

Abstract
Adeno-associated virus (AAV) serotype 1 (AAV1) has been shown to be more effective than the well-studied AAV serotype 2 (AAV2) in muscle gene transfer. Replacement of amino acids 350 to 430 of AAV2 VP1 with the corresponding amino acids from VP1 of AAV1 resulted in a hybrid vector, termed AAV-221-IV, which behaved similarly to AAV1 in vitro and in vivo in muscle. Intramuscular injection of 1 × 1011 vector particles per mouse of hybrid vector carrying a human FIX transgene in CD4 knockout mice resulted in an average level of human FIX in the plasma of 450 ng/ml, 4- to 10-fold higher than in mice injected with an AAV2 vector carrying the same transgene, and 80% of the transgene levels in animals treated with the same dose of AAV1. DNA analysis of injected muscle showed a 10-fold higher copy number after gene delivery by the hybrid vector compared with AAV2. A comparison of total DNA versus DNA from intact virus particles suggests a higher stability of hybrid virus particles. These results suggest that changes in the AAV capsid have an effect on virus-cell receptor interaction, and also influence trafficking and processing of the virus particle in the cell. This “hybrid vector” retains the heparin-binding sites of AAV2 and, therefore, can be purified by passage through a heparin-Sepharose column with the same efficiency as AAV2. When tested in vivo, either in CD4 knockout mice or in a hemophilic mouse model, the heparin-purified hybrid vector showed >10-fold higher activity than similarly purified AAV2. This demonstrates the utility of this hybrid vector in the performance of large-scale heparin column purification to generate a vector with a high expression profile for muscle-directed gene delivery. Initiation of clinical studies with this hybrid vector may be facilitated because it differs from AAV2 by only nine amino acids.
INTRODUCTION
Hemophilia B is a hereditary bleeding disorder caused by the deficiency of circulating functional plasma coagulation factor IX. Because the factor IX gene is located on the X chromosome, hemophilia B affects primarily males. Clinically, it is characterized by frequent spontaneous bleeding into joints and soft tissues, easy bruising, and prolonged bleeding. The consequences of bleeding into critical closed spaces, such as the intracranial or the retroperitoneal space, are severe and can lead to death. The current treatment for hemophilia B is infusion of purified factor IX concentrates. Although protein replacement therapy is effective in controlling the disease, repetitive infusion is necessary for patients with severe disease. Thus, in the long run, gene therapy would be an ideal treatment for this genetic disease (Kay and High, 1999; High, 2004).
Adeno-associated virus (AAV) has been developed as a delivery vehicle for human gene therapy (Muzyczka, 1992; Muzyczka and Berns, 2002). To accommodate the factor IX gene expression cassette, the AAV coding region is removed and only two flanking cis elements, inverted terminal repeats (ITRs, 145 bp each), are preserved. By supplying the trans elements (rep and cap genes) in a different plasmid, high-titer recombinant AAV vectors can be produced (Grimm et al., 1998; Xiao et al., 1998). The current yield is sufficient for animal studies and initial-phase human clinical trials for hemophilia. In both mouse and dog models, AAV vectors have shown great promise in delivering therapeutic levels of the factor IX gene, which often leads to persistent circulating factor IX secretion from targeting tissues such as muscle or the liver (Herzog et al., 1997, 2001; Chao et al., 1999, 2001; Wang et al., 2000; Couto, 2004). Despite successful studies in animals, human clinical trials for hemophilia B have not yet yielded satisfactory results (Kay et al., 2000; Manno et al., 2003).
Although a large number of AAV serotypes of various origin have been identified (Gao et al., 2002, 2003, 2004; Schmidt et al., 2004), AAV serotype 2 (AAV2) has been used in the majority of AAV gene delivery studies including the current human clinical trials. However, extensive studies of alternative AAV serotypes revealed that other AAV serotypes often outperform AAV2 in specific tissues. For example, AAV serotype 1 (AAV1) has been shown to be 10-100 times better than AAV2 in transducing muscle (Xiao et al., 1999; Chao et al., 2000). AAV5 and AAV8 are superior to AAV2 for gene delivery to the liver (Chiorini et al., 1999; Auricchio et al., 2001; Gao et al., 2002; Lebherz et al., 2004; Rabinowitz et al., 2004; Sarkar et al., 2004). Nevertheless, AAV2 is the most studied AAV prototype. The integration frequency mediated by AAV2 ITRs has been characterized, and the primary cellular receptor for AAV2 has been identified as heparan sulfate proteoglycan (HSPG) (Summerford and Samulski, 1998; Qing et al., 1999; Summerford et al., 1999). This has enabled development of purification of AAV2-based vectors by heparin-based chromatography (Gao et al., 2000; Snyder and Flotte, 2002; Liu et al., 2003). Chromatography-based purification offers the advantage of scalability, and this method of purification may result in higher vector purity compared with vectors purified by traditional CsCl gradient centrifugation.
Extensive studies have been carried out to identify key amino acids in the AAV capsid proteins. By site-directed mutagenesis, Wu et al. identified two clusters of amino acids, amino acids 509-522 and 561-591, that are responsible for AAV2 heparin binding (Wu et al., 2000). The Kleinschmidt group further demonstrated that basic amino acids such as R484, R487, R585, and R588 and K532 play a critical role AAV2 heparin binding (Kern et al., 2003). Our previous study discovered that the tissue tropism-determining domain for AAV2 does not overlap with the heparin-binding domain (Hauck and Xiao, 2003). Thus, it is possible to combine the strong affinity of AAV1 for muscle and the high binding activity of AAV2 into one hybrid vector. This novel hybrid vector can, therefore, be purified by heparin chromatography while maintaining high efficiency in muscle cell transduction. In the current study, we have characterized hybrid vector AAV-221-IV for its effectiveness in treating hemophilia B in mice. Because this hybrid vector more closely resembles AAV2 than AAV1 in terms of neutralizing antibodies, and differs from AAV2 by only nine amino acids, AAV-221-IV may be readily developed for clinical use on the basis of the fact that AAV2 has been used in several clinical trials to date and has demonstrated excellent safety.
