serine and cysteine, an additional editing step is required in ThrRS to hydrolyze the misactivated amino acids.
In addition, a highly conserved tryptophan residue W205 is proposed to be responsible for amino acid discrimination and positioning in CysRS (19). In CysRS, a W205Y CysRS mutant exhibited weak activity towards serine (48). This tryptophan residue is strictly conserved in all 64 known CysRS sequences, and exhibits a conformational change upon cysteine binding (19, 53). The corresponding W227 in MshC may play a similar role in amino acid recognition. Significant pockets surrounding W227 suggest a similar flexibility allowing W227 to exist in alternate conformations prior to cysteine and ATP binding. W227 is 3.3 Å away from the thiol group of CSA, suggesting a potential hydrogen bonding interaction. The interactions of cysteine with both the zinc atom and indole ring of W227 likely provide a coordinated mechanism of amino acid discrimination (cysteine vs. serine) in MshC.
An additional structural motif of five conserved residues are at the base of the active site; H228, Q246, H256, H257, and H279. The four histidine imidazole rings interact with each other via hydrogen bonds. H228 is within 2.9 Å of H256, which is one of the zinc ligands, and is 3.9 Å away from H257. H257 is 2.8 Å from H279. The function of this arrangement is unknown, but has been previously observed in the related CysRS, where it was proposed to serve as a possible second zinc binding position for further amino acid discrimination (19).
The MshC structure exhibits an active site architecture which is poised to accept either substrate in the first half-reaction, which is consistent with a random binding of ATP and cysteine. In addition to the structural information, previous steady state kinetic studies suggested that ATP binds to MshC prior to cysteine (11), while later studies suggested that cysteine could bind to free MshC before ATP binding, supporting a random kinetic sequence for ATP and cysteine binding (10). More recent fluorescence data show that both ATP and cysteine can bind to free MshC (data not shown).
CSA is a tight binding inhibitor of MshC, which mimics the cysteineadenylate intermediate formed in the first half of the ligase reaction. The second half-reaction catalyzed by MshC is the nucleophilic attack of the 2-amino group of GlcN-Ins on the enzyme bound cysteineadenylate to form the Cys-GlcN-Ins product and AMP. The structure of CSA bound to MshC provides some insight into the enzyme-ligand coordination just prior to the second ligation reaction. Interactions are observed between the N6 and N1 purine nitrogen atoms of CSA and the carbonyl backbone oxygen and amide of I283, a residue directly adjacent to the proteolized loop (285-297). Based on these interactions and the KMSKS motif observed in the CysRS-tRNAcys structure (1U0B), the proteolyzed loop was modeled onto the MshC structure (Figure 5).
