

Summary
Hemorrhagic trauma leads to organ dysfunction, sepsis and death. There is abnormal production of proinflammatory cytokines by Kupffer cells, tissue hypoxia and liver injury following trauma-hemorrhage. The physiological conditions consequent to trauma-hemorrhage are consistent with factors necessary to initiate endoplasmic reticulum (ER) stress and unfolded protein response. However, the contribution of ER stress to apoptosis and liver injury after trauma-hemorrhage is not known. In the present study ER stress was investigated in mice that underwent trauma-hemorrhage or sham operation. Expression of endoplasmic reticulum stress proteins Bip, ATF6, PERK, IRE1α, and PDI were significantly elevated in the liver after trauma-hemorrhage compared to the controls. The ER stress associated proapoptotic transcription factor CHOP protein expression was also significantly elevated in trauma-hemorrhage group. Consistent with this, enhanced DNA fragmentation was observed, confirming apoptosis, in the liver following trauma-hemorrhage. These results demonstrate the initiation of ER stress and its role in apoptosis and liver injury, subsequent to hemorrhagic trauma.
Keywords: ER stress, trauma, hemorrhage, shock, hypoxia, apoptosis
Introduction
Endoplasmic reticulum (ER) is the first compartment of the protein secretory pathway as this is the site of folding and post-translational modification (N-linked glycosylation, disulphide bond formation and oligomerization) of newly synthesized proteins [1-3]. These processes are facilitated by optimum levels of ATP, Ca2+ and an oxidizing environment together with very sophisticated cellular machinery that senses the protein requirement[1,4]. Perturbations in the metabolic and energy balance causes an accumulation of unfolded or misfolded proteins in the ER leading to ER stress and initiates an unfolded protein response (UPR)[1-4]. At least three ER transmembrane receptors are important in the initial signaling of ER stress to the cell by UPR. These proteins are: activating transcription factor 6 (ATF-6), pancreatic ER kinase (PKR)-like kinase (PERK) and inositol-requiring enzyme 1 (IRE1)[4]. Among the two isoforms of IRE1, IRE1α and IRE1β, the former is ubiquitously expressed where as the latter, IRE1β, is mostly localized in the gut [5]. Bip (immunoglobulin heavy chain binding protein), an ER chaperone, associates with the intraluminal regions of ATF6, PERK and IRE1 in their inactive state and upon ER stress it is dissociated followed by their activation leading to UPR. UPR is a protective cellular strategy to restore the normal functioning of the ER, however in the event that UPR cannot be controlled, the persistent ER stress may lead to cellular dysfunction, death and disease [1,6].
Trauma-hemorrhage induces hypoxia, cellular apoptosis and organ dysfunction [7-14]. Previous extensive studies by our group and others have clearly demonstrated hepatic, pulmonary and cardiac dysfunction following hemorrhagic trauma [7-9,11,13-17]. The Kupffer cells have been shown to produce excessive amounts of proinflammatory cytokines and significant damage to hepatocytes have been observed as indicated by elevated levels of α-GST in the plasma, after hemorrhagic trauma [7,10,14,15,18]. Hypoxia, glucose deprivation and proinflammatory cytokines, hallmarks of post-trauma-hemorrhage condition, can initiate ER stress [19]. So far there had been no studies reporting the role of hepatic ER stress in organ dysfunction following trauma-hemorrhage. In this paper we investigated the activation of ER stress in the liver of mice, 24 hours after hemorrhage and resuscitation.
Materials and Methods
Animals
Male C3H/HeN mice 8 to 12 weeks old and weighing 19 to 25 g were obtained from Charles River Laboratories (Wilmington, MA). The mice were allowed to acclimatize in the animal facility for one week prior to the experiments. All animal experiments were carried out in accordance with the protocol approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and were consistent with the guidelines of the National Institutes of Health.
