

Abstract
Elucidation of the high-resolution structures of folding intermediates is a necessary but difficult step toward the ultimate understanding of the mechanism of protein folding. Here, using hydrogen exchange-directed protein engineering, we populated the folding intermediate of the T. thermophilus ribonuclease H, which forms before the rate-limiting transition state, by removing the unfolded regions of the intermediate, including an α-helix and two β-strands (51 folded residues). Using multi-dimensional NMR, we solved the structure of this intermediate mimic to an atomic resolution (backbone rmsd 0.51 Å). It has a native-like backbone topology and shows some local deviations from the native structure, revealing that the structure of the folded region of an early folding intermediate can be as well defined as the native structure. The topological parameters calculated from the structures of the intermediate mimic and the native state predict that the intermediate should fold on a millisecond time scale or less and form much faster than the native state. Other factors that may lead to the slow folding of the native state and the accumulation of the intermediate before the rate-limiting transition state are also discussed.
A major question in protein folding has been how protein molecules find their native states in a vast possible conformation space on a biologically meaningful time scale.1 One hypothesis suggests that they solve the conformation search problem by folding through partially unfolded intermediates.2 Indeed, a large number of proteins have been reported to have partially unfolded intermediates on their folding pathways.3–6 However, the key features of the folding intermediates and their exact roles in protein folding are still not fully understood. For example, does the folded region of a folding intermediate have a specific structure or comprise an ensemble of very different structures?7–9 Is the folding intermediate native-like or non-native-like? Why do some proteins populate early folding intermediates before the rate-limiting transition states, whereas the intermediates of others exist after the rate-limiting transition states?10 These issues are difficult to resolve without high-resolution structural information on the folding intermediates.
The main obstacle to obtaining high-resolution structural information of folding intermediates is that protein folding is a fast process and folding intermediates can only populate transiently during folding. The available methods for protein structure determination, such as X-ray crystallography and multi-dimensional NMR, are not directly applicable. In addition, partially unfolded intermediates do not populate significantly under equilibrium native conditions because the native state has the lowest free energy. Moreover, partially unfolded intermediates at high protein concentration (~mM), which are required for their structure determination by the current solution NMR methods, tend to aggregate due to the exposure of hydrophobic side chains.
Despite such difficulties, some successes toward determining the structures of folding intermediates have been achieved by using peptide models to represent the folding intermediates associated with the formation of disulfide bonds11 and by populating partially unfolded states at low pH and equilibrium conditions.12 In these cases, secondary structures for the intermediates have been obtained; information for their tertiary structures, however, is still lacking. More recently, the high-resolution 13, structures of the folding intermediates of a redesigned apocyt b562 (Rd-apocyt b562) 13, 14 and T4 lysozyme,15 which exist after the rate-limiting steps, and the early folding intermediate of engrailed homeodomain (En-HD),16 which exists before the rate-limiting step, have been determined. In these three cases, the structures of the folding intermediates were first characterized using hydrogen exchange and mutation studies at the level of residues and subsequently protein engineering was used to populate the intermediates by deleting or mutating the residues in the unfolded regions of the intermediates.17,18–20 A common feature of the structures of these intermediates is that they have native-like backbone topology with local non-native side chain interactions.
Although the high-resolution structure of the early folding intermediate of En-HD has provided significant insights on the early folding events,16 En-HD is small (61 amino acids) and its native state can form on sub-millisecond time scale as the intermediate does, making it difficult to reveal the cause for the population of the intermediate. By contrast, many larger proteins (> 100 amino acids) have sub-millisecond early folding intermediates and fold to the native state on the time scale of seconds. Therefore, a detailed study of the structural features and roles of the intermediates of these proteins may help to reveal the general principles that govern protein folding. One of the well-studied proteins with early folding intermediates is ribonuclease H (RNase H). For the past decade, Marqusee and coworkers have characterized the folding behavior of several homologues of RNase H, including those from Escherichia Coli,21, Thermus thermophilus,22 and HIV-1.23 Several experimental results indicate that these proteins fold similarly with on-pathway early intermediates although the folded regions in the intermediates of these proteins are not identical: (i) the formation of a significant amount of secondary structure was detected in the dead time of the stopped-flow circular dichroism experiments;22,24 (ii) the structures of the intermediates were characterized in the kinetic hydrogen/deuterium exchange pulse-labeling experiments;22,24 (iii) two partially unfolded intermediates were detected in the native-state hydrogen exchange experiments and one of them was similar to that identified in the pulse-labeling experiment for both E. coli and T. Thermophilus RNase H;21,25 (iv) the kinetic intermediate identified in the hydrogen/deuterium exchange method was confirmed by a protein engineering study for E. Coli RNase H;26 (v) a discrete on-pathway intermediate with properties similar to the kinetic intermediate was identified in a single-molecule experiment also for E. coli RNase H.27
Previously, Marqusee and coworkers used hydrogen-exchange-directed protein engineering to populate the folding intermediate of E. Coli RNase H by deleting the regions that are believed to be unfolded in the intermediate.28 This intermediate mimic forms a dimer (Kd = 0.5 μM) at high protein concentrations. Here, we used the similar protein engineering approach to populate the folding intermediate of the cysteine-free T. thermophilus RNase H (RNase H*), which was used in the earlier folding studies.22 This intermediate involves more folded regions than the intermediate of E. Coli RNase H. We found that the intermediate mimic is highly soluble and exists as a monomer at ~1 mM concentration, which allows us to use multi-dimensional NMR to solve its structure to a high resolution, and to address the issues concerning the early intermediates.
