

Abstract
Structural studies of antibiotics not only provide a short cut to medicine allowing for rational structure-based drug design, but may also capture snapshots of dynamic intermediates that become ‘frozen’ after inhibitor binding1,2. Myxopyronin inhibits bacterial RNA polymerase (RNAP) by an unknown mechanism3. Here we report the structure of dMyx—a desmethyl derivative of myxopyronin B4—complexed with a Thermus thermophilus RNAP holoenzyme. The antibiotic binds to a pocket deep inside the RNAP clamp head domain, which interacts with the DNA template in the transcription bubble5,6. Notably, binding of dMyx stabilizes refolding of the β’-subunit switch-2 segment, resulting in a configuration that might indirectly compromise binding to, or directly clash with, the melted template DNA strand. Consistently, footprinting data show that the antibiotic binding does not prevent nucleation of the promoter DNA melting but instead blocks its propagation towards the active site. Myxopyronins are thus, to our knowledge, a first structurally characterized class of antibiotics that target formation of the pre-catalytic transcription initiation complex—the decisive step in gene expression control. Notably, mutations designed in switch-2 mimic the dMyx effects on promoter complexes in the absence of antibiotic. Overall, our results indicate a plausible mechanism of the dMyx action and a stepwise pathway of open complex formation in which core enzyme mediates the final stage of DNA melting near the transcription start site, and that switch-2 might act as a molecular checkpoint for DNA loading in response to regulatory signals or antibiotics. The universally conserved switch-2 may have the same role in all multisubunit RNAPs.
Our data show that myxopyronins efficiently inhibit formation of transcription initiation complexes by both Escherichia coli and T. thermophilus RNAPs (Supplementary Fig. 1). The crystal structure of dMyx bound to the T. thermophilus RNAP holoenzyme (Fig. 1, Supplementary Fig. 2 and Supplementary Table 1) has been refined at 2.7 Å resolution to the final Rfactor/Rfree = 0.240/0.270. Comparison with the apo-RNAP (Protein Data Bank (PDB) accession 2A6E) showed a subtle but systematic dMyx-induced ~2.0–3.7 Å shift of the β’-subunit amino-terminal domain (residues β’1–600) and the σ-subunit in the RNAP–dMyx complex (Methods and Supplementary Fig. 3). This shift, however, did not change either the pattern of the σ/core-RNAP interactions, or the arrangement of and the distance between the σ-regions 2 and 4 which recognize the −10 and −35 promoter elements5. The configuration of the fork formed by the σ-regions 2.3–2.4 and 2.5–3.1 at the upstream entrance to the RNAP main channel (where melting of the DNA duplex is thought to start6-8) is also largely unaffected by dMyx. In the RNAP–dMyx complex, the width of the main channel is reduced by ~4 Å as compared to that in one of the two crystallographically independent molecules in the apo-RNAP. If systematic, this constriction could affect accommodation of the upstream DNA duplex and/or DNA melting. However, the second apo-RNAP molecule also had the narrowed channel, indicating the inherent flexibility of the neighbouring domains and suggesting that the main channel width is unlikely to be a limiting factor for open complex formation (Supplementary Fig. 4).
Figure 1. Structure of the RNAP–Myx complex.