MATERIALS AND METHODS
Cell lines
Human 293, COS, and HeLa cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, streptomycin (100 μg/ml), and penicillin (100 U/ml) (all purchased from Sigma-Aldrich, St. Louis, MO). All cells were maintained in a humidified 37°C incubator with 5% CO2.
Hybrid vector construction
Hybrid vector pH221-IV is based on the AAV2 serotype and contains a small part of the AAV1 capsid representing amino acids 213-423 (Hauck and Xiao, 2003). In detail, VP1 of the hybrid vector contains, from the N terminus to the C terminus, (1) AAV2 amino acids 1-212, (2) AAV1 amino acids 213-423, and (3) AAV2 amino acids 424-736. To generate the plasmid encoding this hybrid capsid, the Expand Long Template PCR system (Roche Diagnostics, Indianapolis, IN) was used to fuse AAV1 and AAV2 plasmids. Amino acids 213-423 from AAV1 were amplified with primers V2850S (GTG GCG CAC CAA TGG CAG ACA ATA ACG) and V3507A (GCG TAG CTG CTG TGG AAA GG). N-terminal and C-terminal amino acids 1-212 and 424-736, from AAV2, were amplified with the following primer pair combinations: B1881S (CGA GTC AGT TGC GCA GCC ATC GAC GTC A) and V2877A (TCG TTA TTG TCT GCC ATT GGT GCG CCA C); and V3488S (CCT TTC CAC AGC AGC TAC GC) and VXbaIA (TCG GGC TTA AAT ACC CAG CGT GAC CAC A), respectively. The obtained fragments overlap at their respective 5′ and 3′ ends and outer primers were used to combine fragments in subsequent polymerase chain reactions (PCRs). The resulting fragment was digested with HindIII and XbaI and replaced the unmodified HindIII-XbaI fragment in pH22. The plasmid was sequenced to ensure integrity of the cap sequence.
Vector production and purification
The AAV vectors were produced by a modified triple-plasmid transfection based on calcium phosphate precipitation, which has been described previously (Cao et al., 2000). The plasmid carrying the human FIX cDNA under the control of the cytomegalovirus (CMV) enhancer/promoter was described previously (Liu et al., 2003). Vectors were purified by two rounds of CsCl gradient centrifugation, dialyzed against phosphate-buffered saline (PBS), and stored at -80°C in PBS with 3% glycerol.
Heparin column purification
The harvested cell pellet was resuspended in 10 mM Tris (pH 8.0)-150 mM sodium chloride and sonicated for 2 min to release the virus. The resulting mixture was then centrifuged at 3000 × g and the debris was discarded. The supernatant was incubated with Benzonase (Sigma-Aldrich) in the presence of 0.5% sodium deoxycholate for 1 hr at 37°C. Heparin-Sepharose resin (1.5 g; GE Healthcare, Uppsala, Sweden) was pretreated according to the manufacturer’s protocol, added to the mixture, and incubated overnight at room temperature with constant agitation. The resin was then loaded onto a minicolumn (Bio-Rad Laboratories, Hercules, CA). The vectors were eluted with a gradient salt solution from 200 to 1000 mM NaCl in 10 mM Tris, pH 8.0, in a volume of 10 ml/fraction. The vectors were concentrated with a Biomax protein concentrator (Millipore, Bedford, MA) and the titer was determined.
Vector titer determination
Genome titers were determined by either slot-blot hybridization, using transgene probes according to the standard protocol, or real-time PCR with a PRISM 7700 sequence detector (Applied Biosystems, Foster City, CA) (Cao et al., 2000). For real-time PCR titration of rAAV preparations, the primer and fluorescent probe sets were selected for the factor IX gene. Primers and internal probe were designed to amplify 134 bp of human factor IX sequence. Oligonucleotides with sequences of 5′-TTC GAT CTA CAA AGT TCA CCA TCT ATA AC-3′ and 5′-AAA CTG GTC CCT TCC ACT TCA G-3′ were used as forward and reverse primers, respectively, and the sequence (5′→3′) 6-FAM-AAT CTC TAC CTC CTT CAT GGA AGC CAG CA-TAMRA, tagged with 6-FAM fluorescent dye at the 5′ end and TAMRA quencher at the 3′ end, was used as a probe. The final reaction mix consisted of primers (900 nM each); 200 nM probe; dATP, dCTP, and dGTP (200 μM each); 400 μM dUTP; 3.5 mM MgCl2; 8% glycerol; and 1 U of uracil N-glycosylase (UNG) in 1 × TaqMan buffer containing the reference dye ROX and 0.25 U of AmpliTaq Gold polymerase (Roche Molecular Diagnostics, Norwalk, CT) in a total volume of 25 μl. The cycling conditions consisted of 40 cycles of 95°C for 15 sec and 60°C for 1 min. The reactions were performed in accordance with the instructions of the manufacturer. AAV samples were prepared by proteinase K digestion overnight, followed by heat inactivation at 100°C for 20 min.
Animals and vector administration
Immunodeficient CD4 knockout mice were purchased from Jackson Laboratory (Bar Harbor, ME). The protocols were approved by the Institutional Animal Care and Use Committee of Children’s Hospital of Philadelphia (Philadelphia, PA). Recombinant AAV vectors were administered to mice by intramuscular injection. Blood was drawn by retroorbital bleeding at various time points. The procedures followed are described elsewhere (Herzog et al., 1997).
Assay for transgene expression
Levels of transgene expression were determined by enzyme-linked immunosorbent assay (ELISA) as described previously (Herzog et al., 1997). Briefly, to measure factor IX, ELISA plates were coated with a mouse anti-human FIX monoclonal antibody (diluted 1:500 in 0.2 M Na2CO3; Sigma). Collected mouse plasma was diluted 1:5 in dilution buffer (6% bovine serum albumin [BSA], 1 mM EDTA, 0.05% Tween 20 in PBS) and incubated at 4°C overnight. A peroxidase-conjugated polyclonal rabbit anti-human FIX antibody (Affinity Biologicals, Ancaster, ON, Canada; used at a 1:2333 dilution) as detection antibody with 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS; Roche Diagnostics, Mannheim, Germany) as substrate was used.