Results
Identification of the unfolded region of the early folding intermediate
The native structure of T. Thermophilus RNase H* is composed of five α-helices (A to E) and five β-strands (I to V) (Fig. 1a). In the earlier pulse-labeling experiment, seven amide protons in strand II are fully labeled in the early intermediate, indicating that strand II is unfolded in the intermediate.22 There are two amide protons in strand III that can be used as probes in the pulse-labeling experiment; both are labeled. No amide protons in the E-helix can be used as probes to monitor its structure in the intermediate because they exchange too fast in the native state. Nevertheless, it is reasonable to conclude that strand III and the E-helix are unfolded in the intermediates since strand III is stabilized by forming hydrogen bonds with strand II and the E-helix mainly packs against strand II. By contrast, several amide protons in helices A and D and strand IV are fully protected in the intermediate, indicating they are folded in the intermediate. Strand I has one amide proton as probe, which is partially protected. Strand V also has one amide proton as probe, which is fully protected. These results indicate that strands I and V cannot be fully unfolded in the intermediate. The amide protons in helices B and C also exchange too fast in the native state to provide probes for characterizing their structures in the intermediate. However, an earlier protein engineering study on the folding of E. Coli RNase H indicates that they are likely folded in the intermediate.26
Fig. 1.
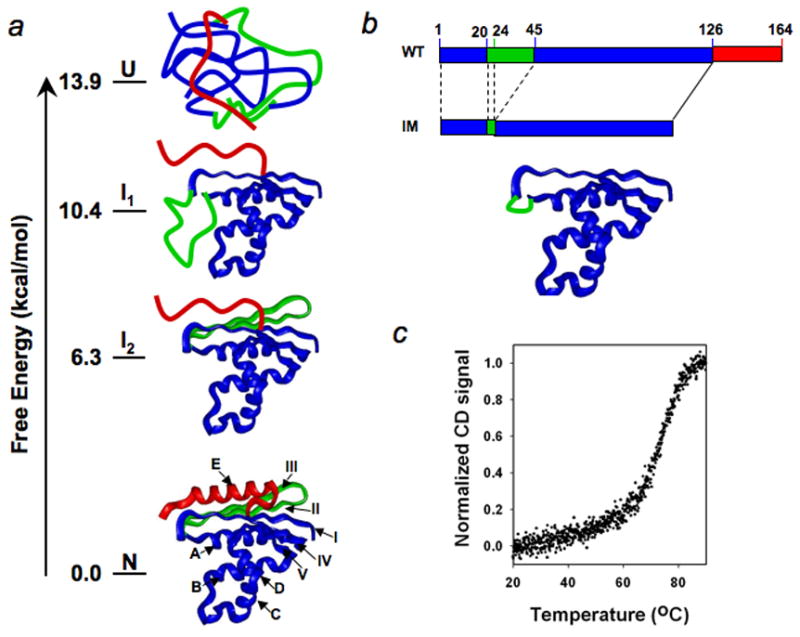
Illustration of the hydrogen exchange-directed protein engineering approach for populating the partially unfolded intermediate. (a), Unfolded, intermediate, and native states and their free energies revealed by the native-state hydrogen exchange method. The ribbon structures are made using Insight II (Accelrys). The pdb code is 1RIL. (b), Populating the folded region of the intermediate mimic by protein engineering. c, Melting curve of the intermediate mimic.
The above conclusions are further supported by the native-state hydrogen exchange experiment, in which two partially unfolded intermediates were identified.25 One intermediate has the E-helix unfolded. The other intermediate has the E-helix and strands II and III unfolded, and is similar to the kinetic intermediate identified in the pulse-labeling experiment (Fig. 1a). In addition, some amide protons in strands I and V are more protected than the amide protons in strands II and III and have m-values of global unfolding, again suggesting that strands I and V could not be fully unfolded in the intermediates.
Population of the kinetic folding intermediate by protein engineering and its characterization
To investigate the structure of the kinetic folding intermediate of T. thermophilus RNase H* at atomic resolution, we used protein engineering to remove the unfolded regions in the kinetic intermediate (I1, Fig. 1a,b), including strands II and III (24–44) and the E-helix (127–164), which allows the folded region of the intermediate to be populated. The heat denaturation experiment showed that this intermediate mimic unfolds cooperatively as temperature increases, with a melting temperature (Tm) of ~75°C (Fig. 1c). The amide 1H-15N hetero-nuclear single quantum correlation (HSQC) spectrum at ~1 mM protein concentration (22°C, 50 mM NaAc, pH 5.2) shows that the cross peaks are well dispersed and can be assigned readily using standard 3D experiments with [15N, 13C]-labeled protein.