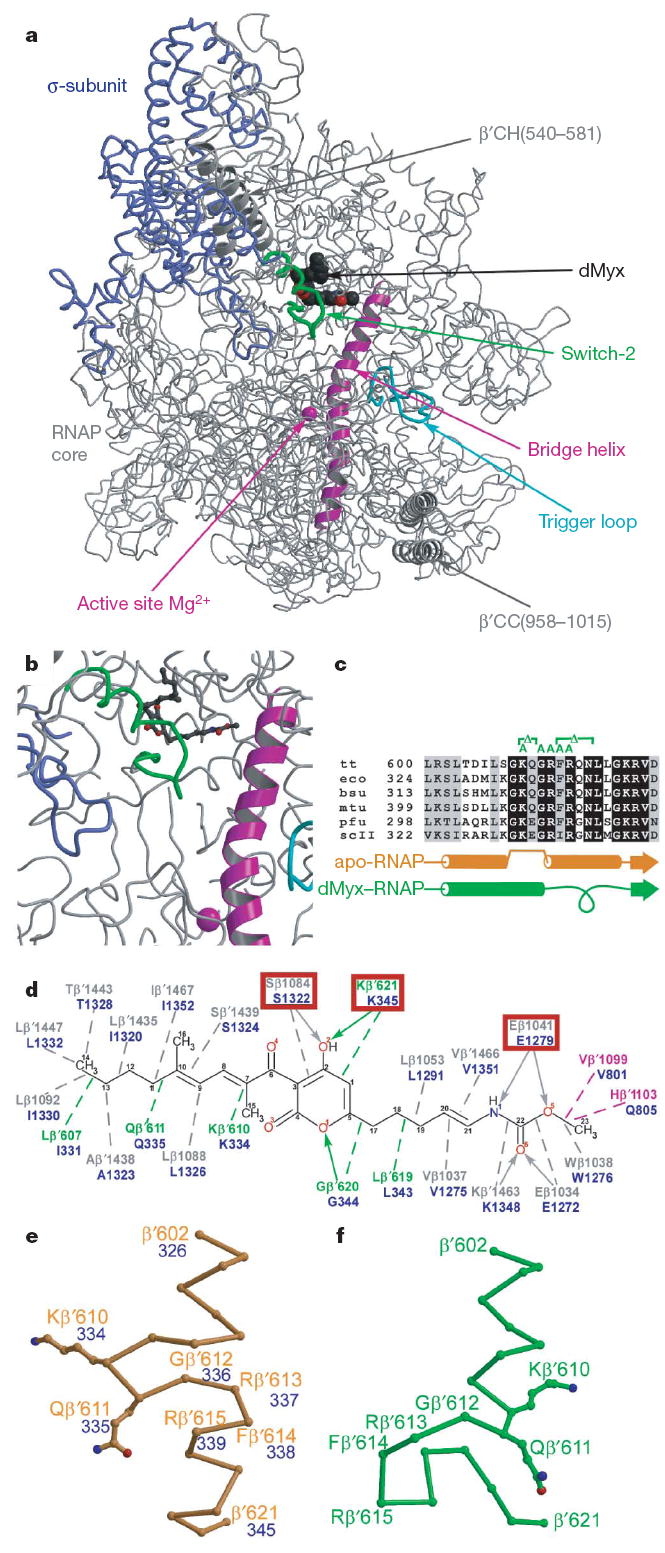
The same colour scheme is used in all figures throughout this manuscript. The σ-subunit, bridge helix, trigger loop and the remainder of the RNAP molecule are in blue, magenta, cyan and grey, respectively. The switch-2 segments in the Myx-free and Myx-bound structures are in orange and green, respectively. dMyx is in black. The Mg2+ ion is shown as magenta sphere. a, The overall view of the complex is shown. CC, coiled–coil; CH, clamp helices. b, Close-up view of the dMyx binding site. c, Sequence alignment of the switch-2 segment from bacterial (bsu, Bacillus subtilis; eco, E. coli; mtu, Mycobacterium tuberculosis; tt, T. thermophilus), archaeal (pfu, Pyrococcus furiosis) and yeast Saccharomyces cerevisiae pol II (scII) enzymes. Substitutions constructed in this work are shown above the sequence in green. d, Schematic drawing of the protein–dMyx interactions. The polar and van der Waals interactions are shown as solid arrows and dashed lines, respectively. The mutated residues are indicated by the red boxes. e, f, Conformations of the switch-2 segment in the Myx-free (e) and Myx-bound (f) holo-RNAP structures. In the panels d and e the E. coli residue numbers are shown in blue.
dMyx binds in the pocket deep inside the RNAP clamp head domain (Fig. 1a, b), which constitutes the wall of the main channel opposite the catalytic centre and forms crucial contacts with the DNA template strand in the EC9,10. Although hydrophobic contacts probably have a dominant role in binding, most of the dMyx polar groups also form specific interactions with the protein (Fig. 1d and Supplementary Fig. 5). The most important and notable change observed in the presence of dMyx is refolding of the highly conserved β’switch-2 segment (β’602–621; Fig. 1c): the α-helix, interrupted in the middle by four flipped-out residues, is straightened, whereas its carboxy-terminal portion (~two-helical turns) unwinds and refolds into a loop (Fig. 1e, f). This loop extends towards the active site, where it approaches the σ hairpin loop11 (σ317–333; Fig. 1a, b).
