

Abstract
Negative motivational symptoms are observed soon after withdrawal from chronic opiate administration, and are thought to mediate dependence. Examination of brain region-specific signaling changes that accompany early withdrawal may shed light on neural mechanisms underlying negative reinforcement and dependence. Thus, we measured alterations in protein phosphorylation in multiple limbic brain regions in rats undergoing 24 hrs spontaneous or naltrexone-precipitated withdrawal from chronic (6 hrs/day) intravenous heroin self-administration. Region-specific increases in cyclic AMP-dependent GluR1S845 phosphorylation were found in the nucleus accumbens shell, basolateral amygdala, hippocampal CA1 and CA3 subregions, and premotor cortex from 12 to 24 hrs of spontaneous withdrawal, and there were no changes in prefrontal cortex, nucleus accumbens core or caudate-putamen. Increased GluR1S845 phosphorylation was detected earlier (12 hrs withdrawal) in the central amygdala and ventral tegmental area. In contrast, prominent increases in extracellular signal-regulated kinase phosphorylation were found in both prefrontal and premotor cortex, and CA1 and CA3 between 12 and 24 hrs withdrawal. Phosphorylation of striatal cyclic AMP response element binding protein increased in the caudate-putamen but not in the nucleus accumbens. Naltrexone administration after 24 hrs withdrawal increased extracellular signal-regulated kinase phosphorylation in the central amygdala, and nucleus accumbens core and shell. Thus, spontaneous withdrawal from heroin self-administration produces region- and time-dependent changes in cyclic AMP and extracellular signal-regulated kinase activity that could contribute to the behavioral manifestation of opiate dependence.
Keywords: protein kinase A, extracellular signal-regulated kinase, cyclic AMP response element binding protein, nucleus accumbens, amygdala, prefrontal cortex
Introduction
The reinforcing effects of opiate drugs like heroin are mediated by opiate receptors in the ventral tegmental area (Bozarth and Wise, 1981), nucleus accumbens (Olds, 1982), and hippocampus (Stevens et al., 1991) through both dopamine-dependent and –independent mechanisms. Following chronic heroin self-administration, neuroadaptations in mesolimbic dopamine neurons and their target regions are thought to play a central role in the maintenance of opiate addiction (Koob and Le Moal, 2001; Nestler and Aghajanian, 1997; Self and Nestler, 1995). Given that opiate receptors are coupled to inhibitory G proteins that reduce cyclic AMP formation (Childers, 1991), a compensatory up-regulation in cyclic AMP-dependent protein kinase (PKA) has been reported in the nucleus accumbens following chronic heroin self-administration (Self et al., 1995). Increased PKA activity in the nucleus accumbens causes elevated drug and alcohol intake (Self et al., 1998; Wand et al., 2001) and exacerbates the aversive aspects of opiate withdrawal (WD) (Valverde et al., 1996). The aversive motivational effects could promote heroin-seeking behavior through negative reinforcement that alleviates dysphoria in dependent subjects (Hutcheson et al., 2001). However, the functional consequence of PKA up-regulation on downstream cyclic AMP-dependent protein phosphorylation in spontaneous WD has not been studied.
In addition to PKA-mediated protein phosphorylation, extracellular signal-regulated kinase (ERK) activity may also be regulated in opiate WD. Phosphorylation of ERK kinase (MEK), an upstream activator of ERK, substantially increases in cortex and striatum with naltrexone-precipitated WD from chronic repeated morphine administration, but no changes are found after 24 hrs of spontaneous WD (Asensio et al., 2006), arguably a more naturalistic context of opiate WD. The functional consequences of either PKA or ERK activity could involve downstream phosphorylation of the transcription factor cyclic AMP response element binding protein (CREB), leading to transcriptional activation (Mattson et al., 2005; Yamamoto et al., 1988). Thus, CREB activation is increased in the nucleus accumbens and amygdala, but decreased in the ventral tegmental area, with naltrexone-precipitated WD from continuous morphine administration (Shaw-Lutchman et al., 2002). Whether a similar activation of CREB would occur during spontaneous WD using a chronic intermittent schedule of self-administered heroin that more closely approximates human drug use patterns is unknown.
In this study, we tracked the development of protein phosphorylation changes from 12 to 24 hrs of spontaneous WD from chronic intermittent heroin self-administration. Animals self-administered heroin in daily 6 hrs sessions since this level of exposure produces tolerance-like escalation in heroin intake, self-injurious behavior, and handling-induced vocalization during spontaneous WD, along with up-regulation in PKA activity in the nucleus accumbens (Lenoir and Ahmed, 2007; Self et al., 1995). We measured cyclic AMP/PKA-specific phosphorylation of the GluR1 AMPA receptor subunit (Ser 845; Mammen et al., 1997) as an in vivo marker of functional increases in PKA activity in multiple limbic and non-limbic brain nuclei. We compared increases in PKA-mediated GluR1S845 phosphorylation with ERK phosphorylation that is increased by either PKA-dependent signaling (Grewal et al., 1999), or by PKA-independent signaling involving glutamate receptor or receptor tyrosine kinase activity associated with neuronal activation (Thomas and Huganir, 2004). In striatal regions, we compared GluR1S845 and ERK phosphorylation with CREBS133 phosphorylation, since ERK can play a crucial downstream role in PKA-mediated CREBS133 phosphorylation in the striatum (Grewal et al., 2000; Zanassi et al., 2001). We also compared changes both with and without a naltrexone challenge after 24 hrs WD to determine whether blockade of residual heroin metabolites would exacerbate WD-induced protein phosphorylation. The functional consequence of GluR1, ERK, and CREBS133 phosphorylation on the behavioral manifestation of opiate dependence is discussed in relation to intracellular signaling and regional neurotransmission.
Materials and Methods
Subjects and surgery
Individually housed, male, Sprague-Dawley rats initially weighing 275−325g (Charles River, Kingston, RI, USA) were maintained in a climate-controlled environment (21° C) on a 12-hr light-dark cycle (lights on at 7:00 AM) in accordance with guidelines established by the National Institute of Health and the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. Prior to heroin self-administration, animals were trained to lever-press for 45 mg sucrose pellets while maintained on a restricted diet at 85% original body weight. Sucrose self-administration was conducted in ventilated operant chambers (Med Associates, Georgia, VT, USA) on a fixed-ratio 1 (FR1) reinforcement schedule until acquisition criteria were achieved (100 pellets self-administered for 3 consecutive days) to facilitate subsequent acquisition of heroin self-administration. Following acquisition of lever-press behavior, animals were fed ad libitum prior to surgical implantation of a chronic indwelling intrajugular catheter as described previously (Edwards et al., 2007b). Catheters were flushed daily with 0.2 ml of heparinized (20 IU/ml) bacteriostatic saline containing gentamycin sulfate (0.33 mg/ml) to prevent clotting and antibiotic ointment was applied daily to the catheter exit wound to prevent infection.
