

Abstract
Rsf-1 interacts with hSNF2H to form a chromatin remodeling complex that participates in several biological processes. We have previously shown that Rsf-1 gene amplification was associated with the most aggressive type of ovarian cancer and cancer cells with Rsf-1 overexpression depended on Rsf-1 to survive. In this report, we determine if formation of the Rsf-1/hSNF2H complex could be one of the mechanisms in contributing tumor cell survival and growth in ovarian carcinomas. Based on immunohistochemistry, we found that Rsf-1 and hSNF2H co-upregulated in ovarian cancer tissues. Ectopic expression of Rsf-1 in SKOV3 ovarian cancer cells with undetectable endogenous Rsf-1 expression enhanced hSNF2H protein levels and promoted SKOV3 tumor growth in a mouse xenograft model. Our studies also indicated that induction of Rsf-1 expression affected the molecular partnership of hSNF2H and translocated hSNF2H into nuclei where it co-localized with Rsf-1. Furthermore, analysis of Rsf-1 deletion mutants demonstrated that Rsf-D4 fragment contained the hSNF2H binding site based on co-immunoprecipitation and in vitro competition assay. As compared to other truncated mutants, expression of Rsf-D4 resulted in remarkable growth inhibition in ovarian cancer cells with Rsf-1 gene amplification and overexpression, but not in those without detectable Rsf-1 expression. The above findings suggest that interaction between Rsf-1 and hSNF2H may define a survival signal in those tumors overexpressing Rsf-1.
Introduction
Gene amplification represents one of the molecular genetic hallmarks in human cancer. Elucidating the molecular mechanisms of how amplified genes maintain malignant phenotypes and propel tumor progression is fundamental to understand the molecular etiology of human cancer and would have therapeutic implications. Previous genome-wide analysis using digital karyotyping (1) has identified a novel amplicon at chromosome 11q13.5 in high-grade serous carcinomas, the most common and malignant type of ovarian cancer (2). 11q13.5 amplification occurs in 13-15% of ovarian serous carcinoma based on fluorescence in situ hybridization analysis (2, 3) and the amplification is significantly associated with a shorter overall survival in patients with ovarian serous carcinoma (2). In addition to ovarian carcinoma, the 11q13.5 region is found to be amplified in other types of neoplastic diseases including breast, bladder, esophageal, and head and neck cancer (4).
Among the genes within the 11q13.5 amplicon, Rsf-1 (also known as HBXAPα) was proposed as a candidate cancer-associated gene because it showed the highest correlation between DNA copy number and RNA copy number in ovarian cancer tissues. Rsf-1 protein partners with hSNF2H (also known as SMARCA5) to form the RSF complex which belongs to the ISWI family of chromatin remodelers (5). In this complex, Rsf-1 functions as a histone chaperone to modulate DNA binding activity of RSF complex, while hSNF2H possesses nucleosome-dependent ATPase and helicase activities for DNA unwinding (5). In addition, Rsf-1 protein contains a novel PHD zinc finger domain which has been shown to participate in protein-protein interaction and transcriptional regulation (6, 7). The Rsf-1/hSNF2H complex (RSF complex) mobilizes nucleosomes to remodel the chromatin structure in response to a variety of growth signals and environmental cues. It has been shown that such nucleosomal remodeling as occurs in many SWI/SNF complexes is essential for transcriptional activation or repression (8-10) , DNA replication (11) and cell cycle progression (12). Recently, a growing body of evidence has accumulated to support a novel role of chromatin remodeling in cancer (13-19). The change in the homeostasis of Rsf-1 and hSNF2H protein networks may be related to the aggressive behavior of tumors with Rsf-1 gene amplification and overexpression.
Although alterations of chromatin structures have been linked to cancer development, the molecular mechanisms underlying how Rsf-1 gene amplification and overexpression contribute to tumor progression are largely unknown. Our previous studies have shown that higher RNA or protein levels of Rsf-1 are associated with the most aggressive type of ovarian cancer (2, 20) and a shorter overall survival in cancer patients (2, 21). Furthermore, Rsf-1 gene knockdown inhibited cell growth in ovarian cancer cells which harbor Rsf-1 amplification, but not in cell lines without Rsf-1 overexpression, suggesting an important role of Rsf-1 amplification in maintaining the survival and growth in ovarian cancer. In this study, we address if interactions between Rsf-1 and hSNF2H proteins are required for the survival and growth of cancer cells.
Materials and Methods
Tissue microarrays and immunohistochemistry
One hundred and sixty-three paraffin-embedded high-grade ovarian serous carcinoma tissues were obtained from the Department of Pathology at the Johns Hopkins Hospital. Acquisition of tissue specimens was approved by an institutional review board. Tissue microarrays (triplicate 1.5 mm cores from each specimen) were prepared to facilitate immunohistochemistry using an EnVision+System peroxidase kit (DAKO, Carpentaria, CA) with an antibody dilution of 1:1,000 for the anti-Rsf-1 antibody (Upstate, Lake Placid, NY) and 1:1,000 for the anti-hSNF2H antibody (Upstate). Immunointensity was independently scored by two investigators based on nuclear immunoreactivity and labeled as negative (0), weakly positive (1+), moderately positive (2+), strongly positive (3+) and intensely positive (4+). For discordant cases, a third investigator scored and the final intensity score was determined by the majority scores.