Trauma-Hemorrhage Procedure
This procedure was performed as described earlier [20,21]. The mice were fasted for 16 hours prior to the procedure but allowed water ad libitum. They were anesthetized with isoflurane (Minrad, Bethlehem, PA) and restrained in a supine position. A 2-cm midline laparotomy was performed, which was closed asceptically in 2 layers with sutures (Ethilon 6/0, Ethicon, Somerville, NJ). Both femoral arteries and the right femoral vein were asceptically cannulated with polyethylene-10 tubing (Becton Dickinson, Sparks, MD). Blood pressure was measured via one of the arterial lines using a blood pressure analyzer (Micro-Med, Louisville, KY). Upon awakening, animals were bled rapidly through the other arterial catheter to a mean arterial blood pressure of 35 ± 5 mm Hg within 10 minutes, and was maintained for 90 minutes. At the end of that period, mice were resuscitated with four times the shed blood volume in the form of lactated Ringer's solution over 30 min. Lidocane was applied to the incision sites, catheters removed and incisions were closed with sutures. The same surgical procedures were conducted on sham-operated animals, but they did not undergo hemorrhage or resuscitation.
Tissue Harvesting
The animals were anesthetized with isoflurane at 24 hours after sham operation or resuscitation in the trauma-hemorrhage group, and liver removed, frozen in liquid nitrogen, and stored at -80°C.
Plasma Levels of α-gluthathione S-transferase (GST) and TNF-α
Plasma was obtained 2 hours after the trauma-hemorrhage or sham procedure. Elevated plasma levels of α-GST is an indicator for hepatic damage [22]. The plasma α-GST level was measured by a sandwich ELISA as per the protocol provided by the manufacturer (Biotrin International, Mount Merrion, Co. Dublin, Ireland). Briefly, samples were incubated in microwells coated with anti-[alpha]-GST IgG for 1 h at room temperature on a shaker, followed by incubation with anti-α-GST IgG conjugated to horseradish peroxidase, and finally with the substrate for 15 min at room temperature. The optical density was read at 450 nm and sample concentrations were calculated. TNF-α concentration in the plasma, was determined with cytometric bead arrays using flow cytometry according to the manufacturer's instructions (BD Pharmingen, San Diego, Calif). Briefly, capture beads were incubated with 50-μL samples for 1 h at 25°C, followed by phycoerythrin detection reagent and analyzed using the LSRII flow cytometer (BD Biosciences, Mountain View, Calif).
Western Blots
Protein expression of Bip, ATF6, PERK, phosphorylated PERK (PERK-p), PDI, IRE-1α and CHOP (C/EBP homologus protein, also known as GADD153) were analyzed by Western Blot. Briefly, total proteins in liver tissue lysates were resolved using 4-12% Nupage gel (Invitrogen, Carlsbad, CA) and transferred to PVDF membranes. The membranes were saturated with blocking buffer (10 mM Tris, 150 mM NaCl, and 0.05% Tween-20 supplemented with 5% dry milk) for 1 h at room temperature and incubated with the respective primary antibodies. The primary antibodies used were: Bip, IRE-1α (Cell Singling Technology, Beverly, CA), CHOP, PERK and PERK-p (Santa Cruz Technology, Santa Cruz, CA) and ATF-6 and β-actin (Abcam, Inc, Cambridge, MA). The antibody to ATF6 recognized both the full length protein (85 kD) and the cleaved N-terminal fragment (50 kD). The membranes were then washed five times with TBST (Tris-buffered saline supplemented with 0.05% Tween-20) followed by incubation with an appropriate secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) conjugated with horseradish peroxidase for 1 h at room temperature. The membranes were again washed five times with TBST and probed using ECL (Amersham, Piscataway, NJ), and autoradiographed. Rabbit polyclonal β-actin antibody was used to detect β-actin as the loading control. The bands on autoradiographs were quantified using ChemiImager 5500 imaging software (Alpha Innotech Corp., San Leandro, CA). In order to assess early activation of proapoptotic protein CHOP, additional Western blots were performed using liver tissues obtained from sham and trauma-hemorrhage mice at 2 hours after the procedure.
Apoptosis
Apoptosis was measured by quantifying the DNA fragmentation in the liver tissue. An ELISA kit, Cell Death detection by Roche Diagnostics (Indianapolis, IN), that provide a quantitative determination of histone-associated DNA fragments was used. The assay used monoclonal antibodies directed against both DNA and histones. Briefly, frozen liver tissues were sonicated and lysed using the lysing buffer provided by the manufacturer, pelleted and the supernatant was used for the assay [23]. The assay was performed as per the manufacturer's protocol.