To verify the dMyx-binding determinants revealed by the structure and to probe the role of switch-2 refolding in the dMyx mechanism, we performed in vitro mutational analysis of E. coli RNAP (Supplementary Information). As anticipated, substitutions of three residues (Ser β1322, Glu β1279 and Lys β’345, numbered as in E. coli β’; Fig. 1d) making crucial interactions with dMyx conferred resistance to the antibiotic (Fig. 2 and Supplementary Fig. 6). On the other hand, after the switch-2 refolding Lys 334 forms only weak van der Waals interactions with dMyx whereas Arg 337 and Arg 339 do not interact with the inhibitor at all (Fig. 1d). Consistently, substitutions of these residues for Ala do not substantially affect inhibition by dMyx (Fig. 2). To design switch-2 variants with altered refolding properties, we used the following considerations. First, we selected residues without essential direct contacts with dMyx. Second, deletion of two flipped out residues, Lys 334 and Gln 335 (the integration of which into an α-helix probably initiates refolding; Fig. 1e), would prevent both opening of the inhibitor-binding site and formation of the C-terminal loop. Third, in the structures without dMyx, Phe 338 is integrated into the hydrophobic core that probably stabilizes the original switch-2 conformation (Supplementary Fig. 7). The Phe/Ala substitution, as well as a deletion of 338–341 residues, would weaken these interactions, thereby presumably favouring refolding. Fourth, although in both conformations Gly 336 is located at the junction between the α-helical and unfolded portions, its main chain angles (φ, ψ) appear in the disallowed and favourable (for the amino acids with side chains) regions of the Ramachadran plot for the original and refolded configurations, respectively, suggesting that its substitution for Ala would favour the refolded conformation. In support of structural considerations, the Δ334–335 was resistant, whereas the Δ338–341, F338A and G336A variants were hypersensitive to dMyx (Fig. 2).
Figure 2. Effect of RNAP mutations on dMyx activity.
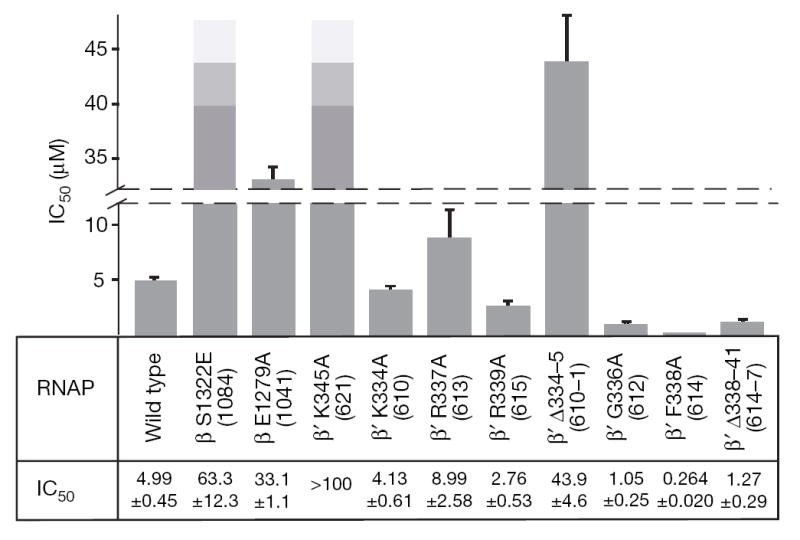
The half-maximal inhibitory concentration (IC50) values were measured in vitro with purified RNAP variants (see Methods). The data is for all variants tested in this study. The IC50 could not be determined for the highly resistant β’ K345A variant. The T. thermophilus residue numbers are shown in brackets. Errors are standard deviations of the best fit estimates for IC50 and were calculated by nonlinear regression of the RNA synthesis measurements versus dMyx concentration assuming a hyperbolic dependence.