Inducible constructs and Rsf-1 inducible cell clones
The full-length Rsf-1 gene was tagged with a V5 epitope at the C-terminal and was then cloned into Tet-off expression vectors, pBI or pTRE-hygro (Clontech, Mountain View, CA). Parental RK3E and SKOV3 cells were transfected with a tTA (tetracycline-controlled transactivator) expression vector. The inducible Rsf-1 expression vectors were constructed and introduced into the RK3E-tTA and SKOV3-tTA cells, and the stable transfectants were selected. Transfection was performed using Lipofectamin (Invitrogen, Carlsbad, CA) according the protocol accompanied by the reagent. To determine the efficiency of Rsf-1 induction in the Tet-off system, we performed Western blots to analyze Rsf-1 protein expression at different time points after induction. The method of Western blot using the anti-Rsf-1 antibody has been previously described (2, 22).
Double immunofluorescence staining
Rsf-1 inducible RK3E cells were used to determine whether Rsf-1 and hSNF2H co-localized in the same cellular compartment. The inducible cells grown on chamber slides (Nunc, Roskilde, Denmark) were transfected with an hSNF2H expressing vector tagged with an Xpress epitope at the N-terminal. Cells were washed with Dox-containing or Dox-free medium to turn-off or turn-on the Rsf-1 gene, respectively. Forty-eight hours after transfection, cells were fixed and incubated with an anti-Xpress antibody (Invitrogen, Carlsbad, CA) to detect hSNF2H, followed by an FITC-conjugated goat anti-V5 antibody (QED Bioscience, San Diego, CA) to detect Rsf-1. The cells were then incubated with a Rhodamin-conjugated anti-mouse antibody (Jackson ImmunoResearch Lab., West Grove, PA).
Co-immunoprecipitation
A series of Rsf-1 deletion mutants (Rsf-D1 to Rsf-D10) were cloned into pcDNA6/V5 which was used to transfect a stable HEK293 cell line constitutively expressing hSNF2H tagged with an Xpress epitope at the N-terminal (Invitrogen, Carlsbad, CA). Forty-eight hours after transfection, cells were lysed and deletion mutant proteins were pulled down by anti-V5 agarose (Sigma, St. Louis, MO), and immunoblotted with an anti-Xpress antibody to detect hSNF2H protein. For the competition assay, recombinant Rsf-1 deletion mutant protein, Rsf-D4, was expressed and purified by a PET27b E.Coli purification system (Novagen, San Diego, CA). Rsf-D4 protein was added into cell lysate at different concentrations from 0 to 50 μg/ml during co-immunoprecipitation to compete with full-length Rsf-1 protein for hSNF2H binding. Co-immunoprecipitation was also performed to assess the hSNF2H binding proteins. The anti-BAZ1A Ab (clone 1F6) was purchased from Abnova (Taipei, Taiwan), and the anti-BAZ-1B (W3614) was purchased from Sigma. The cell lysate was prepared from OVCAR3 cells treated with Rsf-1 shRNA or vector control. Cell lysate was also prepared from SKOV3 cells with Rsf-1 gene turning on or turning off. The lysate was then equally aliquoted into three parts, and the immunoprecipitation was performed using anti-Rsf-1, anti-BAZ1A, anti-BAZ1B antibodies, respectively. The immunoprecipitates were immunoblotted with an anti-hSNF2H antibody.
Quantitative real-time PCR
PCR reactions were performed using an iCycler (Bio-Rad, Hercules, CA), and the threshold cycle numbers (Ct) were obtained using the iCycler Optical system interface software. Mean Ct of the gene of interest was calculated from duplicate measurements and normalized with the mean Ct of a control gene, beta-amyloid precursor gene (APP), for which expression is relatively constant among the SAGE libraries analyzed (23). In cases where no gene expression was observed, a cut-off Ct value of 45 cycles was used. Data were expressed as fold increase or decrease as compared to the gene turned-off or vector control samples. We considered a change exceeding two fold (delta Ct > 1) to be significant in this study.
Cell growth and apoptosis assays
OVCAR3 ovarian cancer cells transfected with Rsf-1 deletion mutants and empty vector were grown in 96-well plates at a density of 3,000 cells per well. To determine similar expression levels of deletion mutants, the deletion mutant constructs were cloned into the pBI vector which co-expressed GFP in transfected cells. We also generated a new OVCAR3-tTA cell clone with a high transfection efficiency using the Amaxa transfection kit (T buffer with program T-16) (Gaithersburg, MD) and this OVCAR3-tTA cells were used for cell growth and apoptosis assays. We observed a similar percentage of transfected cells with equal green fluorescence signals in all transfection groups after second day of transfection, suggesting a similar transfection efficiency for each construct. Cell number was measured 4 days after transfection based on the fluorescence intensity of SYBR green I nucleic acid staining (Molecular Probes, Eugene, OR). Cell growth on Rsf-D4 transfected OC-24 and RK3E cells was monitored daily for 4 consecutive days using the SYBR green I staining. BrdU uptake and staining were performed using a cell proliferation kit (Amersham, Buckinghamshire, UK) as previously described (2). For apoptosis assay, apoptotic cells were detected by staining with Annexin V-FITC (BioVision, Mountain View, CA). The percentage of BrdU-positive and Annexin V–positive cells was determined by counting at least 400 cells from different fields for each experiment. The data was expressed as mean ± SD from triplicates.