Heroin self-administration procedures in rats
Following 1 week of recovery from surgical catheterization, animals were allowed to acquire self-administration by performing a single lever press response (FR1) to receive intravenous injections of heroin (60 μg/kg/100 μl injection) delivered over 5 sec in daily 6-hr sessions conducted over 18 days in the dark cycle. During each injection, the house light was turned off and a cue light above the active lever was illuminated, followed by an additional 10 sec time-out period when the both house and cue lights were off and lever responding produced no programmed consequence. Untreated age- and batch-matched controls were individually housed and remained in their home cages, but were handled daily throughout the testing procedure to habituate potential stress responses prior to euthanization. An additional group of home cage controls was injected with naltrexone hydrochloride (1.0 mg/kg, sc) 1 hr prior to sacrifice to determine potential effects in non-dependent animals.
Measurement of protein phosphorylation in regional brain tissue homogenates
Twelve or 24 hrs after completion of the last heroin self-administration session, animals were gently placed in a plexiglass restrainer and immediately euthanized by microwave irradiation aimed at the head region (5 kW, 1.5 s, Murimachi Kikai Co., LTD. Tokyo, Japan). For determination of spontaneous and naltrexone-precipitated WD effects, self-administering animals withdrawn for 24 hrs were split into two groups that received subcutaneous injections of either naltrexone (1.0 mg/kg) or saline (1.0 ml/kg) 1 hr prior to sacrifice following prior habituation to the injection procedure with 3 daily saline injections. Rat brains were rapidly dissected and regional tissue samples collected with 12−16 gauge punches from chilled coronal brain slices (1.5 mm thick). Tissue samples were immediately homogenized by sonication in lysis buffer (320 mM sucrose, 5 nM HEPES, 50 mM NaF, 1 mM EGTA, 1 mM EDTA, 1% SDS, with Protease Inhibitor Cocktail and Phosphatase Inhibitor Cocktails I and II diluted 1:100; Sigma, St. Louis, MO, USA), boiled for 5 min, and stored at −80 °C until determination of protein concentrations by the Lowry method. Samples of 20 μg protein were subjected to SDS-polyacrylamide gel electrophoresis on 10% acrylamide gels using a Tris/Glycine/SDS buffer (Bio-Rad, Hercules, CA, USA), followed by electrophoretic transfer to PVDF membranes (polyvinylidene fluoride; Amersham, Piscataway, NJ, USA). Membranes were blocked overnight in 5% nonfat milk at 4° C, and incubated in primary antibody for pGluR1S845 (1:2500; Chemicon, Temecula, CA, USA), pERK (1:7500; Cell Signaling, Danvers, MA, USA), or pCREBS133 (1:2500; Cell Signaling) for 24 hrs at 4° C. Membranes were washed and labeled with species-specific peroxidase-conjugated secondary (1:10,000; BioRad) for 1 hr at 25° C. Following chemiluminescence detection (ECL plus; Amersham), blots were stripped and reprobed for total protein levels of GluR1 (1:10K; Chemicon), ERK (1:7500; Cell Signaling), or CREB (1:1500; Chemicon) for determination of phosphorylated/total protein ratios. For initial regional comparison of basal phosphorylated and total GluR1, ERK, and CREB levels, 30 μg samples from each brain region were analyzed in the same blot for individual animals and normalized to β-tubulin (1:250K; Chemicon). Images were captured and immunoreactivity was quantified densitometrically using Scion Image software (Frederick, MD) under conditions linear over at least a 3-fold range of protein content.
Data analysis
Mean daily heroin self-administration was compared by 2-factor ANOVA (study group × test day). Densitized phosphorylated proteins were expressed as a ratio of total protein as an internal standard, and expressed as a percentage of the mean for untreated control values (6−8/blot) to normalize data across blots. Experimental data from 12 and 24 hr WD groups (excluding naïve and naltrexone-treated controls) were compared by 1-factor ANOVA followed by Newman-Keuls posthoc comparisons among groups, or planned t-test comparisons with the 12 hr WD group. Since both phosphorylated and total GluR1 levels were regulated in the VTA, these data were analyzed separately in addition to phosphorylated/total protein ratios.
Results
Animals trained to self-administer heroin developed stable self-administration by the 3rd week of training, with mean daily heroin intake (days 13−18) averaging 2.9 ± 0.4 mg/kg (12 hr WD group), 3.4 ± 0.4 mg/kg (24 hr WD group), and 3.1 ± 0.5mg/kg (24 hr WD + NTX group). Self-administering animals demonstrated a mild but significant escalation of heroin intake over the course of training (effect of test session; F2, 17 = 2.060, p = 0.008), averaging 2.7 ± 0.1 mg/kg/session for the first three self-administration sessions and 3.3 ± 0.2 mg/kg/session for the last three sessions across all study groups (Fig. 1A).
Figure 1.