Apoptosis was further ascertained by TUNEL assay (R&D systems, Minneapolis, MN). TUNEL assay was performed on cryosections of liver tissues harvested from sham and trauma-hemorrhage animals, 24 hours after the procedure. Briefly, cryosections were fixed used 4% formaldehyde, washed permeabilized using proteinase K. The permeabilized sections were TdT labeled and treated with Strep-Fluor solution for detection using fluorescence microscopy as cells containing fragmented nuclear chromatin characteristic of apoptosis will exhibit fluorescent staining after labeling. The slides were observed under florescent microscope using 495 nm filter.
Statistics
Statistical calculations were carried out using GraphPad Prism software (San Diego, CA). Mann-Whitney U test was used to calculated significance.
Results
Expression of ER stress proteins following trauma-hemorrhage
BIP is a heat shock protein and an ER chaperone, that is present in the lumen of the ER and remains bound to the intra-luminal regions of ATF6, PERK and IRE1 and its release from these transmembrane proteins initiates the signaling of ER stress and UPR. Bip protein expression was significantly (p<0.05) increased in the liver of mice following trauma hemorrhage as compared to the sham operated mice (Fig. 2A). Consistent with the increase in Bip, there was a significant increase in ATF6 expression after trauma-hemorrhage (Fig. 2B). Activated ATF6 migrate to Golgi and is cleaved by zinc metalloprotenases, site-1 and site-2 proteases, in the Golgi membrane, so that a 50 kD cytosolic fragment of the ATF6 translocates to nucleus and initiates UPR gene transcription. As shown in Figure 2B, the 50 kD fragment of ATF6 was clearly visible in the liver of trauma-hemorrhage mice as compared to sham.
Fig. 2.
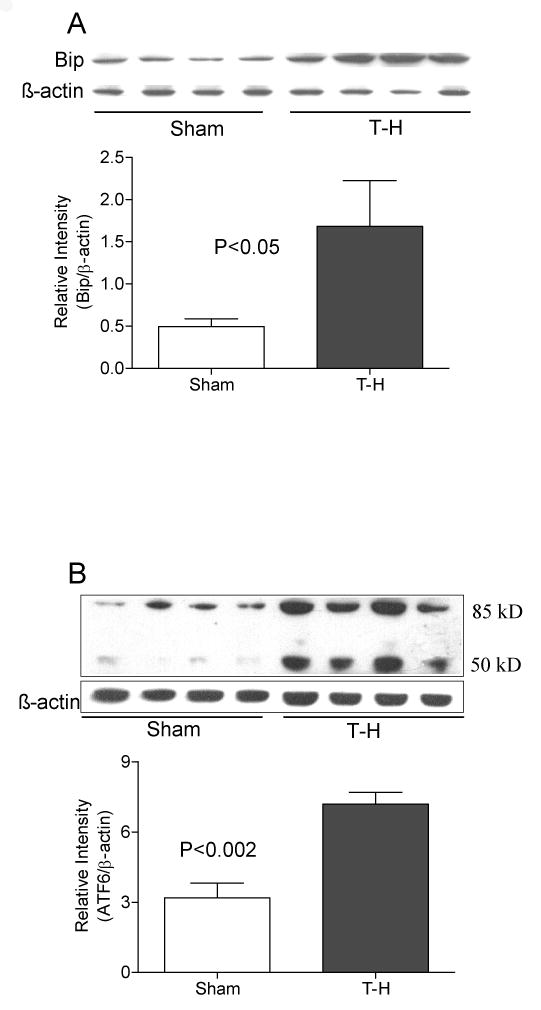
Increased expression of Bip and ATF6 following trauma-hemorrhage (T-H). Mice were subjected to trauma-hemorrhage (T-H, n=8) and resuscitation as described in the methods section, liver isolated, homogenized and total proteins in the homogenates were tested by Western blot. Sham (Sham, n=8) operated mice served as controls. Protein expression of Bip (panel A) and ATF6 (panel B) was found to be increased in mice subjected to T-H compared to sham operated mice. The 50 kD cleaved fragment of ATF6 is visible in lanes of samples from trauma-hemorrhage animals (panel B). Gel picture shows results of four sham and four T-H animals and the bar diagram shows the mean±SEM of the density of these bands (8 sham and 8 T-H animals). β-actin served as loading control. The bar diagram in panel B represents the protein expression of full length ATF6.