The intermediate mimic is a monomer
To obtain the high-resolution structure of the intermediate, it is essential that the intermediate mimic exist as a monomer at the condition (~ 1 mM protein concentration) under which the structure is determined. To examine whether the intermediate mimic exists as a monomer, we performed the gel filtration experiment; the intermediate mimic had an elution volume for a globular protein with a molecular weight of ~13 kDa (Fig. 2a). We also measured the longitudinal and transverse relaxation times (T1 and T2) of backbone 15N (Fig. 2b). The averaged values are ~0.55 s for T1 and ~75 ms for T2. A value of 7.3 for the ratio of T1/T2 leads to a total correlation of time of 9.6 ns, as anticipated for a monomeric state of a globular protein with a molecular weight of ~13 kDa29. Moreover, analytical ultracentrifugation experiments unequivocally showed that a single species with a molecular weight of 12.8 ± 0.4 kDa exists in the range of 7–200 μM (Fig. 2c). Finally, the cross-peaks in the 1H-15N HSQC spectra at 1 mM and 200 μM protein concentrations remain unchanged (Fig. 2d).
Fig. 2.
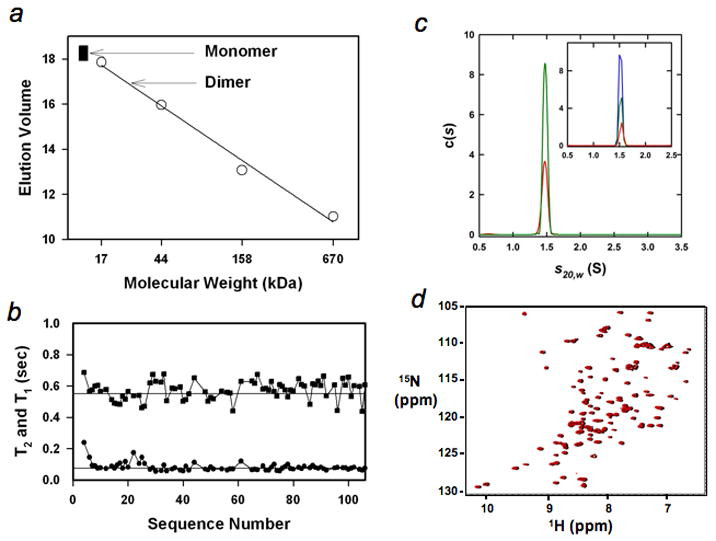
The intermediate mimic is a monomer. (a), Gel filtration (Sephadex G-75) of the intermediate mimic (filled squares, repeated three times at protein concentrations from ~10 μM to ~100 μM) and standards (circles), including horse myoglobin (17 kDa), chicken ovalbumin (44 kDa), bovine γ-globulin (158 kDa), and bovine thyroglobulin (670 kDa). The arrows indicate the anticipated monomeric and dimeric positions. (b), Longitudinal and transverse relaxation times, T1 (square) and T2 (circle). The straight lines illustrate the values of 0.55 s and 75 ms respectively. (c), c(s) distributions based on sedimentation velocity data collected at 60 krpm, 300 nm and 20.0°C are shown for loading concentrations of 100 μM (red) and 200 μM (green). The inset shows the c(s) distributions for sedimentation velocity data collected at 60 krpm, 285 nm, 20.0°C and loading concentrations of 7.1 μM (red), 14.3 μM (green) and 28.6 μM (blue). (d), Overlay of the 1H-15N HSQC spectra of the intermediate mimic at 1 mM (black) and 200 μM (red). The peaks in black were slightly shifted toward right to help to observe them.
NMR structure of the intermediate mimic
The structure of the intermediate mimic was determined using standard multidimensional NMR methods (see Methods), and the statistical parameters of the structure are shown in Table 1. The intermediate mimic has a well-defined structure with rmsd values of 0.51 Å for backbone atoms and 0.84 Å for all heavy atoms. Fig. 3a shows the overlay of Cα atoms of 10 calculated NMR structures. Fig. 3b and c compare the ribbon structures of the native state and the intermediate mimic.
Table 1.
Statistical parameters for the 20 NMR structures of the T. Thermophilic RNase H* intermediate mimic.
| NMR distance and dihedral constraints | |||
| Total NOE | 2499 | ||
| Intra residues NOE | 744 | ||
| Sequential NOE (|i-j|=1) | 602 | ||
| Short-range NOE (|i-j|≤4) | 572 | ||
| Long-range NOE (|i-j|≥5) | 581 | ||
| H-bonds | 63 | ||
| Dihedral angles | 126 | ||
| Structure statistics | |||
| Violations (mean ± s.d.) | |||
| NOE (all) | 0.048 ± 0.002 | ||
| Dihedral angle (°) | 0.35 ± 0.03 | ||
| Maximum distance restraint violations (Å) | 0.3 | ||
| Maximum angle restraint violations (°) | 3.0 | ||
| Deviations from idealized geometry | |||
| Bonds (Å) | 0.0057 ± 0.0005 | ||
| Angles (°) | 0.71 ± 0.04 | ||
| Impropers (°) | 0.55 ± 0.04 | ||
| Average pairwise r.m.s. deviation (Å) | |||
| Backbone atoms | 0.51 | ||
| All heavy atoms | 0.84 | ||
| Ramachandran map statistics (%) | |||
| Most favored | Allowed | Generally allowed | Disallowed |
| 72.8 ± 1.0 | 21.2 ± 1.2 | 4.9 ± 0.5 | 1.1 ± 0.0 |
Fig. 3.