Histochemical analysis
Tissue obtained by muscle biopsy was frozen initially in cooled methylbutane followed by liquid nitrogen. Serial muscle cryosections (10 μm) were stained for hFIX, using a goat anti-hFIX antibody (diluted 1:400; Enzyme Research Laboratories, South Bend, IN), followed by anti-goat rhodamine-conjugated antibody (diluted 1:200; Chemicon International, Temecula, CA).
Clotting assay
Mouse plasma samples for FIX clotting activity were collected by tail clipping and collection of blood into 20 μl of 3.8% sodium citrate solution. Activated partial thromboplastin time (aPTT) was measured with a fibrometer (FibroSystem; BD Diagnostic Systems, Sparks, MD). Plasma samples and aPTT reagent (bioMérieux, Durham, NC) were mixed (50 μl each) and incubated at 37°C for 3 min before addition of 50 μl of 25 mM CaCl2, and measurement of the clotting time with the fibrometer.
Inhibition assays
Heparin and neutralizing antibodies against AAV are assayed in the following way. COS cells (3 × 105 cells per well) were seeded in 24-well plates and infected with rAAV (MOI of 20,000) at 37°C for 1 hr in serum-free DMEM containing 25 mM HEPES buffer. At 1 hr postinfection, cells were washed with PBS and incubated in DMEM with 10% FBS for 48 hr and medium was assayed directly in an ELISA for human factor IX expression. To study the effect of heparin on the infectivity of AAV vectors, AAV vectors were preincubated with heparin (Sigma-Aldrich) at a final concentration of 200 μg/ml in serum-free DMEM containing 25 mM HEPES buffer at 37°C for 1 hr before they were applied to the cells. To test the effect of monoclonal antibody A20 (Progen Biotechnik, Heidelberg, Germany), which recognizes only intact AAV2 particles, the A20 antibody was diluted 1:5 in DMEM and preincubated with the vectors for 30 min at room temperature before infection. To obtain neutralizing antibodies against AAV1 and AAV2, mouse plasma was collected 4 weeks after AAV1 or AAV2 vector was administered intravenously to C57BL/6 mice. The effect of neutralizing antibodies against AAV1 and AAV2 on the hybrid vectors was assayed by preincubating the hybrid vectors with mouse sera containing neutralizing antibodies that were diluted 1:5 in DMEM containing 25 mM HEPES buffer. After incubating the vectors and mouse sera for 30 min at room temperature, the vectors were applied to target cells. Supernatants were harvested 48 hr postinfection and human factor IX in the samples was assayed in duplicate.
DNA analysis
Genomic DNA was extracted from injected murine muscle, using a PUREGENE DNA purification kit (Gentra Systems, Minneapolis, MN). A total of 20 μg of genomic DNA extracted from injected muscle at week 14 was digested with endonuclease EcoRV, separated in a 0.8% agarose gel, and transferred to a positively charged nylon membrane (GE Healthcare, Little Chalfont, UK). Southern blot hybridization was performed with a random-primed labeled probe (1.2 kb) to intron 1 of F.IX, using [α-32P]dCTP and a High Prime kit (Roche Diagnostics, Mannheim, Germany). Vector genomes were identified as a 1.7-kb band. The intensity of bands on autoradiographs was quantitated by densitometric scanning of the exposed X-ray film.
RESULTS
Generation of AAV-221-IV
AAV2 is the most studied serotype for gene delivery even though AAV1 is more effective for gene transfer targeting muscle. AAV2 has been approved for use in several clinical studies, including for muscle and liver administration to evaluate the safety of delivery of human factor IX to treat hemophilia B. To extend our previous study that an AAV1-AAV2 hybrid vector could combine the advantages of purification methods developed for AAV2 and the higher efficacy of AAV1 in transducing muscle, we generated AAV-221-IV-based vectors to express factor IX gene. Details of the capsid sequence of AAV-221-IV are illustrated in Fig. 1. Amino acids 213-423 from AAV1 have replaced the corresponding region in AAV2. Because of the close sequence similarity between these serotypes, there are only nine amino acid differences between AAV-221-IV and AAV2. This hybrid was produced by triple transfection, using a helper plasmid carrying the AAV2 rep gene and a vector plasmid containing AAV2 ITRs.
FIG. 1.

Composition of the AAV-221-IV capsid protein. AAV-221-IV is identical to AAV2 except for nine amino acids. The line illustration shows the VP1 protein for AAV1, AAV2, and AAV-221-IV. Amino acids 212-423 are aligned. The nine amino acids that differ between AAV1 and AAV2 are shaded.
AAV-221-IV vector directs high-level factor IX expression in CD4-deficient mice
To test the performance of AAV-221-IV, we compared factor IX gene expression after administration of CsCl gradientpurified AAV1, AAV-221-IV, and AAV2 vector to murine muscle. All vectors were prepared with a vector plasmid carrying the human FIX (hFIX) gene under the control of a CMV-IE promoter (Liu et al., 2003). To avoid antibody formation against human FIX protein in mouse, CD4 knockout mice were used. Vectors were injected intramuscularly into mice at a dose of 1 × 1011 vector genomes (VG)/mouse. As shown in Fig. 2A, expression levels of circulating human FIX in CD4 mice receiving AAV-221-IV were 4- to 5-fold higher than those receiving AAV2-based vectors, and reached approximately 80% of the hFIX levels achieved in mice injected with AAV1-based vectors over the observed time period (Fig. 2A).
FIG. 2.