Similar increase in the levels was observed for other ER transmembrane molecules PERK and IRE1α upon ER stress (Fig.3 and 4). We examined both phosphorylated PERK and total PERK protein (Fig.3A and B) and found both of them to be significantly increased after trauma-hemorrhage. Another protein critical in ER stress is the enzyme PDI, as disulphide bond formation is an essential step in the proper folding of newly synthesized proteins in the oxidative environment of ER. As shown in Fig. 5, PDI was also significantly increased following trauma-hemorrhage.
Fig. 3.
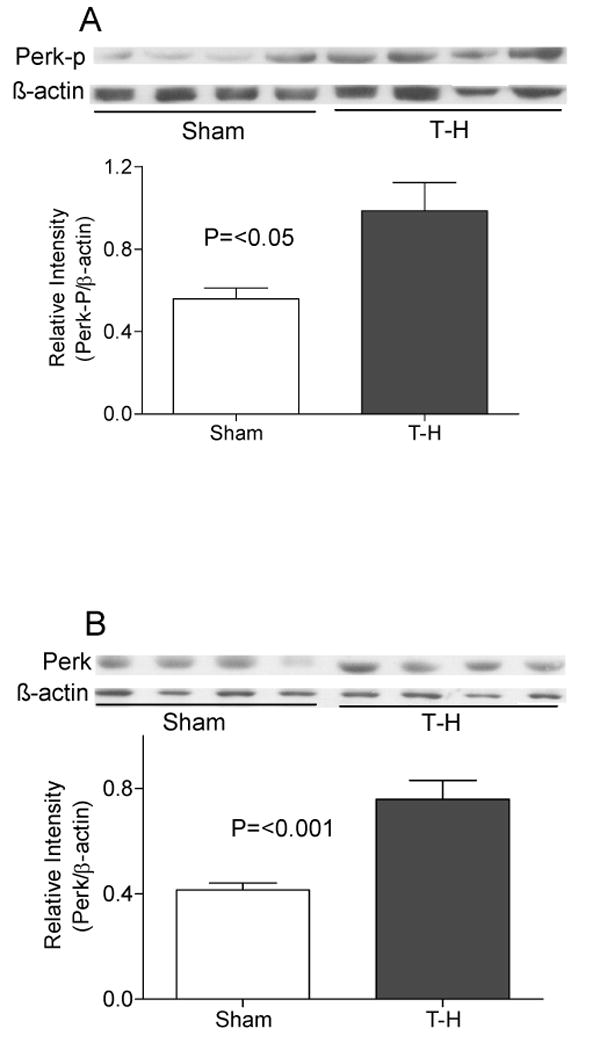
PERK expression and phosphorylation following trauma-hemorrhage (T-H). Protein expression of phosphorylated PERK (panel A) as well as well as total PERK protein (panel B) were found to be increased in the liver of mice subjected to T-H compared to sham operated mice. Gel picture shows results of samples from four sham and four T-H animals and the bar diagram shows the mean±SEM of the density of these bands (8 sham and 8 T-H animals). β-actin served as loading control.
Fig. 4.
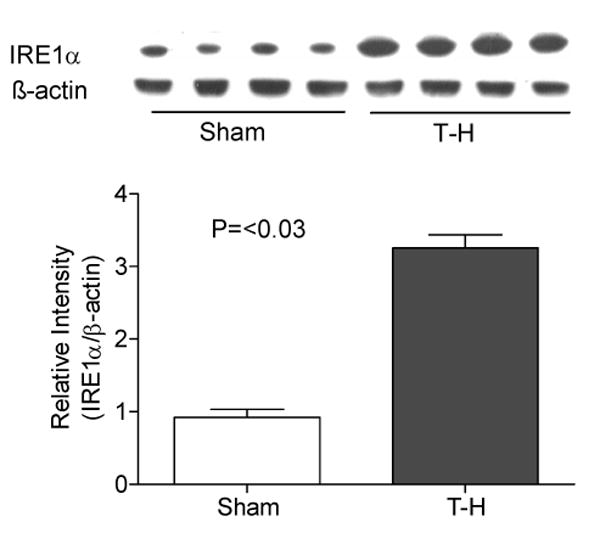
Elevated IRE1α protein expression following trauma-hemorrhage (T-H). Protein expression of IRE1α was significantly increased in the liver of mice subjected to T-H compared to sham operated mice. Gel picture shows results of four sham and four T-H animals and the bar diagram shows the mean±SEM of the density of these bands. β-actin served as loading control.