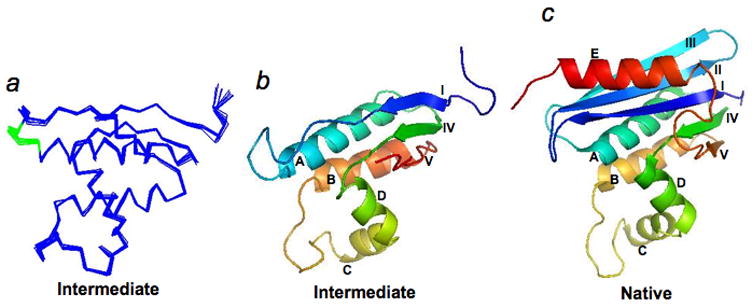
High-resolution NMR structure of the intermediate mimic. (a), Overlay of the structures of the intermediate mimic on their Cα atoms. (b), Ribbon structure of the intermediate mimic. Secondary structures are labeled as in Fig. 1. (c), Ribbon structure of the native state.
Kinetic folding of the intermediate mimic
The kinetic folding intermediate of T. Thermophilus RNase H* folds rapidly (<12 ms) from the unfolded state.22 To determine whether the intermediate mimic also forms quickly, we performed stopped-flow fluorescence experiments in urea solution (25°C, 20 mM NaOAc, 50 mM KCl, pH 5.5) (there are five Trp residues in the hydrophobic core of the folded structure of the intermediate mimic) (Fig. 4a) and equilibrium unfolding experiment (Fig. 4b). We detected significant loss of fluorescence in the dead time (8 ms) of the experiments without observable folding kinetics under folding conditions (< 3M urea, Fig. 4a).
Fig. 4.
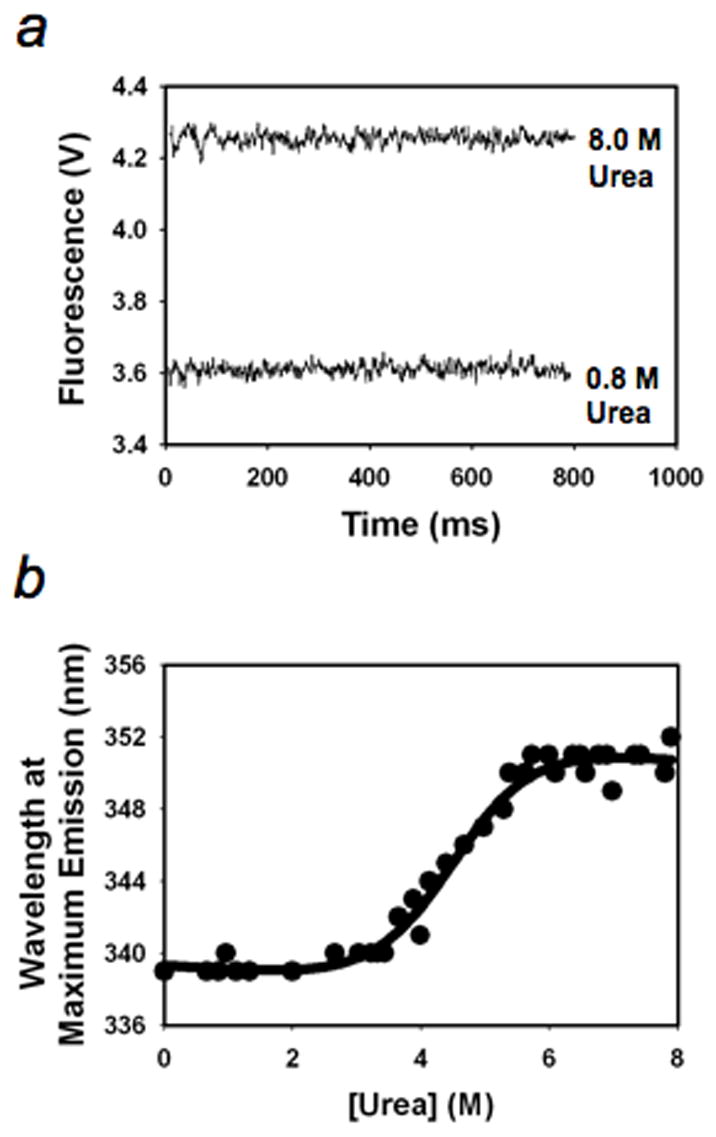
Kinetic folding and equilibrium unfolding of the intermediate mimic. (a), Kinetic traces. (b), Equilibrium unfolding.
Stability test of the intermediate mimic
To test whether the engineered intermediate adequately mimic the folded region of the true kinetic intermediate, we compared the unfolding free energy of the intermediate determined by the burst phase CD signals 25 with that of the intermediate mimic determined by the equilibrium unfolding. (Both experiments were performed in H2O and urea solutions and analyzed with a two-state model.) Equilibrium denaturation of the intermediate mimic by urea showed a cooperative unfolding (Fig. 4b). Fitting of the data to a two-state unfolding model yields an unfolding equilibrium constant of 7.1 × 10−4 and an unfolding free energy of 4.3 ± 1.0 kcal/mol, which is in excellent agreement with the value, 4.5 ± 0.9 kcal/mol, derived from burst phase signals, suggesting that the deleted regions are indeed fully unfolded in the intermediate.