AAV-221-IV vector delivers factor IX to C57BL/6-CD4 knockout mice. Mice were injected intramuscularly with CsCl-purified AAV1, AAV2, or AAV-221-IV (1 × 1011 VG/mouse) carrying an hFIX construct under the control of a CMV promoter/enhancer construct. (A) Human factor IX (hFIX) expression profile after vector administration. hFIX expression was measured by ELISA and average values and standard deviation are shown for each group (n = 3). (B) Immunofluorescence staining for human factor IX in AAV-injected mouse muscle. Shown are samples obtained from mice killed at week 14 after vector delivery. The signal for human factor IX is shown in red. “Untransduced” is from a CD4 mouse that did not receive vector. (C) Southern blot analysis for vector genomes after vector administration. Total cellular DNA was extracted 14 weeks postinjection. After EcoRV digestion, 20 μg of DNA was electrophoresed on a 0.8% gel. The resulting membrane was probed for human factor IX. The viral genome shown here is the 1.7-kb fragment. The reference standards were prepared by adding 0.35, 3.5, and 35 copies of plasmid per murine diploid genome before EcoRV digestion.
To reveal the basis for differences in transduction, we analyzed muscle sections from the treated animals. Immunohistochemical staining for hFIX expression showed that the number of positive fibers in animals receiving AAV-221-IV vectors was similar to those administered AAV1 vectors (Fig. 2B), and were 3-fold higher than in those receiving the same dose of AAV2. In addition, the staining signal resulting from AAV1 and AAV-221-IV injection was stronger than that from AAV2 vectors. To test whether the difference in the capsid also affected vector genome persistence, vector genome copy numbers after vector administration were compared by Southern blot analysis. As shown in Fig. 2C, in mice receiving AAV2 factor IX vector, the copy number of vector genomes is substantially lower than that of AAV1 and AAV-221-IV.
AAV-221-IV vector corrects the phenotype of hemophilia B mice
To demonstrate the performance of this new hybrid vector in a hemophilia model, we intramuscularly administered 1 × 1011 particles of CsCl gradient-purified AAV1-CMV-hFIX, AAV2-CMV-hFIX, or AAV-221-IV-CMV-hFIX to hemophilia B mice with a CD4-deficient background (F.IX-CD4 double-knockout mice). Factor IX expression was determined at weeks 2, 4, 6, 10, and 14. These data are presented in Fig. 3A. In hemophilia B and CD4 double-knockout mice (F.IX-CD4) receiving AAV-221-IV-CMV-hFIX, the expression levels of factor IX were 5- to 6-fold higher than in those receiving AAV2-CMV-hFIX. AAV-221-IV vector expression was slightly lower that that of AAV1 but still at a comparable level to AAV1.
FIG. 3.

CsCl gradient-purified AAV-221-IV vector corrects the hemophilia phenotype. Hemophilia B mice (F.IX-CD4 double knockout) were injected with CsCl gradient-purified AAV1, AAV2, or AAV-221-IV (1 × 1011 VG/mouse) carrying the CMV-hFIX expression cassette. (A) Time course of hFIX expression after vector injection. hFIX expression was measured by ELISA at each time point shown. The average expression levels and standard deviation are shown for each group (n = 3). (B) Activated partial thromboplastin time (aPTT, in seconds) of hemophilic mice as a function of time after intramuscular injection of CsCl-purified vectors. The average aPTT and standard deviation of each group (n = 3) are shown. The aPTT times of hemophilic mice (>60 sec) and of nonhemophilic CD4 knockout mice (35-38 sec) are indicated as horizontal dotted lines. The p value for the difference is less than 0.05.
We also monitored correction of the bleeding phenotype and measured the activated partial thromboplastin time (aPTT) in mice after vector delivery. In the control group of CD4 knockout mice with normal mouse factor IX protein in circulation, the aPTT value was between 35 and 38 sec. Before vector administration, the aPTT time of FIX-CD4 knockout mice was in the range of 68 to 98 sec (Fig. 3B). After 1 × 1011 VG of AAV2-CMV-hFIX was delivered to muscle, the aPTT time of F.IX-CD4 mice was reduced to 58 to 78 sec. In contrast, the aPTT time for mice receiving the AAV-221-IV-CMV-hFIX was 57 to 58 sec, similar to those receiving AAV1 vectors (53 to 62 sec).
Purification and properties of AAV-221-IV particles
AAV2 has a strong affinity for heparan sulfate proteoglycan (HSPG). Heparin can compete with AAV2 and prevent AAV2 transduction. HSPG has been demonstrated to be the primary receptor in its infection process (Summerford and Samulski, 1998). The binding of AAV to heparin has been exploited to develop chromatography-based purification methods for AAV2 vectors, using heparin-Sepharose. This method is highly relevant to purification of clinical-grade vectors because chromatography, unlike gradient ultracentrifugation, can be easily scaled to consistently produce the large amounts of vector required for clinical studies. A chromatographic method may also yield higher purity virus stock compared with CsCl gradient centrifugation (Zolotukhin et al., 1999). AAV1 has poor affinity for heparin and is typically purified by CsCl gradient centrifugation. Because AAV-221-IV maintains the intact C terminus of the AAV2 capsid protein, which has been demonstrated to contain residues important in heparin binding, we attempted to purify AAV-221-IV vector by heparin chromatography. After harvesting transfected cells, clarified cell lysate was incubated with heparin-Sepharose. A salt gradient ranging from 0.2 to 1.0 M NaCl was applied to elute resin-bound vector. For both AAV2 and the hybrid vector AAV-221-IV, vector elution was initially detected at 400 mM NaCl, with peak fractions eluting at 500 mM NaCl (Fig. 4). Using this purification method, the vector yields were comparable to yields obtained by CsCl gradient purification, resulting in comparable amounts of rAAV2 and AAV-221-IV in several independent preparations. This suggested that the hybrid vector modification had no adverse impact on the AAV packaging process.
FIG. 4.