Figure 5.
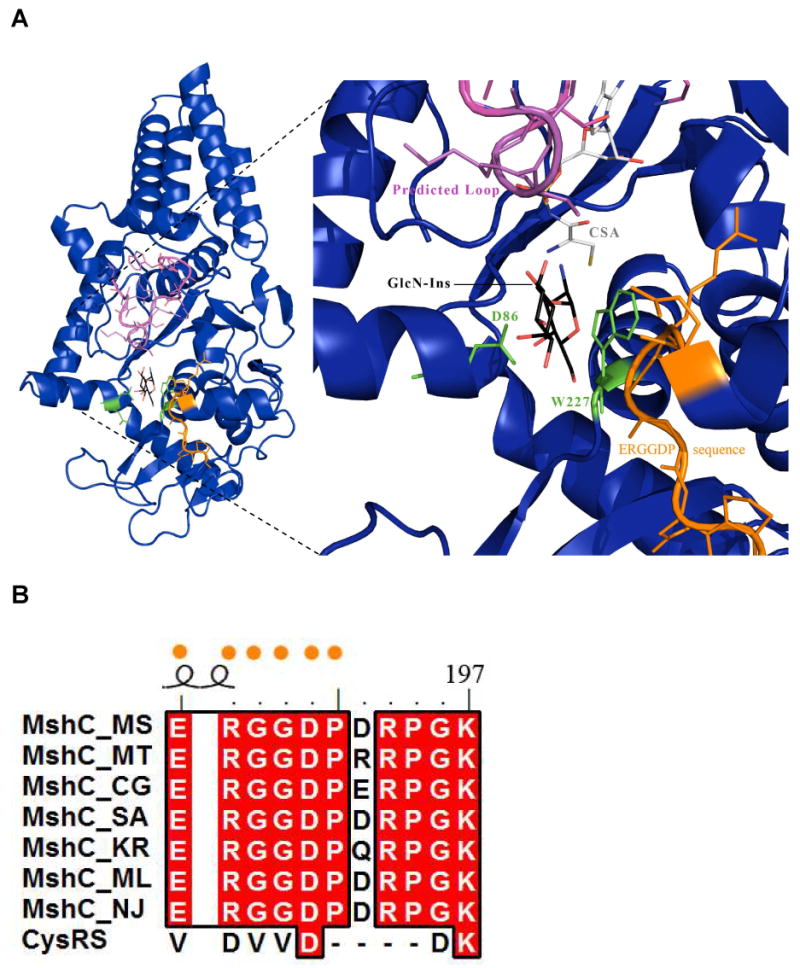
Active site docking of GlcN-Ins as well as modeling of the proteolized MshC loop segment based on the CysRS-tRNAcys (1U0B) structure. A) The modeled conformation of the proteolized loop (pink) on MshC (deep blue). The loop is positioned directly over the GlcN-Ins molecule (carbons colored black). Conserved residues D86 and W227 are shown in green, while the MshC ortholog identifying sequence is displayed in orange. These features likely bind and coordinate the GlcN-Ins molecule in the active site for catalysis. B) Sequence alignment of MshC orthologs and CysRS. The ERGGDP sequence (orange dots) is highly conserved amongst MshC orthologs but absent in the CysRS homologues.
The GlcN-Ins substrate was modeled into the active site in a position that would allow for the attack by the 2-amino group of glucosamine on the cysteineadenylate, as shown in Figure 5. One feature of the model is the stacking interaction of the GlcN ring with W227. As discussed above, W227 is likely to exist in alternate conformations prior to the binding of cysteine and ATP and may also interact with GlcN-Ins in the cysteineadenylate complex. In addition, D86 is a strictly conserved residue amongst MshC orthologs (Figure S1), and is positioned to interact with the GlcN hydroxyls. Finally, a region of strict amino acid conservation unique to MshC orthologs is positioned near the modeled GlcN-Ins. The combined network has the potential to position GlcN-Ins in a favorable catalytic orientation. Since this consensus sequence, E187RGGDP192, is only present in MshC orthologs, this sequence can be used to distinguish between MshC and CysRS sequences during annotation. In addition to these interactions, the docked inositol ring of GlcN-Ins molecule is positioned directly under the predicted proteolyzed loop. While this loop likely plays a role in adenylate formation in the first half reaction, it may also participate in positioning the GlcN-Ins. Two lysine residues and one arginine reside on this loop and are positioned for possible interactions with the inositol ring hydroxyls. After adenylate formation in the first half reaction, interactions with the bound adenylate could act to stabilize the loop in an intermediate conformation, permitting binding by the GlcN-Ins. Engineered constructs of MshC lacking this conserved loop, while being well folded (data not shown), showed no enzymatic activity, suggesting a critical role for the loop in the catalytic mechanism. Structural studies of CysRS have implicated the two lysine residues on the KMSKS motif in binding and positioning the tRNA substrate (19).