Fig. 5.
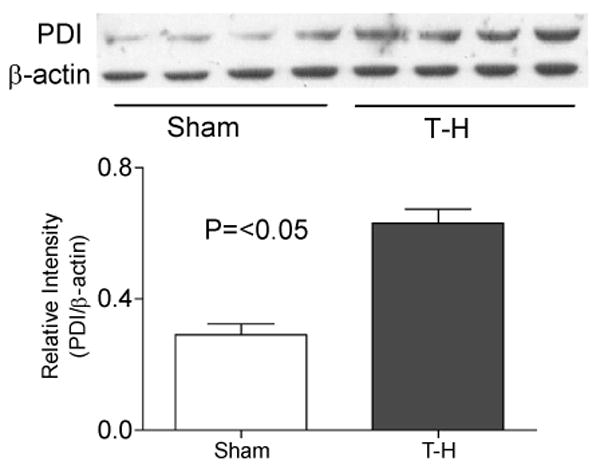
Elevated PDI expression following trauma-hemorrhage (T-H). Protein expression of protein disulphide isomerase (PDI) was significantly increased in the liver of mice subjected to T-H compared to sham operated mice. Gel picture shows results of four sham and four T-H animals and the bar diagram shows the mean±SEM of 8 sham and 8 T-H animals. β-actin served as loading control.
Induction of CHOP and apoptosis following trauma-hemorrhage
As all the tested UPR related genes were consistently upregulated following trauma-hemorrhage, we examined the protein expression of CHOP, a transcription factor that induces apoptosis in mice subjected to trauma-hemorrhage or sham operation. There was a significant increase in CHOP expression in the liver of mice that under-went trauma hemorrhage as compared to the sham-operation (Fig. 6A). We further tested apoptosis in the liver of both trauma-hemorrhage and sham groups. Consistent with the increased expression of CHOP we also found increased apoptosis in trauma-hemorrhage group compared to that in the sham-group (Fig. 6B). Cryosections of liver obtained from sham and trauma-hemorrhage mice, 24 hours after the procedure, were stained by TdT labeling for fragmented nuclear chromatin (TUNEL assay) and we confirmed significant apoptosis observed in the liver from trauma-hemorrhage animals as compared to the sham (Figure 6C and D).
Fig. 6.
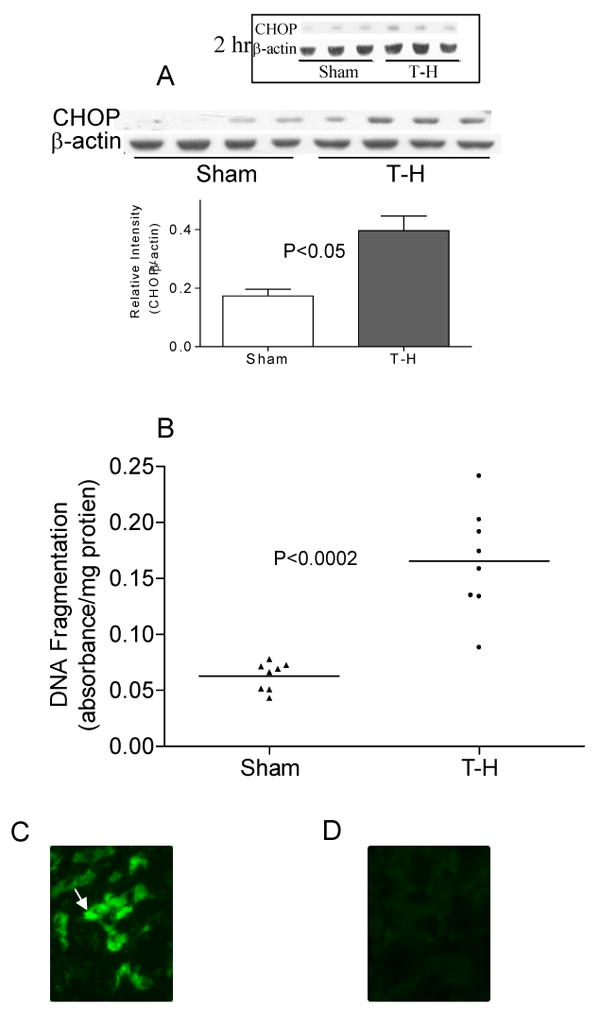
CHOP expression and apoptosis following trauma-hemorrhage. (A) CHOP protein expression was quantified in mouse liver total protein extracts using Western blot. α-actin served as loading control. Gel picture shows results of four sham and four T-H animals and the bar diagram shows the mean±SEM of sham and T-H groups. (B) Apoptosis in the liver following trauma-hemorrhage was quantified by ELISA as described. A significantly increased apoptosis was observed in T-H (n=8) group as compared to sham (n=8) group. Mean of observations is represented as a horizontal bar in each group. (C) and (D) TUNEL staining demonstrates a number of apoptotic nuclei in the liver of mice subjected to trauma-hemorrhage (C) compared to sham (D) controls.
Discussion
Trauma-hemorrhage induces hepatic damage and it has been found that even after fluid resuscitation following trauma-hemorrhage, the hepatic damage is still progressive [16,24]. Associated with a sustained disturbance in hepatic circulation and elevated levels of circulating α-GST levels, hepatocellular apoptosis has also been reported [9,24]. This is accompanied by increased production of IL-1β, IL-6 and TNF-α by the Kupffer cells in the liver and the elevated levels of these cytokines in the serum following trauma-hemorrhage [25,26]. Recent findings have