Tumor xenograft in nude mice
Rsf-1 inducible SKOV3 cells were injected into the subcutaneous tissue of athymic nu/nu mice (5 × 106 cells/injection) (5 mice with 10 injection sites for each group). Doxycyclin (125 μg/mouse) was intraperitoneally injected everyday to suppress Rsf-1 gene expression in the Rsf-1 turned-off control mice. For the Rsf-1 turned-on group, mice were treated with equal volume of phosphate buffer solution. Tumor volume was measured every three days and tumors were excised and weighted at day 51. Tumors from turned-on and -off groups were evaluated for Rsf-1 induction using anti-V5 antibody staining (Invitrogen, Carlsbad, CA) on paraffin-embedded tumor sections.
SAGE data analysis
Analysis of gene expression using the serial analysis of gene expression (SAGE) data has been previous described (24, 25). Briefly, ovarian SAGE libraries including the ovarian surface epithelial cells (OSE4) and ovarian cancer cell lines (MPSC1, ES2, A2780 and OVCAR3) were retrieved from the SAGE database (http://www.hlm.nih.gov/SAGE/) and previously established data (26, 27). The unigene was searched for the protein of interest, and the appropriate match was chosen based on its specificity for the protein and number of sequences. The transcript expression levels were normalized as the number of tags per 100,000 tags in each library.
Results
Co-expression of Rsf-1 and hSNF2H
hSNF2H has been shown to directly interact with Rsf-1 to form a chromatin remodeling complex, RSF (5), but the evidence to support such interaction in cancer cells has not yet been demonstrated. Here, we observed that both Rsf-1 and hSNF2H immunoreactivity was located in the nuclei of tumor cells (Fig. 1A) and showed a significant association (p = 0.048) between Rsf-1 and hNSF2H immunointensity on the same specimens based on χ2 test (Table 1). Second, we performed co-immunoprecipitation and demonstrated that Rsf-1 protein interacted with hSNF2H protein in an ovarian cancer cell line, OVCAR3, which is known to contain Rsf-1 amplification (2) (Fig. 1B). Next, we asked whether induction of Rsf-1 expression enhanced the protein level of hSNF2H. An inducible (Tet-off) Rsf-1 expression system was generated in the SKOV3 cell line which did not harbor Rsf-1 amplification or express a detectable level of endogenous Rsf-1. After induction of Rsf-1, an increased amount of both Rsf-1 and hSNF2H protein levels was found in a time-dependent manner (Fig. 1C). In contrast to the significantly increased protein level, hSNF2H mRNA level as measured at 6 hours after Rsf-1 induction increased only by 1.04 fold. In addition, ectopic expression of hSNF2H in HEK293 cells did not enhance Rsf-1 protein level (Fig. 1 D). The above findings suggest that Rsf-1 proteins may function to stabilize hSNF2H protein level in cancer cells.
Fig. 1.

Co-upregulation of Rsf-1 and hSNF2H expression in ovarian carcinoma cells. A: Immunohistochemistry using anti-Rsf-1 and anti-hSNF2H antibodies on ovarian serous carcinoma tissues. Representative tissue sections with different immunointensity (from 1+ to 4+) of Rsf-1 and hSNF2H are illustrated. For each intensity group, both sections were obtained from similar areas of the same specimen. B: Co-immunoprecipitation was performed to assess if Rsf-1 protein form a complex with hSNF2H protein in OVCAR3 cells. Protein lysate was pulled-down by an anti-Rsf-1 antibody, separated by gel electrophoresis and visualized by silver staining (lane 1). Western blotting was performed to demonstrate the major proteins in the pulled down fraction were Rsf-1 with molecular weight of ∼215 kD (black arrow) and a degradation product of ∼130 kD (gray arrow, lanes 3) and hSNF2H protein with a molecular weight of ∼146 kD (open arrow, lane 5). Protein G alone was used as the control in immunoprecipitation in lanes 2, 4, and 6. C: SKOV3 ovarian cancer line which expresses an undetectable level of endogenous Rsf-1 was engineered to express Rsf-1 controlled by a Tet-off system. Rsf-1 induction increases the hSNF2H protein level in a time-dependent fashion based on Western blotting analysis. D: Western blot analysis showed no increase in Rsf-1 protein level in HEK293 cells which was previously engineered to overexpress hSNF2H (lane 2) as compared to the parental cell control (lane 1). OVCAR3 cells served as the positive control for this assay (lane 3). GAPDH was used as the loading control.