Switch-2 refolding may be pivotal for the dMyx action. Refolding opens the entry to the otherwise inaccessible dMyx-binding site (Supplementary Fig. 8). Also, Arg β’610 and Gln β’611 are flipped out of the helix in the original switch-2 configuration and form hydrogen bonds with the DNA template in the elongation complex (and presumably in the initiation complex, where they may be crucial for stability of the transcription bubble) but lose these contacts on refolding (Fig. 1e, f). This change may inhibit DNA melting beyond the register −3. Furthermore, the newly formed C-terminal loop would clash with the DNA template strand if melting propagates to register +1 (Supplementary Fig. 9). This clash can hardly be avoided: although the upstream DNA (registers −2 to −10, and so on) may show relatively large deviations between the initiation and elongation complexes, the position of the acceptor template (i + 1) is strongly restrained by base pairing with the incoming substrate and thus is probably identical in both states.
To complement the lack of structural information on the DNA conformation in the dMyx-inhibited complex, we tested this model using biochemical approaches. The model suggests that the properly positioned template strand would preclude refolding and thus Myx binding; indeed, dMyx failed to inhibit transcription if added to the preformed open promoter complex (Supplementary Fig. 1b). We then tested the effect of dMyx on RNAP–DNA contacts and the DNA strand separation in λPR promoter complexes using DNaseI and KMnO4 footprinting, respectively. Consistent with published data12-14, the non-template strand thymidine residues at positions −4, −3 and +2 were hypersensitive to KMnO4 modification in the absence of dMyx (Fig. 3a). In the presence of inhibitor, the +2 position became strongly protected. DNaseI probing showed that dMyx induced a loss of protection (4–5 base pairs (bp)) at the downstream footprint boundary (on both DNA strands; Fig. 3a and data not shown). Similar patterns were observed in complexes trapped at intermediate steps of open complex formation7,14,15.
Figure 3. A mechanism of the dMyx action.
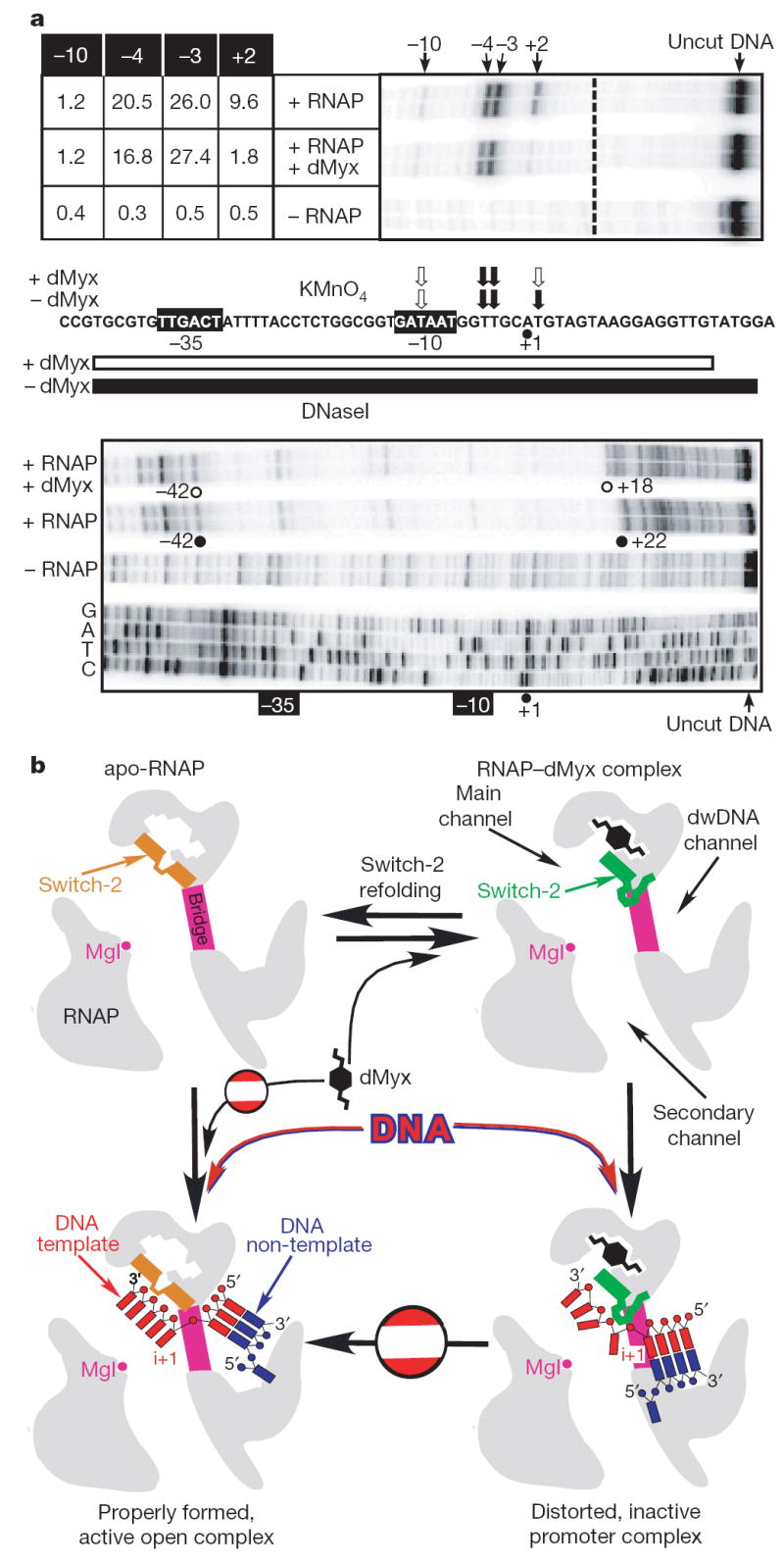