While MshC shares strong overall structural homology with CysRS, the most important features distinguishing these enzymes are also directly connected to their different functional roles. The largest structural difference is the extended anti-codon binding (AB) domain (Figure 6), which is observed in the tRNA-CysRS complex but is not a part of the MshC protein. In CysRS, the extended AB domain provides discrimination for the cysteinyl-tRNA. This structural feature is disordered in the unliganded structure of CysRS (19), but is ordered in the presence of tRNAcys and forms an α/β domain interacting with the anti-codon loop of tRNAcys. The extended anti-codon binding domain is not required in the case of MshC function and has been evolutionarily discarded. A second difference observed in MshC is the conformation of the loop between β7 and β8 of the CP domain (164-202). In CysRS a portion of this loop becomes ordered upon binding tRNAcys, forming mostly random coil and a small 5-residue helix. The loop, approximately 20 Å from the zinc ion, forms the outer edge of a pocket that interacts with the 3′-end of the tRNAcys. In the structure of MshC, this loop is ordered and forms a 10-residue helix (178-187) in addition to a random coil. It is this helix that self associates to form a non-crystallographic dimer axis, placing the two active sites in close proximity (Figure S3). Interestingly, the CysRS and MshC loops show no sequence conservation and both take on different conformations, with the MshC loop making a much closer approach to the zinc ion (∼ 10 Å). In addition, the conformation of this loop places the unique MshC ERGGDP sequence adjacent to the reaction center, suggesting that this loop may participate in binding of GlcN-Ins. MshC has retained many of the structural features of the AaRS's relevant to catalysis of the common first half reaction, amino acid adenylation, while discarding other features to accommodate its different acceptor substrate.
Figure 6.

The structural alignment of MshC with cysteinyl-tRNAcys synthetase (1U0B) using the SCOP (54) database. Left: Shown in blue is MshC and in orange CysRS. The cysteinyl-tRNA extended anti-codon binding (AB) domain is labeled. Right: The L-shaped tRNA from the CysRS-tRNAcys bound structure 1U0B (orange), is modeled as though bound to MshC (blue plus predicted loop in pink). The absence of the extended AB domain conferring tRNA specificity and an alteration in the location and fold of the β7/β8 loop and preformed helix (red star), structurally distinguishes these related enzymes.
Supplementary Material
Three figures containing 1) sequence alignment of MshC and related cysteine synthases, 2) a tpology diagram for MshC and 3) a view of the non-crystallographic dimer in the asymmetric unit. This material is available free of charge at http://pubs.acs.org
Acknowledgments
The authors would like to thank Tinoush Moulaei (NCI) for his suggestion to attempt the proteolysis experiments.
This work was supported by grants from the NIH (AI33696 to J.S.B.) and the Heiser Program for Research in Leprosy and Tuberculosis of the New York Community Trust (to F.F.).
Abbreviations
- GlcN-Ins
1-D-myo-inosityl-2-amido-2-deoxy-α-D-glucopyranoside
- GlcNAc-Ins
1-D-myo-inosityl-2-acetamido-2-deoxy-alpha-D-glucopyranoside
- CSA
5′-O-[N-(L-cysteinyl)-sulfamonyl]adenosine
- MSH
mycothiol
- MshC
cysteine ligase
- PMSF
phenylmethylsulfonylfluoride
- TEA
triethanolamine
- MetRS
methionyl-tRNA syntethase
- CysRS
cysteinyl-tRNA synthase
- AaRS
aminoacyl-tRNA synthetases
References
- 1.Newton GL, Av-Gay Y, Fahey RC. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry. 2000;39:10739–10746. doi: 10.1021/bi000356n. [DOI] [PubMed] [Google Scholar]