Discussion
Early folding intermediates can have well-defined structures
The high-resolution structure of the intermediate mimic of T. thermophilus RNase H* shows that it has a well-defined structure with a native-like backbone topology and some local structure deviations from the native state. The folded region in the intermediate is as well-defined as the native structure; it is not an ensemble of very different structures or a molten globule or a non-specifically collapsed form. The major features of the structure are very similar to those of the intermediates of Rd-apocyt b562 and T4 lysozyme. Thus, proteins may fold through intermediates with specific structures, regardless they exist before or after the rate-limiting transition states.
Clearly, the backbones are highly defined; the structure of the intermediate mimic shows a native-like backbone topology with small local structural deviations from the native state. Similarly, most of the side chains are close to the native conformations. However, a fine comparison of the side chain conformations between the intermediate mimic and the native state is not possible since the resolution of the native structure is low (2.8 Å, pdb:1RIL).
Topology of the intermediate can account for its fast folding
One question concerning the folding behavior of T. thermophilus RNase H is why the intermediate folds so quickly. Earlier interpretations for the fast folding of early intermediates involve molten globule or hydrophobic collapse models in which intermediates fold fast because they are lack of specific tertiary structures whose formation is believed to be intrinsically slow. Since the folded region of the intermediate of T. Thermophilic RNase H* has a well-folded structure and behaves like a small single domain protein, we tested whether its topology can account for the fast folding.30, 31 We found that all of the calculated topological parameters of the intermediate mimic, including contact order,31 long-distance order,32 and total contact distance,33 predict that it should fold in less than 2 ms (see Table 2), suggesting that the topology of the intermediate can indeed explain the fast folding of the intermediate.
Table 2.
Predicted folding times by topology (in unit of millisecond).
| Proteins | Number of residues | CO | LRO | TCD |
|---|---|---|---|---|
| Intermediate mimic | 106 | 0.1 | 2.0 | 0.1 |
| T. thermophilus RNase Ha | 146 | 0.5 | 48 | 3.0 |
| Helix E + Strands II and IIIb | 51 | 86 | 11 | 1.8 |
In the crystal structure of T. thermophilus RNase H, the c-terminal region from 147 to 164 is not observed, presumably unfolded.
The fragments include residues 17-45 (strands II and III) and 126-147 (helix E) of T. Thermophilus RNase H. CO: contact order; LRO: long range order; TCD: total contact distance. The folding times were calculated using an online program (http://sparks.informatics.iupui.edu/index.php? pageLoc=Services). To calculate the topology of the fragments, including Helix E and strands II and III, the two segments are linked together through a pseudo-linker in the order of strands II, III, and helix E since unfolded loops have only small effect on protein folding rates.54,55
Possible factors contributing to the slow folding of the native state
Another question concerning the folding behavior of T. Thermophilus RNase H* is why the native sate folds so slowly (~1s), which leads to the accumulation of the intermediate before the rate-limiting transition state. To see whether topology also plays a role in the slow folding of the native state, we again used topology parameters to predict the folding times of the native state and the region (helix E and strands II and III) that folds after the formation of the intermediate. The predicted folding times for both structures are consistently much longer than those for the formation of the intermediate (Table 2), suggesting that the topology of the structures could also play a role in the slow folding of the native state and the accumulation of the intermediate. This result is further supported by an earlier computer simulation study of the folding of E. Coli RNase H in which Clementi et al.34 identified an intermediate whose structure is similar to the intermediate characterized by the amide hydrogen pulse-labeling experiment and concluded that the existence of intermediate is due to the topological complexity of the protein structure.
Although the topology of the native structure may play a role for the accumulation of the intermediate before the rate-limiting transition state, it is insufficient to account for the folding time of the native state quantitatively; the predicted folding time (< 50 ms) of the native state based on topology is much shorter than the observed folding time (~1 s), suggesting that additional factors may also contribute to the slow folding from the intermediate to the native state.
One possible factor for the slow folding of the native state is misfolding, which might occur in the folding process from the intermediate to the native state. For example, an initial barrier hypothesis for protein folding suggests that early folding intermediates may populate before the rate-limiting step when blocked by later barriers caused by non-obligatory mis-folded reorganization events.10 Indeed, significant non-native-like conformations have been found in the folded regions of the early folding intermediates of β-Lactoglobulin35 and IM7.36 Although the folded region of the intermediate of T. Thermophilus RNase H* is largely native-like, it is still possible that the unfolded region of the intermediate may misfold and create a misfolding barrier in later folding events. Determination of the structures of the second intermediate (I2, Fig. 1a) and the rate-limiting transition state in future studies are needed to examine such a possibility.