suggested that elevated levels of proinflammatory cytokines can enhance ER stress and induce UPR as demonstrated by the increased expression of Bip, CHOP and XBP1s genes in the liver in response to IL-1 and IL-6 [27]. Another ER stress initiating factor is hypoxia, and therefore, the hypoxia seen after trauma-hemorrhage might also be a causative factor for ER stress [28,29]. Trauma-hemorrhage results in a condition manifested with multiple contributory factors of UPR.
The normal function of a cell is adversely affected when misfolding or unfolding of proteins occurs in the ER. The ER stress which induces UPR may be initiated by agents such as brefeldin A, thapsigargin or proinflammatory cytokines, or by conditions such as nutrient deprivation, or disturbance of Ca2+ homeostasis. UPR is initiated as a defensive process to cope with the overload on the ER, but on persistent ER stress, the UPR triggered signaling cascade switch from a protective to apoptotic function resulting in cell death. In the oxidative environment of the ER there is a constant demand for protein disulfide isomerases in catalysis and monitoring disulphide bond formation [19]. PDI is induced by ER stress and plays a crucial role in regulating the proper folding of proteins in the ER by catalyzing disulfide bond formation in order to rescue the ER from stress [30]. We evaluated protein levels of PDI in the liver extracts of mice after trauma-hemorrhage procedure and observed an elevated level consistent with ER stress (Fig.2).
There was a significant increase in the level of the ER chaperone, Bip, protein following trauma-hemorrhage (Fig. 2). Bip, indentical to the 78kDa glucose regulated protein (Grp78), is a peptide-binding molecular chaperone that binds transiently with protein-folding intermediates to prevent protein aggregation [31,32]. Bip is one of the well-characterized ER chaperones and it remains bound with all three ER stress sensors, PERK, ATF6 and IRE1, in maintaining them in inactive state in the absence of ER stress [33]. The release of Bip from stress sensors initiates the activation and transduction of the unfolded protein signals across the ER membrane to the cytosol and the nucleus [33,34]. Under conditions of increased ER stress Bip protein expression increases, and its increased level seen after trauma-hemorrhage is consistent with ER stress. Studies using solid tumors show that glucose starvation and hypoxic conditions also induce Bip expression by activated UPR signaling to ameliorate misfolded protein aggregation in the ER [35,36]. This is tantamount to the post-trauma-hemorrhage condition in many tissues and hence the significant increase observed in the level of Bip/Grp78 following trauma-hemorrhage should also be related to ER stress and activation of UPR.