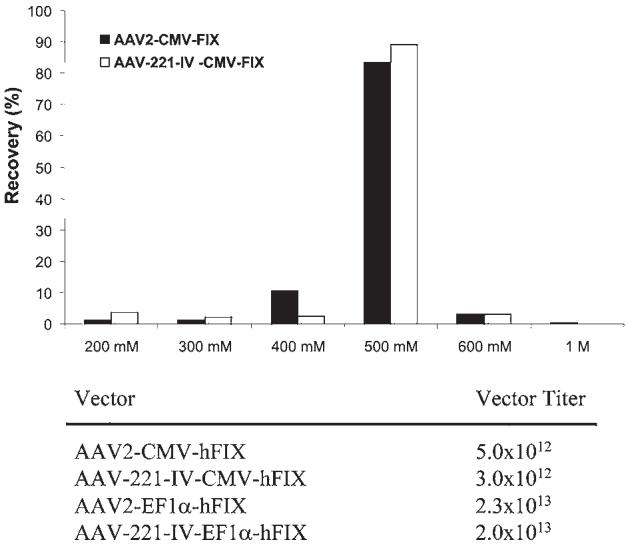
AAV-221-IV production and purification. AAV2 and AAV-221-IV carrying different hFIX transgene constructs (CMV-FIX and Ef1α-FIX) were produced by triple transfection and purified by heparin-Sepharose column chromatography. They were eluted from the column with an NaCl gradient ranging from 200 mM to 1 M (x axis). The amount of vector genomes for each fraction was determined by real-time PCR. The percentage of vector genomes recovered for each fraction is indicated on the y axis. Bottom: Vector yields for two independent vector preparations from 20 plates (150 mm). In both cases AAV2 and AAV-221-IV yielded comparable amounts of vector.
Because AAV-221-IV contains capsid sequences from both AAV1 and AAV2, we tested its immunological properties in vitro. Aliquots of heparin-purified AAV2-CMV-hFIX and AAV-221-IV-CMV-hFIX were preincubated with each of the following: (1) heparin or (2) pooled plasma from immunocompetent mice that had been injected with AAV1 vector, AAV2 vector, or monoclonal antibody A20, which recognizes intact viral particles (Wobus et al., 2000). After these incubations, vectors were tested for in vitro transduction, using COS cells. Both AAV2 and AAV-221-IV transduction was inhibited by heparin pretreatment (Fig. 5). The A20 antibody efficiently blocked AAV2 transduction, but only marginally inhibited AAV-221-IV, which transduced COS cells to a level that was 76% of the untreated control group. Antiserum against AAV1 had no significant effect on AAV2, but was able to reduce hFIX expression from AAV-221-IV vector to 80% of the untreated control group level. AAV2 antiserum inhibited AAV2 transduction to 20% of the control (Fig. 5), but was less efficient in inhibiting AAV-221-IV, which gave 40% of control expression after preincubation with the same concentration of AAV2 antiserum. These results suggest that both AAV1 and AAV2 capsid sequences contribute to the immunological properties of AAV-221-IV vectors.
FIG. 5.
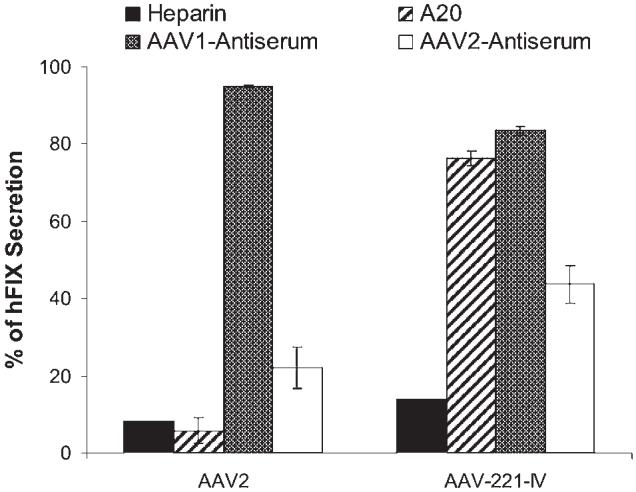
Immunological properties of AAV-221-IV and AAV2 particles. COS cells were infected with heparin-purified AAV2-CMV-hFIX or AAV-221-IV-CMV-hFIX (at an MOI of 20,000). The vectors were either not treated (control) or preincubated with heparin (200 μg/ml), antibody A20, AAV1 antiserum, or AAV2 antiserum before infection. The inhibition of transduction is indicated by the percentage of remaining hFIX secretion into the medium (48 hr postinfection) compared with the nontreated control.
Highly purified AAV-221-IV vector can deliver a therapeutic level of factor IX in vivo
Because AAV-221-IV can also be purified by heparin chromatography, we tested the efficacy of heparin column-purified vectors in vivo. After intramuscular injection of 1 × 1011 VG of heparin column-purified AAV2-CMV-hFIX or AAV-221-IV-CMV-hFIX vector into CD4-deficient mice, hFIX expression levels were measured (Fig. 6). Surprisingly, the hFIX levels in mice receiving AAV2 vectors reached only 20 ng/ml, 7-fold lower than the levels measured in mice receiving the same dose of CsCl gradient-purified AAV-CMV-hFIX (Fig. 2A). Factor IX expression from heparin-Sepharose-purified AAV-221-IV-CMV-hFIX vector was approximately 250 ng/ml, only slightly lower than levels measure with CsCl gradient-purified vectors. Similar results were obtained when these vectors were tested in FIX-CD4 double-knockout mouse. As shown in Fig. 6B, heparin-purified AAV2-CMV-hFIX-injected mice produced circulating FIX at 57 ng/ml, and heparin-purified AAV-221-IV-CMV-hFIX-injected mice produced a plateau value of approximately 200 ng/ml. Determination of the aPTT time resulting from AAV2 vector delivery showed no significant reduction (Fig. 6C). However, in mice receiving AAV-221-IV-CMV-hFIX vector, the aPTT time was reduced to approximately 43 sec and stabilized at 49 sec at later time points.
FIG. 6.