a, Myx alters the contacts between RNAP and λPR promoter DNA. a, A linear DNA fragment encompassing positions −81 to +70 of the λPR promoter was generated by polymerase chain reaction (PCR); the non-template DNA strand was end-labelled with [32P]-γATP (see Methods). The sequence from −44 to +23 is shown. The −35 and −10 hexamers are indicated by black boxes, the start site (+1) is shown by a black dot. The top panel shows probing of the non-template strand by piperidine-induced cleavage of the permanganate-modified T residues. Reactivities of −10, −4, −3 and +3 residues (quantification described in Methods) are shown to the left of the gel and summarized above the promoter sequence where black and white arrows indicate high and low reactivity, respectively. The bottom panel shows protection of the non-template DNA strand from DNaseI digestion. The footprint boundaries in the promoter region shown are indicated on the gel and by black (RNAP alone) and white (RNAP with the inhibitor) bars below the promoter sequence; the dideoxy-sequencing ladder is shown for reference. In the gels shown, independent reaction repeats were analysed for consistency. b, Schematic drawing of the putative mechanism of the dMyx action. dwDNA, downstream DNA.
Our structural data did not show any considerable antibiotic-dependent alterations of the RNAP structure that may affect DNA loading into the main channel at the upstream (−10) promoter region. Consistently, our footprinting analysis demonstrates that dMyx does not prevent RNAP binding to promoter, nucleation of melting at approximately −11, or entry of the double-stranded downstream DNA into the enzyme. The antibiotic imposes a block to DNA melting only beyond register −3, in which the direct interactions with switch-2 are predicted by modelling (assuming that the DNA trajectory is not markedly changed between the initiation and elongation complexes). Moreover, dMyx inhibited transcription on the artificially melted promoters (Supplementary Fig. 10); thus, it precludes the correct loading of the template DNA into the main channel even after complete strand separation. Overall, our present data favour a mechanism (Fig. 3b) in which local dMyx-stabilized refolding of switch-2 disrupts potentially critical interactions with and/or sterically occludes accommodation of the melted template DNA near the transcription start site, resulting in misplaced downstream DNA and inhibition of the strand separation. Further allosteric effects (for example, on the clamp opening/closing) of dMyx binding cannot be ruled out, but our study fails to demonstrate any indications of their importance in antibiotic action. A more detailed understanding of the mechanism awaits the high resolution structures of the RNAP open complex with and without dMyx. Discovery of the dMyx-binding determinants and the mechanism of its action would guide rational design of more potent dMyx derivatives (Supplementary Information).