The release of ATF6 from the ER is followed by its translocation to the Golgi complex and successive cleavage by site 1 and 2 proteases releasing the cytosolic N-terminal portion encoding a basic leucine zipper (bZIP) transcription factor [37,38]. Similarly PERK and IRE1 are also present in non-stressed cells in a Bip-bound form, and upon ER stress, Bip is titrated from these ER transmembrane proteins leading to their activation. We found an increased level of ATF6 protein in the liver of mice following trauma-hemorrhage. Further as an indication of the activation of ATF6, we also detected the cleaved ATF6 fragment (50kD size) in trauma-hemorrhage group comp sham animals. Concomitant to the increase of ATF6, the level of PERK, and IRE-1α were also elevated indicating ER stress. PERK and IRE-1α are also ER transmembrane proteins that remain bound to Bip under non-stressed conditions. Upon ER stress Bip is released from PERK and IRE-1 and these proteins are activated. PERK is a type I transmembrane kinase and its activation is effected by phosphorylation; our experiments confirmed an increase in phosphorylated PERK and total PERK following trauma-hemorrhage (Fig. 4). Though in many instances total PERK remains unchanged, there are reports demonstrating elevated levels of PERK mRNA as well as protein following UPR [39,40]. Activation of PERK is an attempt to rescue the cells from ER overload, as activated PERK oligomerizes and phosphorylates eIF2 which shuts off general translation. PERK also phosphorylates bZIP transcription factor Nrf2, the activated Nrf2 translocates to the nucleus and activate transcription through the ARE (antioxidant response element) [2]. Nevertheless, the trauma-hemorrhage has been documented to cause hypoxia and cellular injury mediated by reactive oxygen species. Therefore increased expression of PERK, likely to have been initiated at very early stage after hemorrhage, is possibly a protective response from ER stress and reactive oxygen species.
IRE1 is a protein kinase and an endoribonuclease with a dimerization domain in the ER lumen and the enzymatic domains in the cytosol. The oligomerization and autophosphorylation of IRE1 result in the activation of ribonuclease activity whereby XBP-1 mRNAs are spliced to generate the spliced variant XBP-1s, a more potent transcription factor. A subset of UPR genes participating in ER protein folding and ER-associated degradation (ERAD) are activated by XBP-1s. Though activation of XBP-1 through IRE1 is a protective mechanism, persisting overexpression of IRE1 is reported to result in the recruitment of TNF-receptor-associated factor 2 (TRAF2) and activate c-Jun N-terminal kinase (JNK) pathway leading to apoptosis [41,42]. It is also suggested that persistent ER stress may attenuate the expression of IRE1 and ATF6 and continued PERK activation may be critical in apoptosis [43]. How the UPR integrates its cytoprotective and proapoptotic outputs to select between life or death cell fates is still unknown [43]. However, PERK signaling, including translational inhibition and induction of proapoptotic transcription regulator CHOP, switches UPR to apoptotic pathway [4]. Our results demonstrate significantly increased expression of CHOP following trauma hemorrhage and also indicate significant hepatocyte apoptosis (Fig 6). During prolonged stress, the PERK and the IRE1 pathways might converge on CHOP, possibly augmenting each other's effect [4]. The significance of CHOP in apoptosis was shown in Chop-/- mice wherein the embryonic fibroblasts revealed that CHOP deficiency provides partial resistance to ER stress-induced apoptosis [4,6]. Therefore, increased expression of ER stress proteins, proapoptotic protein, CHOP, and increased apoptosis are possibly interconnected as shown in Fig. 7. Though we tested ER stress in the liver only 24 h after trauma-hemorrhage, it is very likely that the ER stress response sets in immediately after trauma. We have observed an immediate release of proinflammatory cytokines and induction of hypoxia within an hour after trauma-hemorrhage. A decrease in ATP level is also reported following T-H [8]. Several studies have clearly demonstrated that the major source of proinflammatory cytokines soon after trauma-hemorrhage is Kupffer cells [20,44,45]. Previous studies have also clearly demonstrated mitochondrial dysfunction, apoptosis and hepatocyte damage as evidenced by elevated plasma α-GST levels, increased lipid peroxidation (indicative of oxidative stress) and DNA fragmentation in hepatocytes after trauma-hermorrhage [15,46,47]. It is also known that hepatocytes do undergo ER stress response [15,46-50]. Our present study demonstrates that an ER stress response is initiated following trauma-hemorrhage, and this response may be significant in the apoptosis and organ dysfunction observed in the liver after trauma.