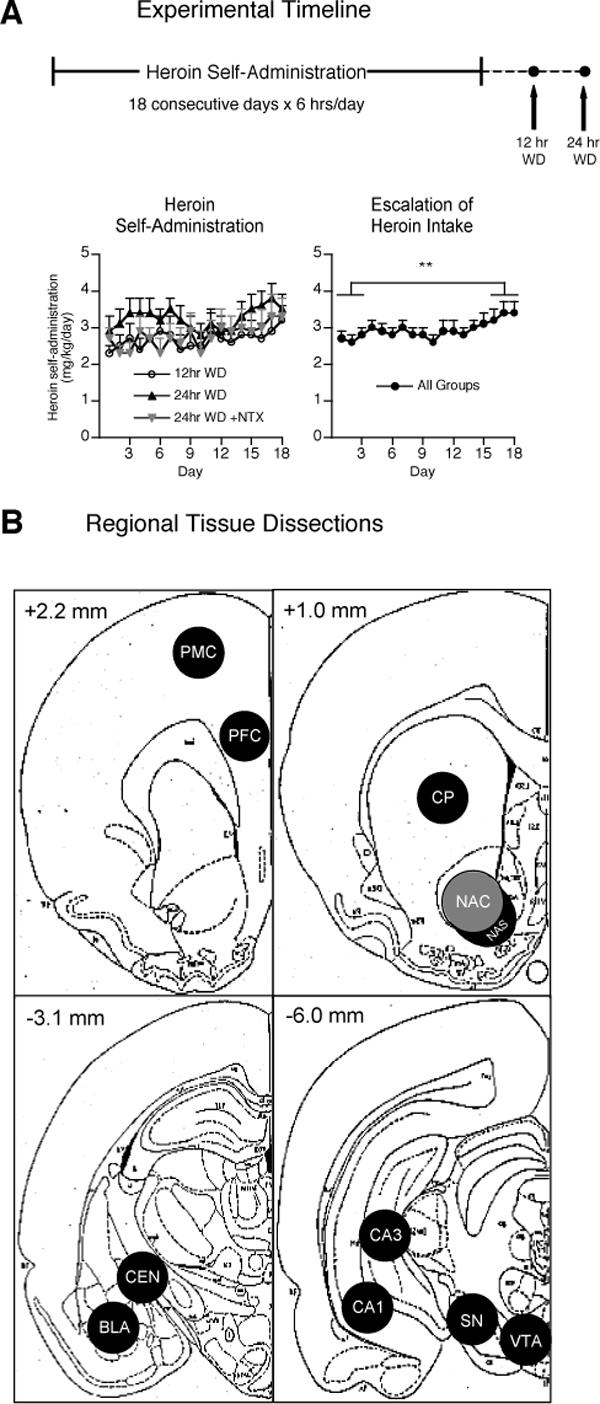
(A) Experimental timeline depicting rats self-administering heroin (60 μg/kg/100 μl injection) over 18 consecutive 6-hr sessions during their dark cycle. Brain tissue was collected at either 12 (n = 8) or 24 hrs of spontaneous withdrawal (WD). Animals in the 24 hr WD group were injected with either saline (n = 12) or 1 mg/kg naltrexone (NTX) (n = 12) and sacrificed 1 hr later. Overall average heroin intake exhibited a mild but significant escalation over the 18 days of self-administration (**p < 0.01). (B) Schematic representation of regional brain samples collected with 12−16 gauge punches from 1.5 mm thick coronal slices. Anterior-posterior coordinates represent posterior sides of brain slices (Paxinos and Watson, 1998). Premotor cortex (PMC), prefrontal cortex (PFC), caudate-putamen (CP), nucleus accumbens core (NAC), nucleus accumbens shell (NAS), basolateral amygdala nucleus (BLA), central amygdala nucleus (CEN), substantia nigra (SN), ventral tegmental area (VTA).
Tissue punches were collected from animals euthanized by microwave fixation according to anatomical regions depicted in Fig. 1B. Microwave fixation was used to preserve in vivo protein phosphorylation status and avoid rapid alterations typically observed following dissection of un-fixed tissue (O'Callaghan and Sriram, 2004). Except where specifically noted in the ventral tegmental area, all of the observed changes in other brain regions involved regulation in phosphorylation states with no regulation in the total amount of GluR1, ERK, or CREB proteins.
Regional comparison of basal protein phosphorylation in drug-naïve animals
We compared basal levels of phosphorylated and total forms of GluR1, ERK, and CREB by calculating optical density ratios of phosphoprotein:tubulin and total protein:tubulin immunoreactivity across brain regions. Relative to the prefrontal cortex, GluR1S845 phosphorylation was ∼50% lower in the caudate-putamen, despite similar total amounts of GluR1 in these regions (Fig. 2A). Similarly, phosphorylated GluR1S845 levels were substantially lower in both the ventral tegmental area and substantia nigra regions, but these reductions were paralleled by markedly lower total amounts of GluR1 protein. In contrast, greater phosphorylated GluR1S845 and total GluR1 levels were found in both CA1 and CA3 hippocampal subregions. Basal phosphorylated ERK levels were lowest in the caudate-putamen, CA3, ventral tegmental area and substantia nigra, while total ERK protein levels were lower only in the premotor cortex and ventral tegmental area relative to other regions (Fig. 2B). In striatal subregions, neither CREBS133 phosphorylation nor total amounts of CREB protein substantially differed between nucleus accumbens core, shell and caudate-putamen (Fig. 2C). Thus, only basal GluR1S845 and ERK phosphorylation in the caudate-putamen were substantially lower independent of total protein levels in striatal subregions, and these changes were not paralleled by reduced CREBS133 phosphorylation. β-tubulin levels were similar across all brain regions indicating equivalent protein content in regional samples (Fig. 2A).
Figure 2.

Regional comparison of phosphorylated (p) and total levels of (A) GluR1, (B) ERK and (C) striatal CREB (n = 3 rats). Optical densities ratios (relative to β-tubulin) were calculated for each phosphorylated and total protein and presented as mean ± sem (see Fig. 1 for abbreviations). Representative blots for phosphorylated and total GluR1, ERK, CREB and β-tubulin are shown.
Dissociation of WD-induced GluR1S845 and ERK phosphorylation in prefrontal but not premotor cortex
In the prefrontal cortex, ERK phosphorylation increased by > 50% from 12 to 24 hrs WD from chronic heroin self-administration (T27 = 2.529, p = 0.018), while there were no changes in PKA-mediated GluR1S845 phosphorylation (Fig. 3A). In non-limbic premotor cortex, both GluR1S845 (F2,20 = 6.925, p = 0.010) and ERK (F2,20 = 4.381, p = 0.026) phosphorylation increased by 31% and 47%, respectively, between 12 and 24 hrs spontaneous WD (Fig. 3B). These changes in protein phosphorylation were related to specifically to spontaneous WD and not chronic heroin exposure or other testing/procedural influences, since they did not occur after only 12 hrs spontaneous WD relative to untreated controls. Naltrexone administration failed to augment WD-induced protein phosphorylation in these cortical subregions.
Figure 3.

Cortical regulation of GluR1S845 and ERK phosphorylation in (A) prefrontal and (B) premotor subregions during spontaneous WD from chronic intermittent heroin self-administration. Microwave-fixed brain tissue was collected after 12 or 24 hrs spontaneous WD, or 1 hr following an injection of 1 mg/kg naltrexone in drug-naïve (NTX) and 24 hr WD (24 hr + NTX) animals. Data are presented as the mean ± SEM of phosphorylated (p)/total protein ratios expressed as a percent change from untreated age and batch-matched controls (n = 8−11/group). Asterisks indicate values differ from 12 hr WD group (*p < 0.05, **p < 0.01) by Newman-Keuls posthoc tests or (+p < 0.05) by Student's t tests. Data from 24 hr WD groups with saline and NTX challenge are pooled where similar.