- 2.Newton GL, Unson MD, Anderberg SJ, Aguilera JA, Oh NN, delCardayre SB, Av-Gay Y, Fahey RC. Characterization of Mycobacterium smegmatis mutants defective in 1-d-myo-inosityl-2-amino-2-deoxy-alpha-d-glucopyranoside and mycothiol biosynthesis. Biochem Biophys Res Commun. 1999;255:239–244. doi: 10.1006/bbrc.1999.0156. [DOI] [PubMed] [Google Scholar]
- 3.Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother. 2002;46:3348–3355. doi: 10.1128/AAC.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchmeier NA, Newton GL, Koledin T, Fahey RC. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol. 2003;47:1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 5.Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderberg SJ, Newton GL, Fahey RC. Mycothiol biosynthesis and metabolism. Cellular levels of potential intermediates in the biosynthesis and degradation of mycothiol in Mycobacterium smegmatis. J Biol Chem. 1998;273:30391–30397. doi: 10.1074/jbc.273.46.30391. [DOI] [PubMed] [Google Scholar]
- 7.Bornemann C, Jardine MA, Spies HS, Steenkamp DJ. Biosynthesis of mycothiol: elucidation of the sequence of steps in Mycobacterium smegmatis. Biochem J. 1997;325(Pt 3):623–629. doi: 10.1042/bj3250623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newton GL, Fahey RC. Mycothiol biochemistry. Arch Microbiol. 2002;178:388–394. doi: 10.1007/s00203-002-0469-4. [DOI] [PubMed] [Google Scholar]
- 9.Newton GL, Ta P, Bzymek KP, Fahey RC. Biochemistry of the initial steps of mycothiol biosynthesis. J Biol Chem. 2006;281:33910–33920. doi: 10.1074/jbc.M604724200. [DOI] [PubMed] [Google Scholar]
- 10.Williams L, Fan F, Blanchard JS, Raushel FM. Positional Isotope Exhange Analysis of Mycobacterium smegmatis Cysteine Ligase (MshC) Biochemistry in press. 2008 doi: 10.1021/bi800327u. [DOI] [PubMed] [Google Scholar]
- 11.Fan F, Luxenburger A, Painter GF, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium smegmatis cysteine ligase (MshC) Biochemistry. 2007;46:11421–11429. doi: 10.1021/bi7011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sareen D, Steffek M, Newton GL, Fahey RC. ATP-dependent L-cysteine:1D-myo-inosityl 2-amino-2-deoxy-alpha-D-glucopyranoside ligase, mycothiol biosynthesis enzyme MshC, is related to class I cysteinyl-tRNA synthetases. Biochemistry. 2002;41:6885–6890. doi: 10.1021/bi012212u. [DOI] [PubMed] [Google Scholar]
- 13.Hausmann CD, Ibba M. Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol Rev. 2008;32:705–721. doi: 10.1111/j.1574-6976.2008.00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng R, Blanchard JS. Steady-state and pre-steady-state kinetic analysis of Mycobacterium tuberculosis pantothenate synthetase. Biochemistry. 2001;40:12904–12912. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 15.Kim YS, Kang SW. Steady-state kinetics of malonyl-CoA synthetase from Bradyrhizobium japonicum and evidence for malonyl-AMP formation in the reaction. Biochem J. 1994;297(Pt 2):327–333. doi: 10.1042/bj2970327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Babbitt PC, Kenyon GL, Martin BM, Charest H, Slyvestre M, Scholten JD, Chang KH, Liang PH, Dunaway-Mariano D. Ancestry of the 4-chlorobenzoate dehalogenase: analysis of amino acid sequence identities among families of acyl:adenyl ligases, enoyl-CoA hydratases/isomerases, and acyl-CoA thioesterases. Biochemistry. 1992;31:5594–5604. doi: 10.1021/bi00139a024. [DOI] [PubMed] [Google Scholar]
- 17.Chang KH, Xiang H, Dunaway-Mariano D. Acyl-adenylate motif of the acyl-adenylate/thioester-forming enzyme superfamily: a site-directed mutagenesis study with the Pseudomonas sp. strain CBS3 4-chlorobenzoate:coenzyme A ligase. Biochemistry. 1997;36:15650–15659. doi: 10.1021/bi971262p. [DOI] [PubMed] [Google Scholar]