Alternatively, the large size of T. Thermophilus RNase H* (164 amino acids) may play a role in the slow folding of the native state. For example, the possible effect of chain length on protein folding rates has been suggested by several studies37–44 and seems to be an important factor for interpreting the folding rates of large proteins, which often have detectable folding intermediates in kinetic folding experiments. Indeed, good correlations between the folding rates and the topology of the native structures modified by the number of residues or chain length of proteins have been reported, which included E. coli RNase H.48
In general, a protein with a size of more than 150 amino acids may include two or more domains. In cases when two of the domains have very different topology, it is likely that a folding intermediate may populate during the process of folding since the domain with simple topology is likely to fold faster than the domain with complex topology. The exact folding rate of the native state may be affected by other factors such as chain length, misfolding, and detailed energetic interactions. These factors could be related rather than completely independent. A more complex topology or a longer chain is likely to increase the chance of misfolding.
Conclusion
The folded region of the early folding intermediate of T. thermophilus RNase H* has a well-defined structure with a native-like backbone topology and some local structural deviations from the native state rather than an ensemble of very different structures. The key features of the structure are similar to those of the folding intermediates of Rd-apocyt b562 and T4 lysozyme, which exist after the rate-limiting transition states. The specific structure of the intermediate provides evidence for the hypothesis that a protein can fold through subdomain-like intermediates in a hierarchical, stepwise manner. The topology of the intermediate of T. thermophilus RNase H* can explain its fast folding. Possible factors that might contribute to the slow folding of the native state and the accumulation of the intermediate before the rate-limiting transition state include topology, chain length, misfolding and detailed energetic interactions.
Material and Methods
Protein sample preparation
The gene of the T. thermophilus RNase H* intermediate mimic was synthesized using short DNA fragments and PCR, as described in an earlier study 45 and cloned into a pET-42b vector using Nde I and Bam HI restriction enzymes. Proteins were over-expressed in BL21(DE3) cells in LB media or in M9 media with 15NH4Cl or 15NH4Cl/13C-D-Glucose as the sole sources for nitrogen and carbon. They were purified using Ni-NTA agarose (Qiagen) and reversed-phase HPLC.
Sedimentation velocity experiments
Sedimentation velocity experiments were conducted in duplicate at 20°C on a Beckman Optima XL-I analytical ultracentrifuge in 100 mM NaOAc (pH = 5.2) using wavelengths of 285 nm (28.6, 14.3 and 7.1 μM) or 300 nm (200 and 100 μM). 150 to 180 scans were acquired at rotor speeds of 60 krpm as single absorbance measurements at 2.8 minute intervals using a radial spacing of 0.003 cm. Data were analyzed in SEDFIT11.33 46. Data analysis in SEDFIT 11.3 (http://www.analyticalultracentrifugation.com/default.htm) was implemented using solution densities ρ and viscosities η calculated using the program SEDNTERP 1.2 (http://www.jphilo.mailway.com/download.htm). The partial specific volume for the protein was also calculated in SEDNTERP (http://leonardo.fcu.um.es/macromol/programs/hydropro/hydropro.htm) and corrected to account for the 15N labeling. c(s) analyses were carried out using an s-value range of 0.02 to 4.0 with a linear resolution of 100 and a confidence level (F-ratio) of 0.68. Both c(s) and single species analyses, implemented using time independent noise corrections, were statistically indistinguishable returning similar root mean square deviation (rmsd) values of 0.0035 to 0.0068 absorbance units for the best fits. All sedimentation coefficients were corrected to s20,w. An analysis of the data in terms of a continuous c(s) distribution returned excellent fits showing the presence of a monodisperse single species (Figure 1). This was confirmed by analysis in terms of a single discrete species (Figure S1), which returned an average sedimentation coefficient of 1.51 ± 0.03 S and a molecular mass of 12.6 ± 0.4 kDa. A careful examination of these data show that the sedimentation coefficient decreases with increasing concentration (Figure S2), and extrapolation to zero concentration returns an experimental sedimentation coefficient of 1.53 S. This corresponds to an experimental molecular mass of 12.8 ± 0.4 kDa, demonstrating that the intermediate mimic of RNase H is a monomer (n = 0.99 ± 0.02) in solution.
NMR experiments
All NMR spectra were acquired at 22°C on Bruker DRX 500 and 700 MHz spectrometers equipped with a pulsed-field gradient unit and triple resonance probes. All samples were prepared at ~1 mM in 100 mM NaAc-d3 buffer (pH 5.2) with 10% (v/v) D2O or 99.9% (v/v) D2O. The backbone and aliphatic side chain signals (1H, 15N, and 13C) were assigned by using three-dimensional triple resonance through-bond scalar correlation experiments (3D CACBNH, CACBCONH, HNCA, HNCOCA, HNCO, HBHA(CO)NH, H(CCO)NH, and HNCANNH). Distance restraints were obtained from 3D 15N-, 13C-edited NOESY and 2D homo-nuclear 1H NOESY experiments with a mixing time of 110 ms.47. T1 and T2 experiments were performed as described before18. All NMR data were processed with NMRPipe48 and analyzed with NMRVIEW49. The intensities of the NOE peaks were calibrated by comparison with the averaged intensity of the NOEs between HN and Hα pairs of the same residues from 26 to 40 in a helical conformation.