Performance of chromatography-purified hybrid vectors in vivo. (A) Expression profile of hFIX protein in CD4 knockout mice after intramuscular injection of heparin-purified AAV2-CMV-hFIX and AAV-221-IV-CMV-hFIX. Mice were injected with heparin-purified vector at 1 × 1011 VG/mouse. hFIX expression was measured by ELISA and average values and standard deviation are shown for each group (n = 3). (B) Time course of hFIX expression in factor IX-CD4 double-knockout mice (C57BL/6 background) after intramuscular injection. Mice were injected with heparin-purified AAV2-CMV-hFIX (n = 4) or AAV-221-IV-CMV-hFIX (n = 6) at 1 × 1011 VG/mouse. Circulating human FIX proteins were measured by ELISA and average values and standard deviation are shown for each group. (C) Activated partial thromboplastin time (aPTT, in seconds) of hemophilic mice as a function of time after intramuscular injection of heparin-purified vectors. Vectors were injected at a dose of 1 × 1011 VG/mouse in each group. The aPTT of plasma samples was determined at each time point shown. The average aPTT and standard deviation of each group are given. The aPTT times of hemophilic mice (>60 sec) and of nonhemophilic CD4 knockout mice (35-38 sec) are indicated as horizontal lines.
DISCUSSION
Adeno-associated virus serotype 2, the first AAV serotype to be cloned and sequenced, is the most widely studied AAV serotype. Almost all studies on AAV replication, packaging, rescue, and integration have used this serotype. For example, the integration preference of AAV2 for human chromosome 19 was discovered and the requirement for such integration requires the AAV2 Rep protein and AAV2 ITRs (Samulski, 1993; Young et al., 2000; Urabe et al., 2003; Weitzman et al., 2003). Whether such properties of AAV2 are conserved among other AAV serotypes remains unclear even though they share a high degree of homology in both trans elements (rep and cap genes) and cis signals (ITRs). Even though AAV2-based vectors are often reported to be relatively inefficient in transduction of many tissues or organs, nevertheless they are the only serotype used in clinical trials to date because they are the best characterized, and have demonstrated excellent safety in clinical studies. On the other hand, it would be highly desirable to use a more efficient vector in muscle, such as AAV1, in clinical trials; AAV1 may be able to achieve therapeutic effects in humans at significantly lower doses compared with AAV2. AAV1 has not yet been characterized to the extent of AAV2 in terms of immune responses and integration frequency. Even though AAV1 and AAV2 share a high degree of amino acid sequence homology, the use of AAV1 vectors clinically is a concern because AAV1 was initially isolated from a monkey cell line, whereasAAV2 is a prevalent and innocuous virus in humans. Here, we report a novel AAV capsid variant that differs by only nine amino acids from AAV2, and demonstrates the superior transduction efficiency of AAV1 vectors in muscle.
The vector AAV-221-IV is identical to AAV2 except for nine amino acids in the amino-terminal region of the VP1 capsid protein. Previous studies have demonstrated that the primary elements responsible for the HSPG affinity of AAV2 reside in the carboxy-terminal region of VP1, and therefore AAV-221-IV was predicted to maintain heparin binding. Here we have demonstrated that this novel hybrid vector can be efficiently purified by heparin chromatography. As shown in Fig. 4, this vector has an affinity for heparin similar to that of AAV2. The nine amino acid changes in AAV-221-IV did not appear to decrease AAV packaging efficiency, which contrasts with results observed with many AAV1-AAV2 hybrids that are deficient in packaging. In our studies, we also unexpectedly observed that heparin-purified AAV2 vectors were less efficient than CsCl gradient-purified vector. This result may be related to the copurification of empty or incompletely filled capsids with bona fide vector particles during column chromatography. These inactive particles may interfere with the efficiency of vector uptake by target cells, or may interfere with transduction through some other currently undefined mechanism. Inhibition of liver transduction in mice by empty capsids in AAV2 vectors prepared by column chromatography has been reported (Parker et al., 2003). A combination of heparin column and CsCl gradient purification, or modifications of chromatographic methods to efficiently remove empty capsids, may be required to obtain optimal vector purity and potency.
Although any amino acid changes in the capsid protein could theoretically creates new antigens, the few (9) changes of the 736 amino acids that represent the difference between the AAV-221-IV and AAV2 vector may reasonably be expected to have a minimal effect on immunogenic potential. Our data suggest that the few changes in AAV2 capsid protein in AAV-221-IV probably had occurred in a region that did not markedly change the antigenic properties of AAV2. Our results confirmed that the AAV-221-IV vector is still reactive to antibodies against AAV2 but not AAV1. However, these nine amino acid changes in AAV-221-IV were sufficient to confer muscle transduction potential similar to that of the highly efficient AAV1-based vectors. When purified by CsCl gradient, the FIX expression level of AAV-221-IV was markedly better than that observed with AAV2 vectors but marginally lower (20%) compared with AAV1. This suggests that there may exist additional elements in AAV1 capsid proteins responsible for its enhanced performance in muscle. Further characterization of AAV-221-IV in hemophilic mice also confirmed that AAV-221-IV could produce levels similar to those achieved by AAV1 direct transgene expression and provide therapeutic level of factor IX expression (Figs. 2 and 5).
In the vectors tested in this study, the cis elements that provide transgene expression and the AAV ITRs were identical. Therefore, the only variations are derived from the capsid proteins. The direct influences of the capsid may help to change AAV infection kinetics. Our immunofluorescence staining demonstrated more transduced cells with AAV1 and AAV-221-IV vectors (Fig. 2B) at the same dose. Analysis of AAV vectors by Southern blot confirmed that more vector genomes entered and/or were stabilized in the host cells with AAV1 and AAV-221-IV vectors. This is a likely explanation for why AAV-221-IV-based vectors were more efficient in delivering the factor IX gene than was AAV2.
Another concern specific to factor IX gene delivery is the decrease in specific activity of factor IX protein when factor IX protein is overproduced. The normal specific activity of human factor IX is approximately 200 units/mg. In our testing, the specific activity of factor IX produced by AAV2 and AAV-221-IV hybrid vectors was approximately 150 units/mg. Therefore the specific activity of FIX that was more efficiently generated by AAV-221-VI did not appear to have reduced specific activity compared with FIX produced by AAV2.