- 18.Scholten JD, Chang KH, Babbitt PC, Charest H, Sylvestre M, Dunaway-Mariano D. Novel enzymic hydrolytic dehalogenation of a chlorinated aromatic. Science. 1991;253:182–185. doi: 10.1126/science.1853203. [DOI] [PubMed] [Google Scholar]
- 19.Newberry KJ, Hou YM, Perona JJ. Structural origins of amino acid selection without editing by cysteinyl-tRNA synthetase. Embo J. 2002;21:2778–2787. doi: 10.1093/emboj/21.11.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reger AS, Wu R, Dunaway-Mariano D, Gulick AM. Structural Characterization of a 140 degrees Domain Movement in the Two-Step Reaction Catalyzed by 4-Chlorobenzoate:CoA Ligase. Biochemistry. 2008 doi: 10.1021/bi800696y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bandarian V, Pattridge KA, Lennon BW, Huddler DP, Matthews RG, Ludwig ML. Domain alternation switches B(12)-dependent methionine synthase to the activation conformation. Nat Struct Biol. 2002;9:53–56. doi: 10.1038/nsb738. [DOI] [PubMed] [Google Scholar]
- 22.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 23.Price NC, Johnson CM. In: Proteolytic enzymes - A practical approach. Beynon RJ, Bond JS, editors. IRL Press; Oxford, UK: 1989. pp. 163–180. [Google Scholar]
- 24.Neurath H. Protein Folding. 1980:501–504. [Google Scholar]
- 25.Gao X, Bain K, Bonanno JB, Buchanan M, Henderson D, Lorimer D, Marsh C, Reynes JA, Sauder JM, Schwinn K, Thai C, Burley SK. High-throughput limited proteolysis/mass spectrometry for protein domain elucidation. J Struct Funct Genomics. 2005;6:129–134. doi: 10.1007/s10969-005-1918-5. [DOI] [PubMed] [Google Scholar]
- 26.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography 1992 [Google Scholar]
- 27.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 28.Perrakis A. wARP: Improvement and Extension of Crystallographic Phases by Weighted Averaging of Multiple-Refined Dummy Atomic Models. Acta Cryst. 1997;D53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 29.Vagin A. MOLREP: an Automated Program for Molecular Replacement. J Appl Cryst. 1997;30:1022. [Google Scholar]
- 30.Emsley P. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallographica Section D - Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst. 1997;D58:1285–1294. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Cavarelli J, Delagoutte B, Eriani G, Gangloff J, Moras D. L-arginine recognition by yeast arginyl-tRNA synthetase. Embo J. 1998;17:5438–5448. doi: 10.1093/emboj/17.18.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cusack S. Aminoacyl-tRNA synthetases. Curr Opin Struct Biol. 1997;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- 34.Cusack S, Yaremchuk A, Tukalo M. The 2 A crystal structure of leucyl-tRNA synthetase and its complex with a leucyl-adenylate analogue. Embo J. 2000;19:2351–2361. doi: 10.1093/emboj/19.10.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/s0092-8674(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 36.Fukai S, Nureki O, Sekine S, Shimada A, Vassylyev DG, Yokoyama S. Mechanism of molecular interactions for tRNA(Val) recognition by valyl-tRNA synthetase. Rna. 2003;9:100–111. doi: 10.1261/rna.2760703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mechulam Y, Schmitt E, Maveyraud L, Zelwer C, Nureki O, Yokoyama S, Konno M, Blanquet S. Crystal structure of Escherichia coli methionyl-tRNA synthetase highlights species-specific features. J Mol Biol. 1999;294:1287–1297. doi: 10.1006/jmbi.1999.3339. [DOI] [PubMed] [Google Scholar]
- 38.Nureki O, Fukai S, Sekine S, Shimada A, Terada T, Nakama T, Shirouzu M, Vassylyev DG, Yokoyama S. Structural basis for amino acid and tRNA recognition by class I aminoacyl-tRNA synthetases. Cold Spring Harb Symp Quant Biol. 2001;66:167–173. doi: 10.1101/sqb.2001.66.167. [DOI] [PubMed] [Google Scholar]