Structure calculations
The NOE-derived restraints were subdivided into four classes, strong (1.8–2.7 Å), medium (1.8–3.3 Å), weak (1.8–5.0 Å), and very weak (1.8–6.0 Å). An extra 0.2 Å was added to the upper distance limit for NOE restraints in the medium- and strong-range that involved NH protons, and 0.5 Å was added to the upper distance limit for restraints involving methyl protons. Backbone dihedral angle restraints (ϕ and ψ angles) were obtained from analysis of 1Hα, HN, 13Cα, 13Cβ, 13CO, and 15N chemical shifts with TALOS.50 Structures were calculated with Xplor-NIH.51 The quality of the structures was checked and analyzed using PROCHECK_NMR.52
Kinetic and equilibrium folding/unfolding of the intermediate mimic
The stopped-flow apparatus (SFM4, Biologic) was used to perform the kinetic fluorescence experiments. The excitation wavelength is at 280 nm. The emission was collected with a filter that cut off the signals below 320 nm. The intermediate mimic was dissolved in 8.0 M urea (20 mM NaAc, pH 5.2). Refolding was initiated by diluting the protein solution in 8 M urea with buffer to various low concentrations of urea. The equilibrium unfolding experiments on the intermediate mimic were monitored by CD (J-720, Jasco) and fluorescence (SLM 8000, SIM Instruments) as the functions of temperature or urea, respectively. The wavelength used for monitoring the heat denaturation is at 222 nm. For urea denaturation experiments, the excitation wavelength is at 280 nm. The wavelength at the maximum emission was collected and plotted as the function of urea concentrations, which were fitted to a two-state unfolding model with floating parameters for pre- and post-transition baselines.53
Supplementary Material
Acknowledgments
We thank Walter Englander and Tobin Sosnick for their critical comments. This work is supported by the intramural research program of NCI and NIDDK, NIH.
Footnotes
PDB accession number
The coordinates for the NMR structures of the intermediate mimic of T. Thermophilus of RNase H* have been deposited in the PDB under ID code 2RPI.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levinthal C. How to fold graciously. Mossbaun Spectroscopy in Biological Systems Proceedings. 1969;41:22–24. [Google Scholar]
- 2.Levinthal C. Are there pathways for protein folding? J Chim Phys. 1968;65:44–45. [Google Scholar]
- 3.Baldwin RL. How does protein folding get started? Trends Biochem Sci. 1989;14:291–294. doi: 10.1016/0968-0004(89)90067-4. [DOI] [PubMed] [Google Scholar]
- 4.Ptitsyn OB, Bychkova VE, Uversky VN. Kinetic and equilibrium folding intermediates. Philos Trans R Soc Lond B Biol Sci. 1995;348:35–41. doi: 10.1098/rstb.1995.0043. [DOI] [PubMed] [Google Scholar]
- 5.Brockwell DJ, Radford SE. Intermediates: ubiquitous species on folding energy landscapes? Curr Opin Struct Biol. 2007;17:30–37. doi: 10.1016/j.sbi.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Englander SW, Mayne L, Krishna MM. Protein folding and misfolding: mechanism and principles. Q Rev Biophys. 2007;40:287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison SC, Durbin R. Is there a single pathway for folding of a polypeptide chain? Proc Natl Acad Sci U SA. 1985;82:4028–4030. doi: 10.1073/pnas.82.12.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolynes PG, Onuchic JN, Thirumalai D. Navigating the folding routes. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 9.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 10.Krantz BA, Mayne L, Rumbley J, Englander SW, Sosnick TR. Fast and slow intermediate accumulation and the initial barrier mechanism in protein folding. J Mol Biol. 2002;324:359–371. doi: 10.1016/s0022-2836(02)01029-x. [DOI] [PubMed] [Google Scholar]
- 11.Oas TG, Kim PS. A peptide model of a protein folding intermediate. Nature. 1988;336:42–48. doi: 10.1038/336042a0. [DOI] [PubMed] [Google Scholar]
- 12.Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat Struct Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 13.Feng H, Takei J, Lipsitz R, Tjandra N, Bai Y. Specific non-native hydrophobic interactions in a hidden folding intermediate: implications for protein folding. Biochemistry. 2003;42:12461–12465. doi: 10.1021/bi035561s. [DOI] [PubMed] [Google Scholar]
- 14.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci U SA. 2005;102:5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato H, Feng H, Bai Y. The folding pathway of T4 lysozyme: the high-resolution structure and folding of a hidden intermediate. J Mol Biol. 2007;365:870–880. doi: 10.1016/j.jmb.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 17.Chu R, Pei W, Takei J, Bai Y. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry. 2002;41:7998–8003. doi: 10.1021/bi025872n. [DOI] [PubMed] [Google Scholar]
- 18.Takei J, Pei W, Vu D, Bai Y. Populating partially unfolded forms by hydrogen exchange-directed protein engineering. Biochemistry. 2002;41:12308–12312. doi: 10.1021/bi026491c. [DOI] [PubMed] [Google Scholar]
- 19.Mayor U, Grossmann JG, Foster NW, Freund SM, Fersht AR. The denatured state of Engrailed Homeodomain under denaturing and native conditions. J Mol Biol. 2003;333:977–991. doi: 10.1016/j.jmb.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 20.Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SM, Alonso DO, Daggett V, Fersht AR. The complete folding pathway of a protein from anoseconds to microseconds. Nature. 2003;421:863–867. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 21.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 22.Hollien J, Marqusee S. Comparison of the folding processes of T. thermophilus and E. coli ribonucleases H. J Mol Biol. 2002;316:327–340. doi: 10.1006/jmbi.2001.5346. [DOI] [PubMed] [Google Scholar]