Various strategies to improve the transduction efficacy of AAV vectors have been explored. One popular approach is to engineer a ligand for a specific cellular receptor to the AAV capsid region to target specific cells or tissues. This strategy depends on the identification of specific cell surface receptors and ligands. Insertion of ligands into capsid sequences may affect vector packaging efficacy. In contrast, the approach reported herein takes advantage of a naturally existing AAV serotype, combining advantageous features of both AAV1 and AAV2 vectors into a single hybrid that differs from the well-characterized AAV2 vector by only nine amino acids. Because of its enhanced performance in muscle transduction, combined with high similarity to AAV2 vectors, which have an excellent safety profile in human clinical studies, the AAV-221-IV hybrid vector may be a highly promising candidate for clinical studies for muscle administration. One consideration is that the high muscle transduction efficiency obtained in animal studies may not be reproduced in humans. Whether the hybrid vector can achieve high muscle cell transduction in vivo in humans will remain unknown until it is tested and confirmed.
ACKNOWLEDGMENTS
The authors thank Drs. Katherine High, Roland Herzog, and Valder Arruda for comments on the manuscript. The authors also thank Marlene Webber, Junwei Sun, and Patrick Dolan for help in manuscript preparation. This study was supported by NIH grant R01HL069051 and by a Muscular Dystrophy Association Research award to W.X.
REFERENCES
- AURICCHIO A, O’CONNOR E, HILDINGER M, WILSON JM. A single-step affinity column for purification of serotype-5 based adeno-associated viral vectors. Mol. Ther. 2001;4:372–374. doi: 10.1006/mthe.2001.0462. [DOI] [PubMed] [Google Scholar]
- CAO L, LIU Y, DURING MJ, XIAO W. High-titer, wild-type free recombinant adeno-associated virus vector production using intron-containing helper plasmids. J. Virol. 2000;74:11456–11463. doi: 10.1128/jvi.74.24.11456-11463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAO H, SAMULSKI R, BELLINGER D, MONAHAN P, NICHOLS T, WALSH C. Persistent expression of canine factor IX in hemophilia B canines. Gene Ther. 1999;6:1695–1704. doi: 10.1038/sj.gt.3301024. [DOI] [PubMed] [Google Scholar]
- CHAO H, LIU Y, RABINOWITZ J, LI C, SAMULSKI RJ, WALSH CE. Several log increase in therapeutic transgene delivery by distinct adeno-associated viral serotype vectors. Mol. Ther. 2000;2:619–623. doi: 10.1006/mthe.2000.0219. [DOI] [PubMed] [Google Scholar]
- CHAO H, MONAHAN PE, LIU Y, SAMULSKI RJ, WALSH CE. Sustained and complete phenotype correction of hemophilia B mice following intramuscular injection of AAV1 serotype vectors. Mol. Ther. 2001;4:217–222. doi: 10.1006/mthe.2001.0449. [DOI] [PubMed] [Google Scholar]
- CHIORINI JA, KIM F, YANG L, KOTIN RM. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUTO LB. Preclinical gene therapy studies for hemophilia using adeno-associated virus (AAV) vectors. Semin. Thromb. Hemost. 2004;30:161–171. doi: 10.1055/s-2004-825630. [DOI] [PubMed] [Google Scholar]
- GAO G, QU G, BURNHAM MS, HUANG J, CHIRMULE N, JOSHI B, YU QC, MARSH JA, CONCEICAO CM, WILSON JM. Purification of recombinant adeno-associated virus vectors by column chromatography and its performance in vivo. Hum. Gene Ther. 2000;11:2079–2091. doi: 10.1089/104303400750001390. [DOI] [PubMed] [Google Scholar]
- GAO GP, ALVIRA MR, WANG L, CALCEDO R, JOHNSTON J, WILSON JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO G, ALVIRA MR, SOMANATHAN S, LU Y, VANDENBERGHE LH, RUX JJ, CALCEDO R, SANMIGUEL J, ABBAS Z, WILSON JM. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO G, VANDENBERGHE LH, ALVIRA MR, LU Y, CALCEDO R, ZHOU X, WILSON JM. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIMM D, KERN A, RITTNER K, KLEINSCHMIDT JA. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 1998;9:2745–2760. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- HAUCK B, XIAO W. Characterization of tissue tropism determinants of adeno-associated virus type 1. J. Virol. 2003;77:2768–2774. doi: 10.1128/JVI.77.4.2768-2774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERZOG RW, HAGSTROM JN, KUNG SH, TAI SJ, WILSON JM, FISHER KJ, HIGH KA. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc. Natl. Acad. Sci. U.S.A. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERZOG RW, MOUNT JD, ARRUDA VR, HIGH KA, LOTHROP CD., Jr. Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol. Ther. 2001;4:192–200. doi: 10.1006/mthe.2001.0442. [DOI] [PubMed] [Google Scholar]
- HIGH KA. Clinical gene transfer studies for hemophilia B. Semin. Thromb. Hemost. 2004;30:257–267. doi: 10.1055/s-2004-825639. [DOI] [PubMed] [Google Scholar]
- KAY MA, HIGH K. Gene therapy for the hemophilias [comment] Proc. Natl. Acad. Sci. U.S.A. 1999;96:9973–9975. doi: 10.1073/pnas.96.18.9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAY MA, MANNO CS, RAGNI MV, LARSON PJ, COUTO LB, McCLELLAND A, GLADER B, CHEW AJ, TAI SJ, HERZOG RW, ARRUDA V, JOHNSON F, SCALLAN C, SKARSGARD E, FLAKE AW, HIGH KA. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet. 2000;24:257–261. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- KERN A, SCHMIDT K, LEDER C, MULLER OJ, WOBUS CE, BETTINGER K, VON DER LIETH CW, KING JA, KLEINSCHMIDT JA. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEBHERZ C, GAO G, LOUBOUTIN JP, MILLAR J, RADER D, WILSON JM. Gene therapy with novel adeno-associated virus vectors substantially diminishes atherosclerosis in a murine model of familial hypercholesterolemia. J. Gene Med. 2004;6:663–672. doi: 10.1002/jgm.554. [DOI] [PubMed] [Google Scholar]