Differential WD-induced regulation of GluR1S845, ERK and CREBS133 phosphorylation in striatal subregions
PKA-mediated GluR1S845 phosphorylation increased by 35% from 12 to 24 hrs spontaneous WD in the nucleus accumbens shell (F2,27 = 9.362, p < 0.001), but not nucleus accumbens core or caudate-putamen (Fig. 4A-C). In contrast, spontaneous WD failed to substantially increase ERK phosphorylation in the nucleus accumbens, but naltrexone challenge at 24 hrs WD increased ERK phosphorylation by 49% in the shell (T17 = 2.132, p = 0.049), and by 58% in the core (T17 = 2.234, p = 0.041), without increasing GluR1S845 or CREBS133 phosphorylation, compared to 12 hrs spontaneous WD. Since naltrexone failed to alter ERK phosphorylation in drug-naïve animals, these changes suggest that significant opiate receptor tone persists after 24 hrs spontaneous WD in the core and shell, or that naltrexone effects in other brain regions produce systems level interactions that influence ERK phosphorylation in the nucleus accumbens (see below). In the caudate-putamen, 24 hr WD-induced ERK phosphorylation (33−42% increase from untreated controls) failed to significantly increase relative to the 12 hr WD group. However, CREBS133 phosphorylation increased by 51% in the caudate-putamen from 12 to 24 hrs WD from heroin self-administration (F2,27 = 3.489, p = 0.0449), but CREBS133 phosphorylation failed to increase in the nucleus accumbens. Thus, while increased CREB phosphorylation occurs in the caudate-putamen, neither PKA-mediated GluR1S845 phosphorylation nor phosphorylated (activated) ERK translate into increased CREBS133 phosphorylation in nucleus accumbens core and shell subregions between 12 and 24 hrs spontaneous WD from heroin self-administration. Overall, there was no regulation of striatal protein phosphorylation 12 hrs after the last heroin self-administration session compared to naïve controls, indicating that increased phosphorylation after 24 hrs was induced specifically by WD and not chronic heroin exposure, prior surgery or testing.
Figure 4.

Striatal regulation of GluR1S845 , ERK and CREB phosphorylation in the (A) caudate-putamen, (B) nucleus accumbens core, and (B) nucleus accumbens shell subregions during spontaneous WD from chronic intermittent heroin self-administration. Microwave-fixed brain tissue was collected after 12 or 24 hrs spontaneous WD, or 1 hr following an injection of 1 mg/kg naltrexone in drug-naïve (NTX) and 24 hr WD (24 hr + NTX) animals. Data are presented as the mean ± SEM of phosphorylated (p)/total protein ratios expressed as a percent change from untreated age and batch-matched controls (n = 8−11/group). Asterisks indicate values differ from 12 hr WD group (*p < 0.05, **p < 0.01) by Newman-Keuls posthoc tests or (+p < 0.05) by Student's t tests. Representative blots for phosphorylated and total GluR1, ERK and CREB protein are shown.
WD time determines differential GluR1S845 and ERK phosphorylation in amygdala subregions
In the basolateral amygdala, GluR1S845 phosphorylation increased by only 23% from 12 to 24 hrs spontaneous WD from heroin self-administration (Fig. 5A), but naltrexone administration exacerbated this increase in GluR1S845 phosphorylation to 45% (F2,24 = 6.585, p = 0.005). In the central amygdala, GluR1S845 phosphorylation increased by 41% after only 12 hrs WD compared to naïve controls, and this effect was maintained after 24 hrs WD (Fig. 5B). Thus, either GluR1S845 phosphorylation occurs early in spontaneous withdrawal in the central amygdala, or is up-regulated due to chronic heroin exposure. Minor 25% increases in ERK phosphorylation were found in the basolateral amygdala between 12 and 24 hrs WD that failed to reach significance, while decreases in ERK phosphorylation after 12 hrs WD in the central amygdala were normalized after 24 hrs spontaneous WD (F2,25 = 10.74, p < 0.001). Interestingly, ERK phosphorylation in the central but not basolateral amygdala substantially increased with naltrexone administration after 24 hrs WD. Thus, spontaneous WD led to late forming increases in GluR1S845 phosphorylation in the basolateral amygdala, but early bi-directional changes in GluR1S845 and ERK phosphorylation in the central amygdala.
Figure 5.

Amygdala regulation of GluR1S845 and ERK phosphorylation in the (A) basolateral and (B) central subregions during spontaneous WD from chronic intermittent heroin self-administration. Microwave-fixed brain tissue was collected after 12 or 24 hrs spontaneous WD, or 1 hr following an injection of 1 mg/kg naltrexone in drug-naïve (NTX) and 24 hr WD (24 hr + NTX) animals. Data are presented as the mean ± SEM of phosphorylated (p)/total protein ratios expressed as a percent change from untreated age and batch-matched controls (n = 8−11/group). Symbols indicate values differ from 12 hr WD group (**p < 0.01, ***p < 0.001), or differs from 24 hr WD + NTX (##p < 0.01) by Newman-Keuls posthoc tests. Representative blots for phosphorylated and total GluR1 and ERK protein are shown.
Both GluR1S845 and ERK phosphorylation increase during spontaneous WD in CA1 and CA3 hippocampal subregions
GluR1S845 phosphorylation increased by 26% in the hippocampal CA1 subregion (F2,20 = 5.895, p = 0.010) and 71% in the CA3 (F2,20 = 17.05, p < 0.001) from 12 to 24 hrs spontaneous WD from chronic heroin self-administration, and neither effect was augmented by naltrexone challenge (Fig. 6A-B). In the CA1 subregion, ERK phosphorylation increased by 43% from 12 to 24 hrs spontaneous WD, and further increased to 71% with naltrexone challenge (F2,20 = 6.980, p = 0.005). In the CA3 subregion, ERK phosphorylation increased similarly by 83−92%, with or without naltrexone challenge (F2,20 = 7.303, p = 0.004). No changes in protein phosphorylation were detected after 12 hrs spontaneous WD in either hippocampal subregion, indicating that increased GluR1S845 and ERK phosphorylation were produced specifically by heroin WD rather than chronic heroin exposure or other procedural effects.
Figure 6.

Hippocampal regulation of GluR1S845 and ERK phosphorylation in the (A) CA1 and (B) CA3 subregions during spontaneous WD from chronic intermittent heroin self-administration. Microwave-fixed brain tissue was collected after 12 or 24 hrs spontaneous WD, or 1 hr following an injection of 1 mg/kg naltrexone in drug-naïve (NTX) and 24 hr WD (24 hr + NTX) animals. Data are presented as the mean ± SEM of phosphorylated (p)/total protein ratios expressed as a percent change from untreated age and batch-matched controls (n = 8−11/group). Asterisks indicate values differ from 12 hr WD group (*p < 0.05, **p < 0.01) by Newman-Keuls posthoc tests. Representative blots for phosphorylated and total GluR1 and ERK protein are shown.