Table 1.
hSNF2H and Rsf-1 immunointensity in high grade ovarian serous carcinomas.
| hSNF2H immunointensity |
|||||||
|---|---|---|---|---|---|---|---|
| 0 | 1+ | 2+ | 3+ | 4+ | Total | ||
| Rsf-1 immunointensity | 0 | 0 | 1 | 3 | 0 | 0 | 4 |
| 1+ | 1 | 17 | 15 | 1 | 0 | 34 | |
| 2+ | 1 | 17 | 35 | 6 | 2 | 61 | |
| 3+ | 1 | 13 | 23 | 10 | 3 | 50 | |
| 4+ | 0 | 1 | 6 | 4 | 3 | 14 | |
| Total | 3 | 49 | 82 | 21 | 8 | 163 | |
| p = 0.048 | |||||||
Induction of Rsf-1 expression promotes tumor growth in SKOV3 xenografts
As an attempt to determine whether Rsf-1 protein upregulation contributes to tumor growth, we used the SKOV3 xenograft model in nude mice. Before subcutaneous injection, we analyzed Rsf-1 mRNA level in SKOV3-Rsf-1 Tet-off cells and found that its expression level gradually increased to a level that was comparable to that in Rsf-1 amplified ovarian carcinoma tissues (Fig. 2A). As compared to Rsf-1 non-induced tumors, Rsf-1 induced tumors grew much faster and had an increased tumor weight (p< 0.01) (Fig. 2B, C and D). Immunohistochemistry of the tumors excised at the time of sacrifice showed diffuse Rsf-1 nuclear staining in tumor cells in the Rsf-1 induced but not in the non-induced group, indicating a sustained Rsf-1 expression in xenografts after Rsf-1 induction. As consistent with increased tumor sizes in Rsf-1 induced tumors, the mitotic count (mitoses/100 tumor cells) in the Rsf-1 induced tumors was 1.67±0.34 which was significantly higher than 0.87±0.27 in non-induced tumors (p< 0.05). We did not observe any significant difference between Rsf-1 induced tumors and non-induced controls regarding tumor cell morphology, blood vessel density and invasiveness into surrounding soft tissues.
Fig. 2.

Rsf-1 expression increases tumor size in SKOV3 tumor xenografts. A: After Rsf-1 induction (6 hours and 12 hours), the Rsf-1 mRNA levels in SKOV3 cells are comparable to the ovarian carcinoma tissues with Rsf-1 amplification and are significantly higher than those without Rsf-1 amplification. B: Tumor xenograft experiment was performed on athymic nu/nu mice by injecting Rsf-1 inducible SKOV3 cells subcutaneously. Induction of Rsf-1 expression in SKOV3 tumors increased tumor volumes as compared to non-induced group. C: Representative photographs show larger tumors in Rsf-1 induced (Rsf-1 on) tumors than the non-induced (Rsf-1 off) tumors. The tumor sections were stained with an anti-V5 antibody to confirm the induction of Rsf-1 proteins. Diffuse nuclear staining of Rsf-1/V5 is observed in induced but not in non-induced tumor sections. D: Tumors were excised and weighted in the end of experiment (day 51).
Rsf-1/hSNF2H complex is the predominant chromatin remodeling complex in Rsf-1 overexpressed cells
SNF2H containing complexes (belonging to the ISWI subfamily) have been reported in mammalian cells and they included RSF, CHRAC, ACF/WCRF, NoRC and WICH (28-32) . Each complex contains subunits which may regulate the specificity or catalytic activity of SNF2H. For example, RSF contains Rsf-1 and SNF2H (5); ACF/WCRF contains BAZ1A (also known as ACF1 or WCRF180) and SNF2H (29), and WICH contains BAZ1B (also known as WSTF) and SNF2H (30). As Rsf-1 is one of the putative binding partners for SNF2H, we assessed the cellular localization of both Rsf-1 and SNF2H in an Rsf-1 inducible cell line, RK3E. Immunofluorescence staining revealed that hSNF2H protein was diffusely distributed in both cytoplasm and nuclear compartments when Rsf-1 was turned off (Fig. 3A). Upon Rsf-1 induction, Rsf-1 protein, which contains several nucleus localization signal (NLS) sites, was overexpressed in the nuclei. Interestingly, hSNF2H was translocated into nuclei and co-localized with Rsf-1 (Fig. 3A).
Fig. 3.