- 39.Nureki O, Vassylyev DG, Katayanagi K, Shimizu T, Sekine S, Kigawa T, Miyazawa T, Yokoyama S, Morikawa K. Architectures of class-defining and specific domains of glutamyl-tRNA synthetase. Science. 1995;267:1958–1965. doi: 10.1126/science.7701318. [DOI] [PubMed] [Google Scholar]
- 40.Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285:1074–1077. [PubMed] [Google Scholar]
- 41.Sugiura I, Nureki O, Ugaji-Yoshikawa Y, Kuwabara S, Shimada A, Tateno M, Lorber B, Giege R, Moras D, Yokoyama S, Konno M. The 2.0 A crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure. 2000;8:197–208. doi: 10.1016/s0969-2126(00)00095-2. [DOI] [PubMed] [Google Scholar]
- 42.Crepin T, Schmitt E, Mechulam Y, Sampson PB, Vaughan MD, Honek JF, Blanquet S. Use of analogues of methionine and methionyl adenylate to sample conformational changes during catalysis in Escherichia coli methionyl-tRNA synthetase. J Mol Biol. 2003;332:59–72. doi: 10.1016/s0022-2836(03)00917-3. [DOI] [PubMed] [Google Scholar]
- 43.Hountondji C, Lederer F, Dessen P, Blanquet S. Escherichia coli tyrosyl- and methionyl-tRNA synthetases display sequence similarity at the binding site for the 3′- end of tRNA. Biochemistry. 1986;25:16–21. doi: 10.1021/bi00349a003. [DOI] [PubMed] [Google Scholar]
- 44.Webster T, Tsai H, Kula M, Mackie GA, Schimmel P. Specific sequence homology and three-dimensional structure of an aminoacyl transfer RNA synthetase. Science. 1984;226:1315–1317. doi: 10.1126/science.6390679. [DOI] [PubMed] [Google Scholar]
- 45.Zhang CM, Hou YM. Domain-domain communication for tRNA aminoacylation: the importance of covalent connectivity. Biochemistry. 2005;44:7240–7249. doi: 10.1021/bi050285y. [DOI] [PubMed] [Google Scholar]
- 46.Kuratani M, S H, Takahashi M, Yanagisawa T, Kobayashi T, Yokoyama S. Crystal structures of tyrosyl-tRNA synthetases from Archaea. J Mol Biol. 2006;355:395–408. doi: 10.1016/j.jmb.2005.10.073. [DOI] [PubMed] [Google Scholar]
- 47.Zhang CM, Perona JJ, Hou YM. Amino acid discrimination by a highly differentiated metal center of an aminoacyl-tRNA synthetase. Biochemistry. 2003;42:10931–10937. doi: 10.1021/bi034812u. [DOI] [PubMed] [Google Scholar]
- 48.Zhang CM, Christian T, Newberry KJ, Perona JJ, Hou YM. Zinc-mediated amino acid discrimination in cysteinyl-tRNA synthetase. J Mol Biol. 2003;327:911–917. doi: 10.1016/s0022-2836(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 49.Fersht AR, Dingwall C. Cysteinyl-tRNA synthetase from Escherichia coli does not need an editing mechanism to reject serine and alanine. High binding energy of small groups in specific molecular interactions. Biochemistry. 1979;18:1245–1249. doi: 10.1021/bi00574a020. [DOI] [PubMed] [Google Scholar]
- 50.Rulisek L, Havlas Z. Theoretical studies of metal ion selectivity. 1. DFT calculations of interaction energies of amino acid chains with selected transition metal ion (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) J Am Chem Soc. 2000;122:10428–10439. [Google Scholar]
- 51.Sankaranarayanan R, Dock-Bregeon AC, Rees B, Bovee M, Caillet J, Romby P, Francklyn CS, Moras D. Zinc ion mediated amino acid discrimination by threonyl-tRNA synthetase. Nat Struct Biol. 2000;7:461–465. doi: 10.1038/75856. [DOI] [PubMed] [Google Scholar]
- 52.Sankaranarayanan R, Dock-Bregeon AC, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D. The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 53.Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. doi: 10.1021/bi00085a006. [DOI] [PubMed] [Google Scholar]
- 54.Murzin AG, B SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Three figures containing 1) sequence alignment of MshC and related cysteine synthases, 2) a tpology diagram for MshC and 3) a view of the non-crystallographic dimer in the asymmetric unit. This material is available free of charge at http://pubs.acs.org