Up-regulation in both phosphorylated and total GluR1 levels in the ventral tegmental area in spontaneous heroin WD
In the ventral tegmental area, GluR1S845 phosphorylation increased by 54% after only 12 hrs spontaneous WD from heroin self-administration, while total GluR1 remained unaltered, compared to naïve controls (Fig. 7A, left panel). However, between 12 and 24 hrs spontaneous WD, total GluR1 protein increased by 31% (T16 = 2.481, p = 0.0246), resulting in a normalization of GluR1S845/GluR1 protein ratios (Fig. 7A, right panel; naltrexone group T15 = 3.202, p = 0.006). Thus, spontaneous WD is associated with an initial increase in PKA-mediated GluR1S845 phosphorylation, followed by an up-regulation in the amount of GluR1 protein, and this effect was not found in adjacent substantia nigra tissue (Fig. 7B). While naltrexone challenge failed to augment GluR1S845 phosphorylation in the ventral tegmental area after WD from heroin self-administration, it actually decreased basal GluR1S845 phosphorylation by 27% in drug-naïve animals (T21 = 2.62, p = 0.016), and decreased ERK phosphorylation by 24−30% in both ventral tegmental area (T21 = 3.352, p = 0.003) and substantia nigra (T21 = 2.217, p = 0.0378). Taken together, these results indicate that naltrexone challenge may actually increase GluR1S845 and ERK phosphorylation in heroin WD relative to phosphorylation decreases in drug-naive controls. Finally, in the substantia nigra, early 12 hr spontaneous WD was associated with a 39% increase in ERK phosphorylation that normalized after 24 hrs WD (T27 = 2.601, p = 0.0149).
Figure 7.

Ventral midbrain regulation of GluR1S845 and ERK phosphorylation in the (A) ventral tegmental area (VTA) and (B) substantia nigra subregions during spontaneous WD from chronic intermittent heroin self-administration. Microwave-fixed brain tissue was collected after 12 or 24 hrs spontaneous WD, or 1 hr following an injection of 1 mg/kg naltrexone in drug-naïve (NTX) and 24 hr WD (24 hr + NTX) animals. Both phosphorylated and total GluR1 levels were increased after 24 hrs WD from heroin self-administration in the VTA (left panel), resulting in no change in phosphorylated (p)/total protein ratios (middle panel) relative to naïve animals. Data are presented as the mean ± SEM of phosphorylated/total protein ratios expressed as a percent change from untreated age and batch-matched controls (n = 8−11/group). Symbols indicate values differ from 12 hr WD group (+p < 0.05, ++p < 0.01) or between drug-naïve and NTX challenge (δp < 0.05, δδp < 0.01) by Student's t tests. Data from 24 hr WD groups with saline and NTX challenge are pooled where similar. Representative blots for phosphorylated and total GluR1 and ERK protein are shown.
Discussion
In this study, we measured changes in protein phosphorylation status from 12 to 24 hrs of spontaneous WD from chronic intravenous heroin self-administration in daily 6 hr sessions. Heroin itself is metabolized to morphine, and then converted to morphine-3-glucuronide (an inactive compound) and morphine-6-glucuronide, a potent opioid agonist (Milne et al., 1966; Ulens et al., 2001). Repeated exposure to heroin increases the rate of morphine-6-glucuronide formation, and elevated morphine-6-glucuronide levels persist for up to 24 hrs after heroin administration (Antonilli et al., 2003). Therefore, we administered the opiate receptor antagonist naltrexone after 24 hrs to determine whether changes in protein phosphorylation would intensify with blockade of residual heroin metabolites. Despite the presence of active metabolites, however, 24 hrs of spontaneous WD is sufficient to induce negative motivational symptoms (place aversion) in morphine-dependent rats (Bechara et al., 1995). Moreover, 24 hrs spontaneous WD from heroin self-administration triggers relapse to heroin seeking behavior, while antagonist-precipitated WD is ineffective (Shaham et al., 1996).
Most increases in protein phosphorylation were evident after 24 but not 12 hrs, indicating an acute response to spontaneous WD from heroin self-administration, rather than a sustained up-regulation produced by chronic heroin exposure, or other potential influences related to surgery and testing procedures. Thus, both PKA-mediated GluR1S845 phosphorylation and ERK phosphorylation increased in the premotor cortex and CA1 and CA3 hippocampal subregions between 12 and 24 hrs spontaneous WD. In contrast, GluR1S845 (but not ERK) phosphorylation increased in the nucleus accumbens shell and basolateral amygdala, while ERK (but not GluR1S845) phosphorylation increased in the prefrontal cortex and caudate-putamen in spontaneous WD. While there were no increases in GluR1S845 and ERK phosphorylation the nucleus accumbens core or central amygdala from 12 to 24 hrs spontaneous WD, naltrexone challenge increased ERK phosphorylation in these regions, indicating the presence of residual heroin metabolites after 24 hrs WD.
After only 12 hrs spontaneous WD, GluR1S845 and ERK phosphorylation differentially increased in dopamine cell body regions of the ventral tegmental area and substantia nigra. These changes failed to persist after 24 hrs, potentially reflecting higher levels of morphine-6-glucuronide at 12 hrs that would excite dopamine neuron activity via well-characterized opiate receptor-mediated inhibition of GABAergic interneurons (Johnson and North, 1992). In the central amygdala nucleus, GluR1S845 phosphorylation also was increased at 12 hrs WD, but persisted after 24 hrs, which may reflect a neuroadaptation to chronic heroin rather than an acute response to WD. However, increased GluR1S845 phosphorylation after 12 hrs WD cannot be explained by surgical or procedural influences, since we found no changes in either the central amygdala (Edwards et al., 2007a) or ventral tegmental area (K-H. Choi and D.W. Self, unpublished observations) in animals self-administering saline compared to naïve animals.
Chronic opiate use leads to a dysregulation of cellular and synaptic function in limbic brain regions that regulate incentive motivation and responses to stress, and these changes are thought to exacerbate the behavioral manifestation of opiate WD (De Vries and Shippenberg, 2002; Robinson and Berridge, 1993). Both continuous morphine exposure (Terwilliger et al., 1991) and chronic intermittent heroin self-administration (Self et al., 1995) up-regulate molecular components of the cyclic AMP signaling pathway in the nucleus accumbens. In addition, continuous morphine exposure has been shown to up-regulate this pathway in the amygdala (Terwilliger et al., 1991). Such up-regulation in adenylate cyclase and/or PKA levels could contribute to rebound super-activation of cyclic AMP-PKA-mediated GluR1S845 phosphorylation in the nucleus accumbens shell and basolateral amygdala in our study. Consistent with this notion, GluR1S845 phosphorylation failed to increase in the prefrontal cortex, caudate-putamen, ventral tegmental area (independent from total GluR1) and substantia nigra after 24 hrs WD, regions where chronic heroin self-administration and/or continuous morphine exposure fails to up-regulate adenylate cyclase or PKA levels (Duman et al., 1988; Nestler and Tallman, 1988; Self et al., 1995; Terwilliger et al., 1991). These previous studies also failed to find up-regulation in adenylate cyclase or PKA levels in whole hippocampus with continuous morphine exposure, possibly suggesting that increased GluR1S845 phosphorylation in CA1 and CA3 reflects WD-induced increases in hippocampal norepinephrine release (Done et al., 1992) and activation of beta-adrenergic receptors coupled to stimulatory G proteins.