- 23.Kern G, Handel T, Marqusee S. Characterization of a folding intermediate from HIV-1 ribonuclease H. Protein Sci. 1998;7:2164–2174. doi: 10.1002/pro.5560071014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 25.Hollien J, Marqusee S. Structural distribution of stability in a thermophilic enzyme. Proc Natl Acad Sci U SA. 1999;96:13674–13678. doi: 10.1073/pnas.96.24.13674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: a protein engineering study. Nat Struct Biol. 1999;6:825–831. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 27.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain AK, Fischer KF, Reardon D, Handel TM, Marqusee AS. Folding of an isolated ribonuclease H core fragment. Protein Sci. 1999;8:2251–2257. doi: 10.1110/ps.8.11.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: applications to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 30.Sosnick TR, Mayne L, Englander SW. Molecular collapse: the rate-limiting step in two-state cytochrome c folding. Proteins. 1996;24:413–426. doi: 10.1002/(SICI)1097-0134(199604)24:4<413::AID-PROT1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 31.Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 32.Gromiha MM, Selvaraj S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: application of long-range order to folding rate prediction. J Mol Biol. 2001;310:27–32. doi: 10.1006/jmbi.2001.4775. [DOI] [PubMed] [Google Scholar]
- 33.Zhou H, Zhou Y. Folding rate prediction using total contact distance. Biophys J. 2002;82:458–463. doi: 10.1016/S0006-3495(02)75410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 35.Kuwata K, Shastry R, Cheng H, Hoshino M, Batt CA, Goto Y, Roder H. Structural and kinetic characterization of early folding events in beta-lactoglobulin. Nat Struct Biol. 2001;8:151–155. doi: 10.1038/84145. [DOI] [PubMed] [Google Scholar]
- 36.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: misfolding on a path to the native state. Nat Struct Biol. 2002;9:209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 37.Thirumalai D. From minimal models to real proteins: Timescales for protein folding kinetics. J Phys. 1995;5:1457–1469. [Google Scholar]
- 38.Finkelstein AV, Badretdinov AY. Rate of protein folding near the point of thermodynamic equilibrium between the coil and the most stable chain fold. Fold Des. 1997;2:115–121. doi: 10.1016/s1359-0278(97)00016-3. [DOI] [PubMed] [Google Scholar]
- 39.Koga N, Takada S. Roles of native topology and chain-length scaling in protein folding: a simulation study with a Go-like model. J Mol Biol. 2001;313:171–180. doi: 10.1006/jmbi.2001.5037. [DOI] [PubMed] [Google Scholar]
- 40.Galzitskaya OV, Garbuzynskiy SO, Ivankov DN, Finkelstein AV. Chain length is the main determinant of the folding rate for proteins with three-state folding kinetics. Proteins. 2003;51:162–166. doi: 10.1002/prot.10343. [DOI] [PubMed] [Google Scholar]
- 41.Shao H, Peng Y, Zeng ZH. A simple parameter relating sequences with folding rates of small alpha helical proteins. Protein Pept Lett. 2003;10:277–280. doi: 10.2174/0929866033478906. [DOI] [PubMed] [Google Scholar]
- 42.Kamagata K, Arai M, Kuwajima K. Unification of the folding mechanisms of non-two-state and two-state proteins. J Mol Biol. 2004;339:951–965. doi: 10.1016/j.jmb.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Naganathan AN, Munoz V. Scaling of folding times with protein size. J Am Chem Soc. 2005;127:480–481. doi: 10.1021/ja044449u. [DOI] [PubMed] [Google Scholar]
- 44.Ouyang Z, Liang J. Predicting protein folding rates from geometric contact and amino acid sequence. Protein Sci. 2008 doi: 10.1110/ps.034660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Z, Feng H, Bai Y. Detection of a hidden folding intermediate in the focal adhesion target domain: Implications for its function and folding. Proteins. 2006;65:259–265. doi: 10.1002/prot.21107. [DOI] [PubMed] [Google Scholar]
- 46.Shuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bax A, Grzechnik SK. Methodological advances in protein NMR. Acc Chem Res. 1993;26:131–138. [Google Scholar]
- 48.Delaglio F, Grzechnik SK, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multi-dimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 49.Johnson BA, Blevins RA. NMEView: A computer program for the visulization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 50.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 51.Schwieters CD, Kuszewski J, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA, Antoon J, Rullmann C, MacArthur MW, Kaptein R, Thorton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 53.Chu RA, Takei J, Barchi JJ, Jr, Bai Y. Relationship between the native-state hydrogen exchange and the folding pathways of barnase. Biochemistry. 1999;38:14119–14124. doi: 10.1021/bi991799y. [DOI] [PubMed] [Google Scholar]
- 54.Scalley-Kim M, Minard P, Baker D. Low free energy cost of very long loop insertions in proteins. Protein Sci. 2003;12:197–206. doi: 10.1110/ps.0232003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bai Y, Zhou H, Zhou Y. Critical nucleation size in the folding of small apparently two-state proteins. Protein Sci. 2004;13:1173–1181. doi: 10.1110/ps.03587604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.