Our results, together with conformational transitions in switch-2 observed in eukaryotic RNAP16,17, suggest that this region is inherently flexible and may influence the open complex formation in the absence of inhibitors. To test this hypothesis, we used KMnO4 footprinting analysis of the λPR promoter complexes formed by switch-2 variants with altered refolding properties (Fig. 4). In contrast to the wild-type RNAP, which formed a stable complex with the melted region extended to the +2 position, the two mutants lacking the Phe 338 side chain (that probably stabilizes the original switch-2 configuration) demonstrated prominent melting defects. The Δ338–341 enzyme alone behaves as the wild-type RNAP in the presence of dMyx: DNA melting at +2 is blocked, DNaseI footprint is shortened, and the complex is heparin sensitive (Supplementary Fig. 11). In F338A RNAP, the pattern of permanganate sensitivity is shifted further upstream: the +2 position is not melted and the −10 position becomes unprotected. Notably, addition of dMyx shifted all complexes into the same state (Fig. 4). Taken together our data indicate that changes in the switch-2 designed to promote its refolding stabilize promoter intermediates in different states from the properly formed open complex, but are active on addition of substrates. Judging by the similarity of KMnO4 footprints it is tempting to suggest that these states structurally resemble initiation intermediates of wild-type RNAP trapped by altering reaction conditions7,12,14.
Figure 4. Mutations in switch-2 affect the open complex formation.
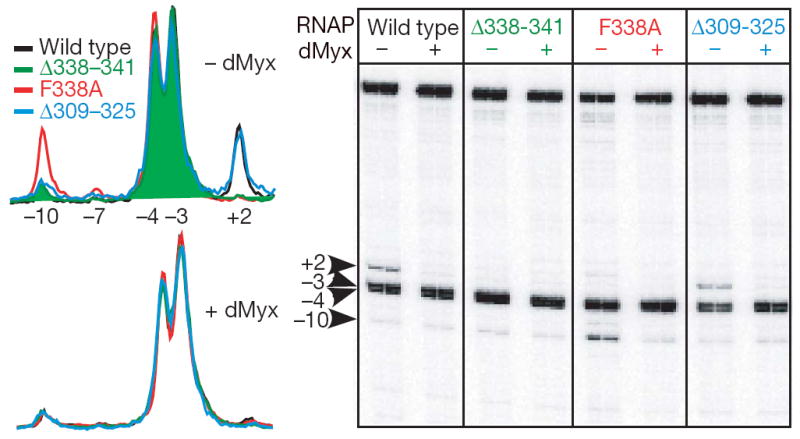
Accessibility of the non-template DNA (blue) strand residues to permanganate modification probed as in Fig. 3a. Wild-type and mutant RNAPs differ in their patterns of reactivity in the absence of dMyx (top traces) but are nearly identical in the presence of 10 μM dMyx (bottom traces). Notably, β’Δ309–325 that removes the entire rudder loop (which is inserted in the same helix as switch-2, but is unlikely to interfere with the nucleic acids) has no effect on DNA melting, suggesting that a melting defect of a different rudder deletion19 might be due to changes in the adjacent switch-2 instead.
Although several arguments favour a hypothesis that the switch-2 state observed in the presence of dMyx resembles a ‘physiological’ (dMyx-independent) intermediate as opposed to an antibiotic-induced dead-end complex (Supplementary Information), we cannot definitively prove this notion. Thus, the detailed understanding of the effects of the substitutions used in this work requires further structural and biochemical characterization. However, our present findings support a mechanism of open complex formation that involves sequential bending of the promoter DNA upstream and downstream of the RNAP main channel as the pivotal transitions. The upstream bend is thought to originate from the σ-subunit–DNA contacts and induces upstream DNA melting ~60Å away from the catalytic centre7,11,13. In contrast, the subsequent step is probably σ-independent and is set near the active site, in which interactions of DNA with the core enzyme introduce a sharp kink in the template strand9,10 that may facilitate opening of the DNA duplex at the downstream edge, thereby finalizing the opening of the transcription bubble. Although the mechanisms of initiation of DNA melting are vastly different between bacterial and eukaryotic enzymes, the final (core-mediated) step of the DNA melting and the template strand loading into the active site is presumably fundamentally conserved, to give rise to essentially identical ‘final’ states observed in the structures of the active transcription complexes.
METHODS SUMMARY
dMyx was synthesized as described previously4. Crystallization of the complex was carried out under essentially the same conditions as for the apo-RNAP holoenzyme18. The structure was refined to a final Rfactor/Rfree of 0.240/0.270 at 2.7Å resolution (Supplementary Fig. 2 and Supplementary Table 1). Escherichia coli rpoB and rpoC mutant vectors were constructed by side-directed mutagenesis and purified as described previously18. Footprinting analysis was performed using standard protocols6,12. See Methods for detailed experimental procedures.