Similarly, increased ERK phosphorylation in the prefrontal cortex in the absence of GluR1S845 phosphorylation could reflect hyperactivity in excitatory afferent input from amygdala, hippocampus or other regions activated by opiate WD (Frenois et al., 2002), rather than intracellular adaptations in ERK signaling. Previous reports showing elevated glutamate, dopamine and acetylcholine release in the prefrontal cortex during opiate WD would support this notion (Bassero et al., 1995; Huang et al., 1997; Rada et al., 1996). In contrast, both spontaneous (48 hrs) and naltrexone-precipitated WD from chronic morphine increases the amount of protein kinase C (PKC) in the frontal cortex (Escriba and Garcia-Sevilla, 1999; Ventayol et al., 1997) that could activate MEK-ERK signaling through stimulation of Raf-1 independent from increased neurotransmitter levels.
Extrinsic mechanisms involving dopamine D1 receptor-stimulated cAMP formation cannot account for increased GluR1S845 phosphorylation in the nucleus accumbens shell, since spontaneous WD from both continuous and intermittent opiate exposure profoundly reduce extracellular nucleus accumbens dopamine levels (Diana et al., 1999; Pothos et al., 1991; Rossetti et al., 1992). Since GluR1S845 phosphorylation is mediated exclusively by PKA, these findings more likely reflect the functional consequence in vivo of up-regulation in PKA observed in vitro (Self et al., 1995; Terwilliger et al., 1991). These changes could contribute to the negative motivational symptoms of opiate dependence and tolerance to opiate reward, since experimental activation of PKA in the nucleus accumbens produces conditioned place aversion in drug naïve animals and tolerance to morphine place preference (M. Janik and D.W. Self, unpublished observations), and is related to increased drug intake (Self et al., 1998; Self, 2004). Conversely, local D2 dopamine receptor stimulation in the nucleus accumbens that inhibits cAMP signaling, or infusions of PKA inhibitors, attenuates the somatic and aversive symptoms of opiate WD (Harris and Aston-Jones, 1994; Valverde et al., 1996). Interestingly, 24 hrs WD from chronic cocaine self-administration also increased PKA-mediated GluR1S845 phosphorylation in the nucleus accumbens shell (Edwards et al., 2007a), but not core, consistent with common up-regulation in PKA levels with multiple drugs of abuse (Freeman et al., 2001; Hope et al., 2007; Lu et al., 2003; Self et al., 1995; Terwilliger et al., 1991).
The nucleus accumbens receives substantial glutamatergic innervation from cortical, hippocampal, amygdala, and thalamic regions that could contribute to ERK phosphorylation via an NMDA receptor-mediated increase in intracellular calcium. However, neither increased PKA-mediated GluR1S845 phosphorylation in the nucleus accumbens shell, nor increased ERK phosphorylation in core and shell (with naltrexone challenge) were reflected by increased CREB phosphorylation. In contrast, previous studies have found that precipitated WD from continuous morphine exposure increases CREB phosphorylation (Chartoff et al., 2006), and CRE (cyclic AMP response element) activity in both the nucleus accumbens and caudate-putamen (Shaw-Lutchman et al., 2002), potentially reflecting a higher degree of opiate dependence with continuous exposure. It should be noted that Shaw-Lutchman et al. were unable to detect striatal CRE activation with intermittent opiate injections, a temporal pattern that more closely resembles daily heroin self-administration in our study.
We measured tissue levels of GluR1S845 phosphorylation that do not necessarily reflect membrane expression, but membrane insertion of GluR1 is dependent on PKA-mediated phosphorylation (Esteban et al., 2003). In this sense, a recent study found increased membrane expression of GluR1 receptors in basolateral amygdala dendrites after chronic morphine self-administration (Glass et al., 2005). These effects would enhance glutamatergic neurotransmission in the amygdala that mediates many somatic signs of opiate WD (Taylor et al., 1998). In addition, basolateral amygdala activity is involved in the ability of conditioned cues to elicit heroin-seeking behavior (Fuchs and See, 2002), and ERK signaling in the central amygdala mediates relapse to cocaine-seeking behavior (Lu et al., 2005). Thus, WD-induced increases in GluR1S845 and ERK phosphorylation could potentially contribute to heroin relapse. The central amygdala also is involved in the expression of dysphoria that occurs during early phases of opiate WD through release of corticotrophin releasing factor (CRF) (Heinrichs et al., 1995; Koob, 1999). Increased ERK phosphorylation could enhance CRF levels via downstream CREB activity that occurs in CRF-containing amygdala neurons (Shaw-Lutchman et al., 2002; Spengler et al., 1992). Thus, repeated daily cycles of heroin self-administration and WD-induced activation of ERK and CREB in central amygdala neurons could facilitate CRF synthesis and release in response to stress, an event also involved in relapse to heroin seeking (Shaham et al., 1997).
We found a positive correlation between GluR1845 phosphorylation in the CA1 subregion after 24 hrs spontaneous WD and average preferred levels of heroin intake (r = 0.800, p = 0.017), suggesting potentially greater dependence in high intake animals. Even larger WD-induced increases in both GluR1S845 and ERK phosphorylation were found in the CA3 subregion. This co-regulation may suggest coupling in PKA and ERK signaling, since cAMP signaling is known to shunt Rap1 to membrane-associated ERK in the hippocampus to regulate synaptic plasticity and spatial memory (Morozov et al., 2003). A previous study found that WD-induced PKA activity in the CA1 occludes the induction of long-term potentiation (LTP), leading to impaired spatial learning (Pu et al., 2002). Our results suggest that increased PKA-mediated GluR1S845 phosphorylation could contribute to this effect by producing a generalized increase in synaptic GluR1 trafficking that reportedly occurs after chronic opiate exposure (Moron et al., 2007).
Chronic heroin self-administration increased the amount of total GluR1 protein in the ventral tegmental area, consistent with previous findings with continuous morphine exposure (Fitzgerald et al., 1996). However, in self-administering animals, these increases developed after 24 but not 12 hrs WD, indicating a dynamic process where total GluR1 is normalized by heroin use. Our results suggests that a concomitant increase in the amount of phosphorylated GluR1S845 could reflect functional increases in membrane AMPA receptors in the ventral tegmental area that contribute to the induction of opiate sensitization (Carlezon et al., 1997). In contrast to dopamine terminal regions, the lack of increases in GluR1S845 (independent of increased GluR1 levels) or ERK phosphorylation after 24 hrs WD agrees with less excitation of dopamine cells in opiate WD (Manzoni and Williams, 1999).