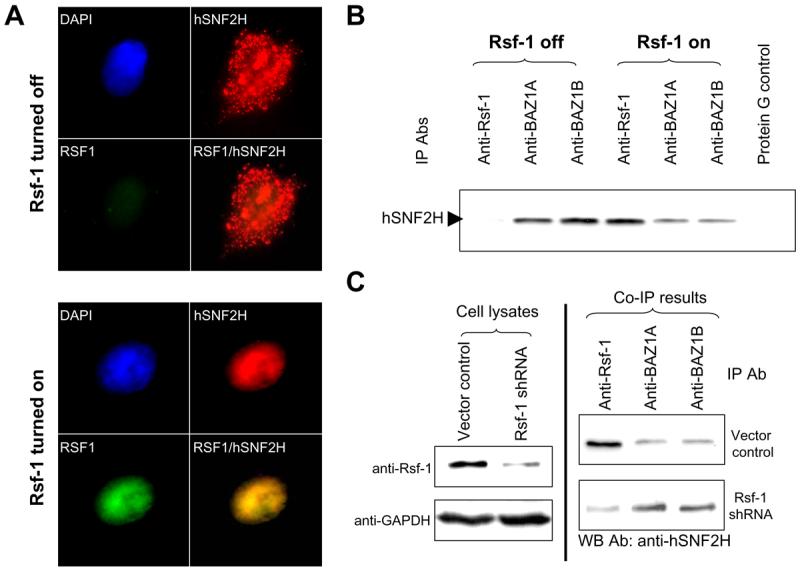
Analysis of interaction between hSNF2H and its binding partners. A: Immunofluorescence staining of Rsf-1 and hSNF2H proteins shows that RSf-1 co-localizes with hSNF2H in the nuclei. Rsf-1 inducible RK3E cells were transiently transfected with hSNF2H gene fused with an Xpress tag. Double immunofluorescence staining was performed to detect Rsf-1 (green fluorescence; anti-V5) and hSNF2H (red fluorescence; anti-Xpress) proteins, respectively. B: Immunoprecipitation was performed in SKOV3 cells stably transfected with a Tet-off inducible Rsf-1 expression construct. The protein level of hSNF2H that was co-immunoprecipitated with Rsf-1 was increased in Rsf-1 induced (Rsf-1 on) cells but its level co-immunoprecipitated with BAZ1A and BAZ1B significantly reduced as compared to the control SKOV3 cells without Rsf-1 induction (Rsf-1 off). Equal protein amount was used in immunoprecipitation for each antibody. C: Immunoprecipitation assays were performed to compare the binding capability between hSNF2H and different binding partners. Rsf-1 gene expression was knocked down by shRNA in Rsf-1 amplified OVCAR3 cells (left panel). The cell lysate of equal protein amount from Rsf-1 shRNA and control vector transfected cells was then immunoprecipitated with anti-Rsf-1, anti-BAZ1A and anti-BAZ1B antibodies (Abs), respectively (right panel). The immunoprecipitated complex was separated by SDS-PAGE and blotted with an anti-hSNF2H antibody to determine the amount of co-immunoprecipitated hSNF2H binding partners including Rsf-1, BAZ1A and BAZ1B.
To determine the gene expression of the candidate binding partners of hSNF2H in ovarian cancer cells, we analyze the serial analysis of gene expression (SAGE) database. Among those binding partners, we found that Rsf-1, BAZ1A, BAZ1B and BAZ2A were abundantly expressed in ovarian cancer cell lines including OVCAR3, A2780, ES2 and MPSC1 while their expression levels were relatively low in the OSE4 cell line which was derived from normal ovarian surface epithelium (supplemental Fig. 1). Of notice, Rsf-1 represented the major binding partner for hSNF2H expressed in OVCAR3 cells that amplified the Rsf-1 locus.
Since Rsf-1 was found to be overexpressed in tumor cells, it is possible that in those cells, excessive Rsf-1 molecules compete with other SNF2H binding partners for SNF2H binding. In order to address this possibility, we performed immunoprecipitation to determine the association of representative SNF2H binding partners including BAZ1A, BAZ1B and Rsf-1 with hSNF2H upon Rsf-1 induction in SKOV3 cells and Rsf-1 knockdown in OVCAR3 cells. BAZ2A was not analyzed here as the antibody reacting to BAZ2A was not currently available. In SKOV3 cells with Rsf-1 induction, the protein level of hSNF2H that was co-immunoprecipitated with Rsf-1 was significantly increased while its level co-immunoprecipitated with BAZ1A and BAZ1B was significantly reduced as compared to the SKOV3 cells without Rsf-1 induction (Fig. 3B). In Rsf-1 shRNA approach, downregulation of Rsf-1 significantly reduced the level of Rsf-1/hSNF2H complex and enhanced the formation of BAZ1A/hSNF2H and BAZ1B/hSNF2H complexes (Fig. 3C). The above results indicated that Rsf-1 overexpression as occurred in Rsf-1 amplified tumors “hijacked” hSNF2H from other partners to the Rsf-1 complex.
Mapping of hSNF2H binding domain on Rsf-1
We have previously shown that cell growth and survival depended on Rsf-1 expression in ovarian cancer cells with Rsf-1 gene amplification and overexpression (2). In order to determine whether the formation of Rsf-1/hSNF2H complex is required for cell survival, we applied a dominant negative approach by generating an Rsf-1 deletion mutant that competed with wild-type (full-length) Rsf-1 for hSNF2H binding. First, we generated a series of Rsf-1 deletion mutants (from Rsf-D1 to Rsf-D10) that contained different protein motifs (PHD, DDT, Glu-rich, etc.) (Fig. 4A). These fragments were cloned into expression vectors which were used to transfect HEK293 cells in order to assess their ability to co-immunoprecipitate with hSNF2H (Fig. 4B). We found that among all deletion mutants, only the Rsf-D4 fragment (a.a. 1-973) could robustly co-immunoprecipitate with hSNF2H while others did not show detectable co-immunoprecipitate (Fig. 4C). The Rsf-D4 contains DDT, Glu-rich and PHD motifs but lacks the C-terminal Rsf-1 domain (Fig. 4D). By increasing the amount of recombinant Rsf-D4 protein during co-immunoprecipitation, we observed that the amount of hSNF2H immunoprecipitate decreased in a dose-dependent fashion, indicating that Rsf-D4 competed with the endogenous full-length Rsf-1 for interacting with hSNF2H (Fig. 5A).
Fig. 4.

