

Abstract
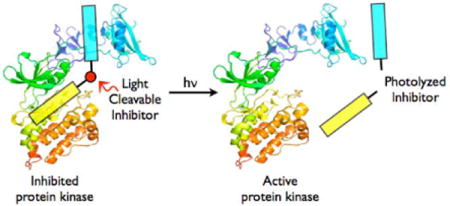
Light-activatable (“caged”) proteins have been used to correlate, with exquisite temporal and spatial control, intracellular biochemical action with global cellular behavior. However, the chemical or genetic construction of caged proteins is nontrivial, with subsequent laborious introduction into living cells, potentially problematic competition with natural endogenous counterparts, and challenging intracellular incorporation at levels equivalent to the natural enzymes. We describe the design, synthesis, and characterization of small molecular equivalents of a caged Src kinase. These compounds are easy to prepare and function by inhibiting the action of the natural unmodified enzyme.
Protein kinases serve as key participants in signaling pathways by catalyzing phosphoryl transfer from ATP to the serine, threonine, and/or tyrosine residues of protein substrates. The eventual cellular response to a stimulus is often dependent upon when and where a particular kinase is activated within the cell. Consequently, the use of constitutively active protein kinases, or various defective mutants thereof, can only partly address the relationship between the action of specific signaling enzymes and ensuing cellular behavior. The creation of “caged” protein kinases, enzymes whose activities can be rapidly unleashed upon exposure to light, has been reported.1 The photo-stimulation of caged proteins inside living cells has also been described, which provides exquisite control over where and when the enzyme is activated.2 However, there are many challenges associated with the application of caged proteins3 to cellular systems. First, chemically modified proteins are primarily introduced into cells via microinjection, a process that limits subsequent behavioral studies to single cell analysis. Second, enzymes often suffer a series of posttranslational modifications during their intracellular lifetime. These modifications can be difficult to replicate in large-scale bacterial enzyme preparations, which are often necessary for the acquisition of sufficient quantities for caging experiments. Third, the intracellular introduction of unnatural enzymes, whether by microinjection or genetic expression, is unlikely to recapitulate the normal endogenous levels of the wild type enzyme. Finally, the action of the caged protein might be difficult to interpret in the presence of its natural intracellular counterpart. We describe a conceptually different strategy for the acquisition of light-activated enzymes, an approach that targets the endogenous enzyme and therefore has the potential to circumvent the limitations outlined above.
We recently reported the design and construction of a potent peptide-derived inhibitor 1 (Ki = 26 ± 4 nM; IC50 = 36 ± 2 nM) for the Src tyrosine kinase.4 Src is comprised of 3 domains, which play various roles in catalysis (SH1), regulation (SH2 and SH3), or substrate recognition (SH1, SH2 and SH3) (Figure 1).5 Compound 1, a “bivalent” species, simultaneously associates with the active site (SH1 domain) and the SH2 domain. A key feature of the inhibitor is that the SH1- and SH2-directed components, which are linked via a β-Ala tether, exhibit approximately equal affinities for their respective binding sites on the target protein. The latter property furnishes an inhibitor with a Src kinase affinity that significantly exceeds (∼50-fold) the affinities of the individual components alone (e.g. SH1 domain-targeted monovalent inhibitor 2; IC50 = 1.9 ± 0.3 μM) (Table 1).4 We reasoned that a bivalent inhibitor, containing a photolabile site positioned at or near the β-Ala tether, would retain its ability to inhibit the target enzyme (Figure 1). However, photolysis should split the inhibitor and thereby curtail its effectiveness. Such a photodeactivatable inhibitor, which is functionally equivalent to a photoactivatable Src kinase, can potentially address the challenges associated with caged proteins outlined above.
Figure 1.
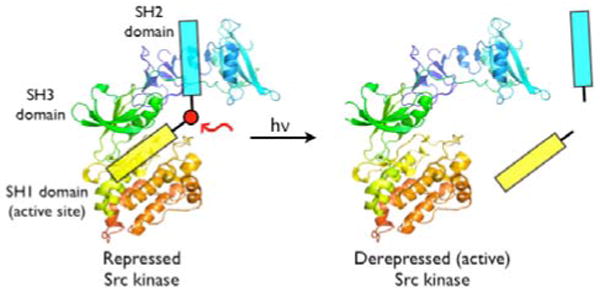
A Photocleavable Caged Protein Equivalent.
Table 1.