In summary, this study shows prominent but region-specific increases in PKA-mediated GluR1S845, ERK, and CREB phosphorylation in a naturalistic context of spontaneous WD from chronic intermittent heroin self-administration. Our results firmly establish the functional consequence of cAMP-PKA up-regulation and other events on downstream protein modifications that reflect the neuronal state of heroin WD. These data also provide biological relevance for other studies showing that experimental manipulation of GluR1, ERK, and CREB mediate several important addiction-related behavioral changes.
Acknowledgements
This work is supported by the United States Public Health Service Grants DA 010460, DA 008227, DA 18743, DA 016472 (S.E.), DA 016857 (D.L.G.), and by the Wesley Gilliland Professorship in Biomedical Research (UTSW).
Abbreviations
- WD
withdrawal
- PKA
protein kinase A
- PKC
protein kinase C
- ERK
extracellular signal-regulated kinase
- MEK
ERK kinase
- CREB
cyclic AMP response element binding protein
- CRE
cyclic AMP response element
References
- Antonilli L, Suriano C, Paolone G, Badiani A, Nencini P. Repeated exposures to heroin and/or cadmium alter the rate of formation of morphine glucuronides in the rat. J Pharmacol Exp Ther. 2003;307:651–660. doi: 10.1124/jpet.103.055467. [DOI] [PubMed] [Google Scholar]
- Asensio VJ, Miralles A, Garcia-Sevilla JA. Stimulation of mitogen-activated protein kinase kinases (MEK1/2) by mu-, delta- and kappa-opioid receptor agonists in the rat brain: regulation by chronic morphine and opioid withdrawal. Eur J Pharmacol. 2006;539:49–56. doi: 10.1016/j.ejphar.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bassareo V, Tanda G, Di Chiara G. Increase of extracellular dopamine in the medial prefrontal cortex during spontaneous and naloxone-precipitated opiate abstinence. Psychopharmacology. 1995;122:202–205. doi: 10.1007/BF02246097. [DOI] [PubMed] [Google Scholar]
- Bechara A, Nader K, van der Kooy D. Neurobiology of withdrawal motivation: evidence for two separate aversive effects produced in morphine-naive versus morphine-dependent rats by both naloxone and spontaneous withdrawal. Behav Neurosci. 1995;109:91–105. doi: 10.1037//0735-7044.109.1.91. [DOI] [PubMed] [Google Scholar]
- Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area. Life Sci. 1981;28:551–555. doi: 10.1016/0024-3205(81)90148-x. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Boundy VA, Haile CN, Lane SB, Kalb RG, Neve RL, Nestler EJ. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–814. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- Chartoff EH, Mague SD, Barhight MF, Smith AM, Carlezon WA., Jr. Behavioral and molecular effects of dopamine D1 receptor stimulation during naloxone-precipitated morphine withdrawal. J Neurosci. 2006;26:6450–6457. doi: 10.1523/JNEUROSCI.0491-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Shippenberg TS. Neural systems underlying opiate addiction. J Neurosci. 2002;22:3321–3325. doi: 10.1523/JNEUROSCI.22-09-03321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana M, Muntoni AL, Pistis M, Melis M, Gessa GL. Lasting reduction in mesolimbic dopamine neuronal activity after morphine withdrawal. Eur J Neurosci. 1999;11:1037–1041. doi: 10.1046/j.1460-9568.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- Done C, Silverstone P, Sharp T. Effect of naloxone-precipitated morphine withdrawal on noradrenaline release in rat hippocampus in vivo. Eur J Pharmacol. 1992;215:333–336. doi: 10.1016/0014-2999(92)90052-6. [DOI] [PubMed] [Google Scholar]
- Duman RS, Tallman JF, Nestler EJ. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther. 1988;246:1033–1039. [PubMed] [Google Scholar]
- Edwards S, Graham DL, Bachtell RK, Self DW. Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci. 2007a;25:2201–2213. doi: 10.1111/j.1460-9568.2007.05473.x. [DOI] [PubMed] [Google Scholar]
- Edwards S, Whisler KN, Fuller DC, Orsulak PJ, Self DW. Addiction-related alterations in D1 and D2 dopamine receptor behavioral responses following chronic cocaine self-administration. Neuropsychopharmacology. 2007b;32:354–366. doi: 10.1038/sj.npp.1301062. [DOI] [PubMed] [Google Scholar]
- Escriba PV, Garcia-Sevilla JA. Parallel modulation of receptor for activated C kinase 1 and protein kinase C-alpha and beta isoforms in brains of morphine-treated rats. Br J Pharmacol. 1999;127:343–348. doi: 10.1038/sj.bjp.0702555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WM, Nader MA, Nader SH, Robertson DJ, Gioia L, Mitchell SM, Daunais JB, Porrino LJ, Friedman DP, Vrana KE. Chronic cocaine-mediated changes in non-human primate nucleus accumbens gene expression. J Neurochem. 2001;77:542–549. doi: 10.1046/j.1471-4159.2001.00252.x. [DOI] [PubMed] [Google Scholar]
- Frenois F, Cador M, Caille S, Stinus L, Le Moine C. Neural correlates of the motivational and somatic components of naloxone-precipitated morphine withdrawal. Eur J Neurosci. 2002;16:1377–1389. doi: 10.1046/j.1460-9568.2002.02187.x. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, See RE. Basolateral amygdala inactivation abolishes conditioned stimulus- and heroin-induced reinstatement of extinguished heroin-seeking behavior in rats. Psychopharmacology. 2002;160:425–433. doi: 10.1007/s00213-001-0997-7. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Kruzich PJ, Colago EE, Kreek MJ, Pickel VM. Increased AMPA GluR1 receptor subunit labeling on the plasma membrane of dendrites in the basolateral amygdala of rats self-administering morphine. Synapse. 2005;58:1–12. doi: 10.1002/syn.20176. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJ. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature. 1994;371:155–157. doi: 10.1038/371155a0. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L. Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol. 1995;6:74–80. [PubMed] [Google Scholar]
- Hope BT, Nagarkar D, Leonard S, Wise RA. Long-term upregulation of protein kinase A and adenylate cyclase levels in human smokers. J Neurosci. 2007;27:1964–1972. doi: 10.1523/JNEUROSCI.3661-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang NK, Tseng CJ, Wong CS, Tung CS. Effects of acute and chronic morphine on DOPAC and glutamate at subcortical DA terminals in awake rats. Pharmacol Biochem Behav. 1997;56:363–371. doi: 10.1016/s0091-3057(96)00236-5. [DOI] [PubMed] [Google Scholar]