Supplementary Material
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Acknowledgments
We thank T. Townes for critical reading of the manuscript and R. Saecker for many stimulating discussions. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research under contract No. W-31-109-Eng-38. This work was supported by National Institutes of Health grants to I.A. and D.G.V.
References
- 1.Brueckner F, Cramer P. Structural basis of transcription inhibition by α-amanitin and implications for RNA polymerase II translocation. Nature Struct Mol Biol. 2008;15:811–818. doi: 10.1038/nsmb.1458. [DOI] [PubMed] [Google Scholar]
- 2.Vassylyev DG, et al. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007;448:163–168. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 3.Irschik H, Gerth K, Hofle G, Kohl W, Reichenbach H. The myxopyronins, new inhibitors of bacterial RNA synthesis from Myxococcus fulvus (Myxobacterales) J Antibiot (Tokyo) 1983;36:1651–1658. doi: 10.7164/antibiotics.36.1651. [DOI] [PubMed] [Google Scholar]
- 4.Lira R, et al. Syntheses of novel myxopyronin B analogs as potential inhibitors of bacterial RNA polymerase. Bioorg Med Chem Lett. 2007;17:6797–6800. doi: 10.1016/j.bmcl.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Paget MS, Helmann JD. The σ70 family of sigma factors. Genome Biol. 2003;4:203. doi: 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artsimovitch I, Kahmeyer-Gabbe M, Howe MM. Distortion in the spacer region of Pm during activation of middle transcription of phage Mu. Proc Natl Acad Sci USA. 1996;93:9408–9413. doi: 10.1073/pnas.93.18.9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YF, Helmann JD. DNA-melting at the Bacillus subtilis flagellin promoter nucleates near −10 and expands unidirectionally. J Mol Biol. 1997;267:47–59. doi: 10.1006/jmbi.1996.0853. [DOI] [PubMed] [Google Scholar]
- 8.Li XY, McClure WR. Stimulation of open complex formation by nicks and apurinic sites suggests a role for nucleation of DNA melting in Escherichia coli promoter function. J Biol Chem. 1998;273:23558–23566. doi: 10.1074/jbc.273.36.23558. [DOI] [PubMed] [Google Scholar]
- 9.Kettenberger H, Armache KJ, Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 10.Vassylyev DG, Vassylyeva MN, Perederina A, Tahirov TH, Artsimovitch I. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007;448:157–162. doi: 10.1038/nature05932. [DOI] [PubMed] [Google Scholar]
- 11.Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- 12.Craig ML, et al. DNA footprints of the two kinetically significant intermediates in formation of an RNA polymerase-promoter open complex: evidence that interactions with start site and downstream DNA induce sequential conformational changes in polymerase and DNA. J Mol Biol. 1998;283:741–756. doi: 10.1006/jmbi.1998.2129. [DOI] [PubMed] [Google Scholar]
- 13.Davis CA, Bingman CA, Landick R, Record MT, Jr, Saecker RM. Real-time footprinting of DNA in the first kinetically significant intermediate in open complex formation by Escherichia coli RNA polymerase. Proc Natl Acad Sci USA. 2007;104:7833–7838. doi: 10.1073/pnas.0609888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suh W-C, Ross W, Record MT., Jr Two open complexes and a requirement for Mg2+ to open the λ PR transcription start site. Science. 1993;259:358–361. doi: 10.1126/science.8420002. [DOI] [PubMed] [Google Scholar]
- 15.Severinov K, Darst SA. A mutant RNA polymerase that forms unusual open promoter complexes. Proc Natl Acad Sci USA. 1997;94:13481–13486. doi: 10.1073/pnas.94.25.13481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 ångstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 17.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 Å resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- 18.Belogurov GA, et al. Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol Cell. 2007;26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuznedelov K, Korzheva N, Mustaev A, Severinov K. Structure-based analysis of RNA polymerase function: the largest subunit’s rudder contributes critically to elongation complex stability and is not involved in the maintenance of RNA–DNA hybrid length. EMBO J. 2002;21:1369–1378. doi: 10.1093/emboj/21.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.