Stable (1-2) and Photolytically Sensitive (4 – 11) Src Inhibitors.
| # | Peptide |
IC50 (nM) |
|---|---|---|
| 1 | Ba-EEEIFGEF-Dap(Hna)-βA3-pYEEIEXa | 35 ± 2 |
| 2 | Ba-EEEIFGEF-Dap(Hna)X + AcpYEEIE-NH2 (3) | 1900 |
| 4 | Ba-EEEIFGEF-Dap(Hna)-βA-I-βA-pYEEIEX | 115 ± 9 |
| 5 | Ba-EEEIFGEF-Dap(Hna)-βA-II-βA-pYEEIEX | 38 ± 4 |
| 6 | Ba-EEEIFGEF-Dap(Hna)-βA-III-βA-pYEEIEX | 18 ± 5 |
| 7 | Ba-EEEIFGE-II-Dap(Hna)-βA3-pYEEIEX | 220 ± 10 |
| 8 | Ba-EEEI-III-GEF-Dap(Hna)-βA3-pY-III-EIEX | 28000 |
| 9 | Ba-EEEIFGE-III-Dap(Hna)-βA3-pY-III-EIEX | 7500 |
| 10 | Ba-EEEIFGEF-Dap(Hna)-βA-III-βA-pY-III-EIEX | 1600 |
| 11 | Ba-EEEIFGEF-Dap(Hna)C-III-βA-pYEEIE-NH2 | 56 ± 4 |
![]()
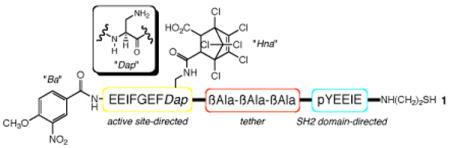
We prepared a small collection of bivalent inhibitors (Table 1), each of which contains one of three different photocleavable groups (I - III). In order to ensure the release of enzymatic activity upon photolysis, the bivalent analog must be a potent inhibitor relative to compound 2. The photocleavable group III furnished the most powerful pre-photolysis inhibitor (6) in the series 4 – 6. We also synthesized several peptides that have two photocleavable sites (8 - 10) using, as a guiding principle, the notion that double photolytic cleavage should destroy all inhibitory activity. Unfortunately, these compounds proved to be exceedingly poor inhibitors. Presumably the additional photosensitive site disrupts SH1 binding (8 – 9) and/or SH2 targeting (8 - 10). With these features in mind, 6 was selected for further study.
We first addressed whether 6 simultaneously associates with the active site and SH2 regions as envisioned (Fig. 1). Ac-pTyrGluGluIle-amide is a validated SH2 domain ligand with a KD of 1 – 5 μM.6 Inhibitory efficacy of 6 is progressively and significantly compromised as a function of SH2 ligand concentration (e.g. IC50 = 325 ± 60 nM @ 320 μM Ac-pTyrGluGluIle-amide), the expected result for an SH2 domain-dependent, active site-targeted inhibitor (Supporting Information).
Src kinase activity is completely blocked in the presence of 1.9 μM 6 (Fig. 2). Photolysis of compound 6 releases kinase activity as measured by a previously described real time fluorescence assay.7 Furthermore, longer photolysis times generate higher rates of substrate phosphorylation. This response is precisely analogous to the direct photoactivation of a “caged” enzyme, in which a photocleavable moiety is removed from a key residue required for activity. In both instances, enhanced photolysis times create larger quantities of active enzyme. Under optimized conditions, photolysis restores up to 90% of the activity displayed by Src in the presence of compound 2 (1.9 μM; i.e. the by-product of photolysis) or up to 50% of Src activity in the absence of compound 2 (Supporting Information). These results compare favorably with those obtained for previously described caged protein kinases1 and phosphatases8. An analogous series of experiments were performed using lysates from COS-1 cell overexpressing wild type Src kinase (Supporting Information).
Figure 2.

Enzymatic activity (ΔF vs time) as a function of irradiation time of compound 6 (1.9 μM). Assays were performed as previously described.7 Experimental conditions: A (0 min hv), B (2 min hv), C (15 min hv), D (120 min hv), and E (0 min hv + 1.9 μM compound 2).
Light-mediated activation of caged inhibitors, activators, and enzymes allows the investigator to control when the agent of interest is switched on. In addition, light exposure time, intensity, or the number of laser pulses, provides a means to control the amount of active species generated. By contrast, spatial control of activity (i.e. localized release) via spot illumination is generally reserved for high molecular weight species with slow diffusion rates, such as proteins.2 Small molecules are unlikely candidates for experimentally meaningful localized release since they commonly exhibit high diffusion rates. Nevertheless, small molecules can be rendered slowly diffusible via anchoring to relatively immobile substrates (e.g. proteins, organelles, etc) using targeting substituents (e.g. amino acid sequences, fatty acids, etc). With this in mind, we explored the following question: can an analog of 6 be spatially affixed to a high molecular object and still function in a light-sensitive fashion?