Mapping for the hSNF2H binding domain(s) on Rsf-1 protein. A: A series of Rsf-1 deletion mutants, Rsf-D1 to Rsf-D10, were generated and cloned into the pcDNA6/V5 vector. The locations of each mutant are shown in the parenthesis. ▲ indicates the predicted nucleus localization signal site; DEAD/DEAH box is a conserved motif for ATP-dependent helicases. Helica indicates the helicase catalytic domain. SANT domain is a conserved DNA-binding domain for SANT SWI3, ADA2, N-CoR and TFIIIB proteins. DDT is a conserved domain of DNA-binding homeobox-containing proteins; PHD is the Plant Homeodomain type zinc finger Domain; Glu indicates the glutamine-rich region and Arg indicates the arginine-rich region. B: Deletion mutants, Rsf-D1 to Rsf-D10, and pcDNA6 (V) were transfected into hSNF2H expressing HEK293 cells and their protein expression was confirmed by Western blot analysis using an anti-V5 antibody. C: Co-immunoprecipitation was performed to determine the minimal domain(s) of Rsf-1 protein that interacts with hSNF2H. The hSNF2H protein was tagged with Xpress and the Rsf-1 mutants were tagged with V5 to facilitate immunoprecipitation. The proteins pulled down by anti-V5 antibody were immunoblotted with an anti-Xpress antibody. In addition to the full-length Rsf-1 (R), Rsf-D4 is the only mutant that co-immunoprecipitated with hSNF2H (arrow head). D: Summary of different motifs on Rsf-1 deletion mutants and their hSNF2H binding capacity based on co-immunoprecipitation (I.P.).
Fig. 5.

Effects of Rsf-D4 expression on OVCAR3 ovarian cancer cells. A: In a co-immunoprecipitation assay, recombinant Rsf-D4 proteins show a does-dependent inhibition in the binding of Rsf-1 and hSNF2H. GAPDH was used as a loading control. B: Western blotting demonstrates a robust Rsf-1 protein expression in OVCAR3 cell line and a moderate expression in the A2780 cell line as compared to other ovarian carcinoma cell lines. C: The effects of deletion mutants were assessed by comparing the cell number of the OVCAR3 cells after transient transfection of Rsf-1 to Rsf-10 constructs. The cell number was measured at day 4 using SYBR green I incorporation and the data were normalized to the empty vector control. D: The cellular proliferating activity in the transfected OVCAR3 cells was determined by the percentage of cells with positive BrdU staining. E: The percentage of apoptotic cells in each treatment was determined by annexin V-FITC staining.
Effects of Rsf-D4 expression on cell growth and apoptosis in ovarian cancer cells
To determine the biological effects of Rsf-D4 on ovarian cancer cells, we expressed deletion mutants from Rsf-D1 to Rsf-D10 individually in OVCAR3 cells that expressed the highest levels of Rsf-1 and hSNF2H proteins (2) (Fig. 5B), then the cell number was determined. As shown in Fig. 5C, the most significant decrease in cell number was observed in Rsf-D4-transfected cells and to a lesser extent in Rsf-D3-transfected cells (Fig. 5C). Furthermore, expression of Rsf-D4 suppressed cellular proliferation based on BrdU uptake (Fig. 5D) and increased the percentage of annexin-V labeled cells (Fig. 5E). In contrast, Rsf-D4 expression did not lead to any significant decrease in cell number in OC24 and RK3E cells with undetectable Rsf-1 expression (p> 0.05) (supplemental Fig. 2).
Discussion
Our previous studies have demonstrated the clinical significance of Rsf-1 gene amplification and overexpression in ovarian carcinomas as increased gene copy number and expression level are associated with the most aggressive type of ovarian cancer (2, 20, 21). Furthermore, we have shown that Rsf-1 amplified tumors depend on Rsf-1 proteins to survive. In this study, we provide new evidence that induction of Rsf-1 expression is associated with hSNF2H protein expression and a higher copy number of Rsf-1/hSNF2H complexes increases tumor size in a tumor xenograft mouse model. We further demonstrate that expression of an Rsf-1 dominant negative protein that contains the hSNF2H binding motif is sufficient to suppress cell growth in cancer cells with Rsf-1 overexpression but not in cells with undetectable Rsf-1 expression levels. These findings provide new insights into the tumor-promoting functions of Rsf-1 and suggest that the formation of the RSF chromatin remodeling complex may be one of the mechanisms contributing to survival and growth in ovarian cancer cells.
Because Rsf-1 forms a chromatin remodeling complex with hSNF2H, it is likely that both proteins are co-expressed in tissues. Thus, we first asked if both Rsf-1 and hSNF2H proteins were co-upregulated. Based on immunostaining on ovarian serous carcinoma tissues, we were able to demonstrate that this was the case. To determine whether the co-upregulation is merely a co-incident event or a result of Rsf-1 overexpression, we established an Rsf-1 inducible ovarian cancer cell line, SKOV3, which showed hSNF2H expression but with undetectable Rsf-1 expression. We observed that induction of Rsf-1 expression in SKOV3 increased protein but not mRNA level of hSNF2H, suggesting that Rsf-1 stabilized the hSNF2H proteins probably through the complex formation that prevented its protein degradation. Therefore, it is likely that in Rsf-1 amplified or overexpressed carcinomas, the amount of hSNF2H also increases as a result of Rsf-1 upregulation to form the RSF chromatin remodeling complex. Besides, we compared the growth of SKOV3 xenografts in mice between Rsf-1 induction and control (non-induction) groups and found that the size of SKOV3 tumors in mice significantly increased upon Rsf-1 induction as compared to those without induction. This data highly suggests that Rsf-1 overexpression promotes tumor growth.