- LIU YL, WAGNER K, ROBINSON N, SABATINO D, MARGARITIS P, XIAO W, HERZOG RW. Optimized production of high-titer recombinant adeno-associated virus in roller bottles. Biotechniques. 2003;34:184–189. doi: 10.2144/03341dd07. [DOI] [PubMed] [Google Scholar]
- MANNO CS, CHEW AJ, HUTCHISON S, LARSON PJ, HERZOG RW, ARRUDA VR, TAI SJ, RAGNI MV, THOMPSON A, OZELO M, COUTO LB, LEONARD DG, JOHNSON FA, McCLELLAND A, SCALLAN C, SKARSGARD E, FLAKE AW, KAY MA, HIGH KA, GLADER B. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- MUZYCZKA N. Use of adeno-associated virus as a general transduction vector for mammalian cells. Curr. Top. Microbiol. Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- MUZYCZKA N, BERNS KI. Parvoviridae: The viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. Lippincott Williams & Wilkins; Baltimore, MD: 2002. [Google Scholar]
- PARKER A, NAGY D, VARGAS J, ANAND V, QU G, SOMMER J, WRIGHT F, COUTO L. In vivo performance of AAV-2 vectors purified by CsCl gradient centrifugation or column chromatography. Mol. Ther. 2003;7:S390. [Google Scholar]
- QING K, MAH C, HANSEN J, ZHOU S, DWARKI V, SRIVASTAVA A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2 [see comments] Nat. Med. 1999;5:71–77. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- RABINOWITZ JE, BOWLES DE, FAUST SM, LEDFORD JG, CUNNINGHAM SE, SAMULSKI RJ. Cross-dressing the virion: The transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J. Virol. 2004;78:4421–4432. doi: 10.1128/JVI.78.9.4421-4432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMULSKI RJ. Adeno-associated virus: Integration at a specific chromosomal locus. Curr. Opin. Genet. Dev. 1993;3:74–80. doi: 10.1016/s0959-437x(05)80344-2. [DOI] [PubMed] [Google Scholar]
- SARKAR R, TETREAULT R, GAO G, WANG L, BELL P, CHANDLER R, WILSON JM, KAZAZIAN HH., JR. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- SCHMIDT M, KATANO H, BOSSIS I, CHIORINI JA. Cloning and characterization of a bovine adeno-associated virus. J. Virol. 2004;78:6509–6516. doi: 10.1128/JVI.78.12.6509-6516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SNYDER RO, FLOTTE TR. Production of clinical-grade recombinant adeno-associated virus vectors. Curr. Opin. Biotechnol. 2002;13:418–423. doi: 10.1016/s0958-1669(02)00369-5. [DOI] [PubMed] [Google Scholar]
- SUMMERFORD C, SAMULSKI RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUMMERFORD C, BARTLETT JS, SAMULSKI RJ. αvβ5 integrin: A co-receptor for adeno-associated virus type 2 infection [see comments] Nat. Med. 1999;5:78–82. doi: 10.1038/4768. [DOI] [PubMed] [Google Scholar]
- URABE M, KOGURE K, KUME A, SATO Y, TOBITA K, OZAWA K. Positive and negative effects of adeno-associated virus Rep on AAVS1-targeted integration. J. Gen. Virol. 2003;84:2127–2132. doi: 10.1099/vir.0.19193-0. [DOI] [PubMed] [Google Scholar]
- WANG L, NICHOLS TC, READ MS, BELLINGER DA, VERMA IM. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol. Ther. 2000;1:154–158. doi: 10.1006/mthe.2000.0031. [DOI] [PubMed] [Google Scholar]
- WEITZMAN MD, YOUNG SM, JR., CATHOMEN T, SAMULSKI RJ. Targeted integration by adeno-associated virus. Methods Mol. Med. 2003;76:201–219. doi: 10.1385/1-59259-304-6:201. [DOI] [PubMed] [Google Scholar]
- WOBUS CE, HUGLE-DORR B, GIROD A, PETERSEN G, HALLEK M, KLEINSCHMIDT JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: Epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU P, XIAO W, CONLON T, HUGHES J, AGBANDJE-McKENNA M, FERKOL T, FLOTTE T, MUZYCZKA N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIAO W, CHIRMULE N, BERTA SC, McCULLOUGH B, GAO G, WILSON JM. Gene therapy vectors based on adeno-associated virus type 1. J. Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIAO X, LI J, SAMULSKI RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOUNG SM, JR., McCARTY DM, DEGTYAREVA N, SAMULSKI RJ. Roles of adeno-associated virus Rep protein and human chromosome 19 in site-specific recombination. J. Virol. 2000;74:3953–3966. doi: 10.1128/jvi.74.9.3953-3966.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZOLOTUKHIN S, BYRNE B, MASON E, ZOLOTUKHIN I, POTTER M, CHESNUT K, SUMMERFORD C, SAMULSKI R, MUZYCZKA N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
This article has been cited by
- 1.Vandenberghe LH, Wilson JM, Gao G. Tailoring the AAV vector capsid for gene therapy. Gene Therapy. 2009 doi: 10.1038/gt.2008.170. [DOI] [PubMed] [Google Scholar]
- 2.Youjin Shen, Jun Yin. The treatment of hemophilia A: from protein replacement to AAV-mediated gene therapy. Biotechnology Letters. 2008 doi: 10.1007/s10529-008-9869-0. [DOI] [PubMed] [Google Scholar]
- 3.Shi Q, Fahs SA, Wilcox DA, Kuether EL, Morateck PA, Mareno N, Weiler H, Montgomery RR. Syngeneic transplantation of hematopoietic stem cells that are genetically modified to express factor VIII in platelets restores hemostasis to hemophilia A mice with preexisting FVIII immunity. Blood. 2008;112(7):2713–2721. doi: 10.1182/blood-2008-02-138214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Büning Hildegard, Perabo Luca, Coutelle Oliver, Quadt-Humme Sibille, Hallek Michael. Recent developments in adeno-associated virus vector technology. The Journal of Gene Medicine. 2008;10(7):717–733. doi: 10.1002/jgm.1205. [DOI] [PubMed] [Google Scholar]