Bivalent peptide 11, which contains a Cys-for-βAla substitution in the tether region, serves as an effective Src kinase inhibitor (Table 1). Compound 11 was covalently attached to Pierce UltraLink® beads and incubated with varying concentrations of Src kinase to generate a standard binding curve (Supporting Information). Illumination affords approximately 50% non-bound active enzyme as assessed by two criteria. First, photolysis affords 45 ± 4 % Src kinase activity in the supernatent (i.e. following removal of beads), as assessed using a real time fluorescence assay (Supporting Information). Second, 50 ± 4% of Src kinase is physically retained by the beads following photolysis (i.e. 50% is present in solution), as determined by fluorescent imaging (Fig. 2). These results are consistent with solution studies described above and support the notion that protein activity and location can be controlled with light using appropriately designed inhibitory species. Application to cell-based systems is underway.
Supplementary Material
Experimental details of peptide synthesis, structure, characterization, and photolytic behavior. This material is free of charge via the Internet at http://pubs.acs.org.
Figure 3.
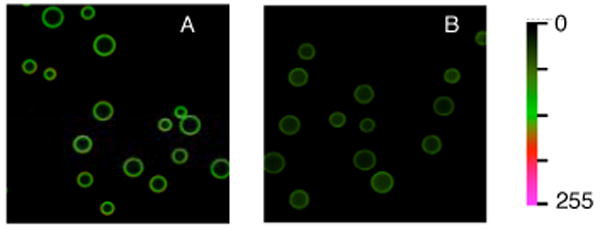
Presence of Src kinase on (A) nonirradiated and (B) irradiated UltraLink® beads modified with bivalent inhibitor 11 as assessed by exposure to an Alexa Fluo488-labeled monoclonal Src antibody.
Acknowledgments
We acknowledge financial support by the NIH (CA079954).
References
- 1.(a) Chang Cy, Fernandez T, Panchal R, Bayley H. J Amer Chem Soc. 1998;120:7661–2. [Google Scholar]; (b) Curley K, Lawrence DS. J Amer Chem Soc. 1998;120:8573–4. [Google Scholar]; (c) Zou K, Cheley S, Givens RS, Bayley H. J Amer Chem Soc. 2002;124:8220–9. doi: 10.1021/ja020405e. [DOI] [PubMed] [Google Scholar]
- 2.For example, see Ghosh M, Song S, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Science. 2004;304:743–6. doi: 10.1126/science.1094561.
- 3.(a) Lawrence DS. Curr Opin Chem Biol. 2005;9:570–5. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]; (b) Xie J, Schultz PG. Cur Opin Chem Biol. 2005;9:548–54. doi: 10.1016/j.cbpa.2005.10.011. [DOI] [PubMed] [Google Scholar]; (c) Petersson EJ, Brandt GS, Zacharias NM, Dougherty DA, Lester HA. Methods Enzymol. 2003;360:258–73. doi: 10.1016/s0076-6879(03)60114-x. [DOI] [PubMed] [Google Scholar]; (d) Marriott G, Roy P, Jacobson K. Methods Enzymol. 2003;360:274–88. doi: 10.1016/s0076-6879(03)60115-1. [DOI] [PubMed] [Google Scholar]
- 4.Hah JM, Sharma V, Li H, Lawrence DS. J Amer Chem Soc. 2006;128:5996–7. doi: 10.1021/ja060136i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sicheri F, Kuriyan J. Curr Opin Struc Biol. 1997;7:777–85. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 6.Lee TR, Lawrence DS. J Med Chem. 1999;42:784–7. doi: 10.1021/jm980663f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Wang Q, Dai Z, Cahill SM, Blumenstein M, Lawrence DS. J Amer Chem Soc. 2006;128:14016–7. doi: 10.1021/ja065852z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Q, Cahill SM, Blumenstein M, Lawrence DS. J Amer Chem Soc. 2006;128:1808–9. doi: 10.1021/ja0577692. [DOI] [PubMed] [Google Scholar]
- 8.Arabaci G, Guo XC, Beebe KD, Coggeshall KM, Pei D. J Amer Chem Soc. 1999;121:5085–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details of peptide synthesis, structure, characterization, and photolytic behavior. This material is free of charge via the Internet at http://pubs.acs.org.