Based on deletion mapping, co-immunoprecipitation study and competition assay, we were able to define the hSNF2H binding domain (Rsf-D4) on the Rsf-1 molecule which contains both DDT and PHD motifs at the N-terminal region. It appears that both DDT and PHD domains are the required motifs for hSNF2H interaction as other deletion mutants containing only one of them did not show a robust co-immunoprecipitation with hSNF2H. Further structure biology studies should be helpful to disclose the precise physical interaction between DDT/PHD domains on Rsf-1 and hSNF2H proteins. The dominant negative effects of Rsf-D4 in ovarian cancer cells with Rsf-1 gene amplification and overexpression but not in cells with low or undetectable Rsf-1 expression suggest that the complex formation of full-length Rsf-1 and hSNF2H is essential for tumor growth and survival for those tumor cells that molecularly “addict” to Rsf-1 gene amplification or upregulation. Furthermore, the lack of growth suppressive effects in cells with undetectable Rsf-1 expression indicates that the Rsf-D4 effects are not likely due to a non-specific cytotoxic effect associated with the Rsf-D4 protein. Although the above represents our preferred view, alternative interpretations should be pointed out. For example, like other dominant negative approaches that are used to modulate the activity of an endogenous protein by interfering normal protein-protein interactions, the Rsf-D4 approach used in this study may not be entirely specific to interrupt the interaction between Rsf-1 and hSNF2H. It is possible that the Rsf-D4 may complex with other protein(s) besides hSNF2H and overexpression of Rsf-D4 can potentially interfere the binding of these protein(s) to full-length endogenous Rsf-1. 1
How does Rsf-1 overexpression and thereby, the increased Rsf-1/hSNF2H complex formation contribute to tumor cell survival and growth? There are at least two possibilities based on the current study. First, It is plausible that an increased number of RSF complexes may facilitate remodeling of chromatin structures or modifying the functions of oncogenes and tumor suppressors that interact with the complexes (32), thus promoting tumorigenesis in cells. Second, over-expression of Rsf-1, as occurs in ovarian carcinoma cells, can alter cellular distribution and the partnership of hSNF2H. hSNF2H is known to interact with several proteins other than Rsf-1 and the hSNF2H containing ISWI complexes have diverse cellular functions. Thus, excessive Rsf-1 molecules may sequester hSNF2H, leading to loss or decrease in the abundance of other hSNF2H-containing complexes such as hSNF2H/BAZ1A and hSNF2H/BAZ1B. Since several protein members in the SNF family have been reported as tumor suppressors and have been found downregulated or inactivated in cancer tissues (16, 18), it is possible that reduction of these hSNF2H complexes with tumor suppressor potential by excessive Rsf-1 contributes to the observed growth-stimulating effects in cancer cells. However, our findings demonstrate that expressing Rsf-D4 deletion mutant which holds avid hSNF2H binding activity can induce growth suppression in tumor cells. This observation suggests that sequestering hSNF2H from other complexes by Rsf-D4 mutant alone was not able to promote cell growth as seen in full-length Rsf-1. Therefore, the tumor promoting phenotype mediated by full-length Rsf-1 is more likely due to an increase of RSF complex formation rather than a decrease of other hSNF2H containing complexes. Further studies are needed to demonstrate the detailed mechanisms underlying how Rsf-1/hSNF2H complexes contribute to tumor development.
In conclusion, the current study attempts to explore the functional roles of a chromatin remodeling protein, Rsf-1, in promoting ovarian cancer. Our data provides evidence that the interaction of Rsf-1 and hSNF2H proteins to form the chromatin remodeling complex is essential for cell survival and growth in ovarian cancer. Our results should help understand the pathogenesis of ovarian cancer development and may have translational implications for new cancer therapy.
Supplementary Material
Acknowledgements
This study was supported by grants from NIH (CA129080), DoD (OC0400600), the Johns Hopkins-Weizmann Institute of Science Research Collaboration Fund and the Johns Hopkins-China Medical University Research Collaboration Fund (CMU95-286). The authors appreciate the technical assistance from David Chu.