- Hutcheson DM, Everitt BJ, Robbins TW, Dickinson A. The role of withdrawal in heroin addiction: enhances reward or promotes avoidance? Nat Neurosci. 2001;4:943–947. doi: 10.1038/nn0901-943. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. The role of the striatopallidal and extended amygdala systems in drug addiction. Ann NY Acad Sci. 1999;877:445–460. doi: 10.1111/j.1749-6632.1999.tb09282.x. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Lenoir M, Ahmed SH. Heroin-induced reinstatement is specific to compulsive heroin use and dissociable from heroin reward and sensitization. Neuropsychopharmacology. 2007;32:616–624. doi: 10.1038/sj.npp.1301083. [DOI] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Shaham Y, Hope BT. Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem. 2003;85:1604–1613. doi: 10.1046/j.1471-4159.2003.01824.x. [DOI] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–219. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. Journal of Biological Chemistry. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson BJ, Bossert JM, Simmons DE, Nozaki N, Nagarkar D, Kreuter JD, Hope BT. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. Journal of Neurochemistry. 2005;95:1481–1494. doi: 10.1111/j.1471-4159.2005.03500.x. [DOI] [PubMed] [Google Scholar]
- Milne RW, Nation RL, Somogyi AA. The disposition of morphine and its 3- and 6-glucuronide metabolites in humans and animals, and the importance of the metabolites to the pharmacological effects of morphine. Drug Metab Rev. 1996;28:345–472. doi: 10.3109/03602539608994011. [DOI] [PubMed] [Google Scholar]
- Moron JA, Abul-Husn NS, Rozenfeld R, Dolios G, Wang R, Devi LA. Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: a proteomics study focusing on endocytic proteins. Mol Cell Proteomics. 2007;6:29–42. doi: 10.1074/mcp.M600184-MCP200. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Tallman JF. Chronic morphine treatment increases cyclic AMP-dependent protein kinase activity in the rat locus coeruleus. Mol Pharmacol. 1988;33:127–132. [PubMed] [Google Scholar]
- O'Callaghan JP, Sriram K. Focused microwave irradiation of the brain preserves in vivo protein phosphorylation: comparison with other methods of sacrifice and analysis of multiple phosphoproteins. J Neurosci Meth. 2004;135:159–168. doi: 10.1016/j.jneumeth.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Olds ME. Reinforcing effects of morphine in the nucleus accumbens. Brain Res. 1982;237:429–440. doi: 10.1016/0006-8993(82)90454-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson GC. The Rat Brain in Stereotaxic Coordinates. 4th ed Academic Press; New York: 1998. [Google Scholar]
- Pothos E, Rada P, Mark GP, Hoebel BG. Dopamine microdialysis in the nucleus accumbens during acute and chronic morphine, naloxone-precipitated withdrawal and clonidine treatment. Brain Res. 1991;566:348–350. doi: 10.1016/0006-8993(91)91724-f. [DOI] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G. Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci. 2002;22:1914–1921. doi: 10.1523/JNEUROSCI.22-05-01914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada PV, Mark GP, Taylor KM, Hoebel BG. Morphine and naloxone, ip or locally, affect extracellular acetylcholine in the accumbens and prefrontal cortex. Pharmacol Biochem Behav. 1996;53:809–816. doi: 10.1016/0091-3057(95)02078-0. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Hmaidan Y, Gessa GL. Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol. 1992;221:227–234. doi: 10.1016/0014-2999(92)90706-a. [DOI] [PubMed] [Google Scholar]
- Self DW. Regulation of drug-taking and -seeking behaviors by neuroadaptations in the mesolimbic dopamine system. Neuropharmacology. 2004;47:242–255. doi: 10.1016/j.neuropharm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Ann Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, McClenahan AW, Beitner-Johnson D, Terwilliger RZ, Nestler EJ. Biochemical adaptations in the mesolimbic dopamine system in response to heroin self-administration. Synapse. 1995;21:312–318. doi: 10.1002/syn.890210405. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Funk D, Erb S, Brown TJ, Walker CD, Stewart J. Corticotropin-releasing factor in stress-induced relapse to heroin-seeking in rats. J Neurosci. 1997;17:2605–2614. doi: 10.1523/JNEUROSCI.17-07-02605.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Rajabi H, Stewart J. Relapse to heroin-seeking in rats under opioid maintenance: the effects of stress, heroin priming, and withdrawal. J Neurosci. 1996;16:1957–1963. doi: 10.1523/JNEUROSCI.16-05-01957.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22:3663–3672. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler D, Rupprecht R, Van LP, Holsboer F. Identification and characterization of a 3′,5′-cyclic adenosine monophosphate-responsive element in the human corticotropin-releasing hormone gene promoter. Mol Endocrinol. 1992;6:1931–1941. doi: 10.1210/mend.6.11.1480179. [DOI] [PubMed] [Google Scholar]
- Stevens KE, Shiotsu G, Stein L. Hippocampal mu-receptors mediate opioid reinforcement in the CA3 region. Brain Res. 1991;545:8–16. doi: 10.1016/0006-8993(91)91263-z. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Punch LJ, Elsworth JD. A comparison of the effects of clonidine and CNQX infusion into the locus coeruleus and the amygdala on naloxone-precipitated opiate withdrawal in the rat. Psychopharmacology. 1998;138:133–142. doi: 10.1007/s002130050655. [DOI] [PubMed] [Google Scholar]
- Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Ulens C, Baker L, Ratka A, Waumans D, Tytgat J. Morphine-6beta-glucuronide and morphine-3-glucuronide, opioid receptor agonists with different potencies. Biochem Pharmacol. 2001;62:1273–1282. doi: 10.1016/s0006-2952(01)00761-4. [DOI] [PubMed] [Google Scholar]
- Valverde O, Tzavara E, Hanoune J, Roques BP, Maldonado R. Protein kinases in the rat nucleus accumbens are involved in the aversive component of opiate withdrawal. Eur J Neurosci. 1996;8:2671–2678. doi: 10.1111/j.1460-9568.1996.tb01562.x. [DOI] [PubMed] [Google Scholar]
- Ventayol P, Busquets X, Garcia-Sevilla JA. Modulation of immunoreactive protein kinase C-alpha and beta isoforms and G proteins by acute and chronic treatments with morphine and other opiate drugs in rat brain. Naunyn Schmiedeberg's Arch Pharmacol. 1997;355:491–500. doi: 10.1007/pl00004974. [DOI] [PubMed] [Google Scholar]
- Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci. 2001;21:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto KK, Gonzalez GA, Biggs WH, 3rd, Montminy MR. Phosphorylation-induced binding and transcriptional efficacy of nuclear factor CREB. Nature. 1988;334:494–498. doi: 10.1038/334494a0. [DOI] [PubMed] [Google Scholar]
- Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem. 2001;276:11487–11495. doi: 10.1074/jbc.M007631200. [DOI] [PubMed] [Google Scholar]