References
- 1.Wang TL, Maierhofer C, Speicher MR, et al. Digital karyotyping. Proc Natl Acad Sci U S A. 2002;99:16156–61. doi: 10.1073/pnas.202610899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shih Ie M, Sheu JJ, Santillan A, et al. Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci U S A. 2005;102:14004–9. doi: 10.1073/pnas.0504195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakayama K, Nakayama N, Jinawath N, et al. Amplicon profiles in ovarian serous carcinomas. Int J Cancer. 2007;120:2613–7. doi: 10.1002/ijc.22609. [DOI] [PubMed] [Google Scholar]
- 4.Schwab M. Amplification of oncogenes in human cancer cells. Bioessays. 1998;20:473–9. doi: 10.1002/(SICI)1521-1878(199806)20:6<473::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 5.Loyola A, Huang J-Y, LeRoy G, et al. Functional Analysis of the Subunits of the Chromatin Assembly Factor RSF. Mol. Cell. Biol. 2003;23:6759–68. doi: 10.1128/MCB.23.19.6759-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shamay M, Barak O, Doitsh G, Ben-Dor I, Shaul Y. Hepatitis B virus pX interacts with HBXAP, a PHD finger protein to coactivate transcription. J Biol Chem. 2002;277:9982–8. doi: 10.1074/jbc.M111354200. [DOI] [PubMed] [Google Scholar]
- 7.Shamay M, Barak O, Shaul Y. HBXAP, a novel PHD-finger protein, possesses transcription repression activity. Genomics. 2002;79:523–9. doi: 10.1006/geno.2002.6717. [DOI] [PubMed] [Google Scholar]
- 8.Holstege FC, Jennings EG, Wyrick JJ, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–28. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 9.Vignali M, Hassan AH, Neely KE, Workman JL. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang JY, Shen BJ, Tsai WH, Lee SC. Functional interaction between nuclear matrix-associated HBXAP and NF-kappaB. Exp Cell Res. 2004;298:133–43. doi: 10.1016/j.yexcr.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Flanagan JF, Peterson CL. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res. 1999;27:2022–8. doi: 10.1093/nar/27.9.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cosma MP, Tanaka T, Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 13.Roberts CWOrkin SH. The SWI/SNF complex--chromatin and cancer. Nat Rev Cancer. 2004;4:133–42. doi: 10.1038/nrc1273. [DOI] [PubMed] [Google Scholar]
- 14.Mohrmann LVerrijzer CP. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim Biophys Acta. 2005;1681:59–73. doi: 10.1016/j.bbaexp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Wolffe AP. Chromatin remodeling: why it is important in cancer. Oncogene. 2001;20:2988–90. doi: 10.1038/sj.onc.1204322. [DOI] [PubMed] [Google Scholar]
- 16.Klochendler-Yeivin A, Muchardt C, Yaniv M. SWI/SNF chromatin remodeling and cancer. Curr Opin Genet Dev. 2002;12:73–9. doi: 10.1016/s0959-437x(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 17.Wong AK, Shanahan F, Chen Y, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000;60:6171–7. [PubMed] [Google Scholar]
- 18.Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1:500–6. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guidi CJ, Sands AT, Zambrowicz BP, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21:3598–603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao TT, Hsu C-Y, Yen MJ, et al. Expression of Rsf-1, a chromatin-remodeling gene, in ovarian and breast carcinoma. Hum Pathol. 2006;37:1169–75. doi: 10.1016/j.humpath.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Davidson B, Trope CG, Wang TL, Shih Ie M. Expression of the chromatin remodeling factor Rsf-1 is upregulated in ovarian carcinoma effusions and predicts poor survival. Gynecol Oncol. 2006;103:814–9. doi: 10.1016/j.ygyno.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 22.Kobel M, Pohl G, Schmitt WD, Hauptmann S, Wang TL, Shih Ie M. Activation of mitogen-activated protein kinase is required for migration and invasion of placental site trophoblastic tumor. Am J Pathol. 2005;167:879–85. doi: 10.1016/s0002-9440(10)62059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buckhaults P, Zhang Z, Chen YC, et al. Identifying tumor origin using a gene expression-based classification map. Cancer Res. 2003;63:4144–9. [PubMed] [Google Scholar]
- 24.Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12:827–31. doi: 10.1158/1078-0432.CCR-05-1397. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama K, Nakayama N, Davidson B, et al. A BTB/POZ protein, NAC-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0604083103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hough CD, Sherman-Baust CA, Pizer ES, et al. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Res. 2000;60:6281–7. [PubMed] [Google Scholar]
- 27.Pohl G, Ho CL, Kurman RJ, Bristow R, Wang TL, Shih Ie M. Inactivation of the mitogen-activated protein kinase pathway as a potential target-based therapy in ovarian serous tumors with KRAS or BRAF mutations. Cancer Res. 2005;65:1994–2000. doi: 10.1158/0008-5472.CAN-04-3625. [DOI] [PubMed] [Google Scholar]
- 28.Poot RA, Dellaire G, Hulsmann BB, et al. HuCHRAC, a human ISWI chromatin remodelling complex contains hACF1 and two novel histone-fold proteins. Embo J. 2000;19:3377–87. doi: 10.1093/emboj/19.13.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bochar DA, Savard J, Wang W, et al. A family of chromatin remodeling factors related to Williams syndrome transcription factor. Proc Natl Acad Sci U S A. 2000;97:1038–43. doi: 10.1073/pnas.97.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bozhenok L, Wade PA, Varga-Weisz P. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. Embo J. 2002;21:2231–41. doi: 10.1093/emboj/21.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakimi MA, Bochar DA, Schmiesing JA, et al. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature. 2002;418:994–8. doi: 10.1038/nature01024. [DOI] [PubMed] [Google Scholar]
- 32.Neely KE, Workman JL. The complexity of chromatin remodeling and its links to cancer. Biochim Biophys Acta. 2002;1603:19–29. doi: 10.1016/s0304-419x(02)00067-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.