Figure 7.

Trauma-hemorrhage induced ER stress causes apoptosis and organ dysfunction. Excessive production of proinflammatory cytokines, hypoxia and insulin resistance (low glucose availability) are observed following trauma-hemorrhage. Any or all of these conditions may trigger ER stress and UPR in order to protect the cell from ER overload. However, persistent ER stress leads to expression of pro-apoptotic molecules (eg, CHOP) resulting in cellular apoptosis and organ dysfunction.
In conclusion, the experimental results described above clearly demonstrate the activation of ER stress and induction of UPR in mice after trauma-hemorrhage. We further observed the induction of CHOP and apoptosis in the liver of the mice subjected to trauma-hemorrhage as compared to that in the sham group. Trauma-hemorrhage is followed by hypoxia, insulin resistance and excessive secretion of inflammatory cytokines, mainly from the Kupffer cells in the liver. All or any of these conditions can trigger the activation of ER stress and UPR. Based upon these results we hypothesize a possible role for ER stress in apoptosis and organ damage following trauma-hemorrhage. Further studies employing techniques such as siRNA to knockout ER stress protein genes may shed more light onto the role of ER stress in trauma-hemorrhage-mediated physiological dysfunction and sepsis.
Fig.1.

Elevated levels of α-GST (A) and TNF-α (B) following trauma-hemorrhage. Significantly increased levels of α-GST and TNF-α were observed in the plasma of mice subjected to trauma-hemorrhage (n=4) compared to sham (n=4), two hours after trauma-hemorrhage and resuscitation.
Acknowledgments
Authors acknowledge the help of Dr Wenhong Kan in her assistance in the ELISA assay. Supported by NIH grant R21AG031440
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kincaid MM, Cooper AA. ERADicate ER stress or die trying. Antioxid Redox Signal. 2007;9:2373–2387. doi: 10.1089/ars.2007.1817. [DOI] [PubMed] [Google Scholar]
- 2.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 3.van Anken E, Braakman I. Endoplasmic reticulum stress and the making of a professional secretory cell. Crit Rev Biochem Mol Biol. 2005;40:269–283. doi: 10.1080/10409230500315352. [DOI] [PubMed] [Google Scholar]
- 4.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frink M, Hsieh YC, Hsieh CH, Pape HC, Choudhry MA, Schwacha MG, Chaudry IH. Keratinocyte-derived chemokine plays a critical role in the induction of systemic inflammation and tissue damage after trauma-hemorrhage. Shock. 2007;28:576–581. doi: 10.1097/shk.0b013e31814b8e0d. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol. 2006;41:511–521. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Hsu JT, Hsieh YC, Kan WH, Chen JG, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Role of p38 mitogen-activated protein kinase pathway in estrogen-mediated cardioprotection following trauma-hemorrhage. Am J Physiol Heart Circ Physiol. 2007;292:H2982–H2987. doi: 10.1152/ajpheart.01303.2006. [DOI] [PubMed] [Google Scholar]
- 10.Jarrar D, Wang P, Knoferl MW, Kuebler JF, Cioffi WG, Bland KI, Chaudry IH. Insight into the mechanism by which estradiol improves organ functions after trauma-hemorrhage. Surgery. 2000;128:246–252. doi: 10.1067/msy.2000.107376. [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki T, Fujimi S, Lederer JA, Hubbard WJ, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Trauma-hemorrhage induces depressed splenic dendritic cell functions in mice. J Immunol. 2006;177:4514–4520. doi: 10.4049/jimmunol.177.7.4514. [DOI] [PubMed] [Google Scholar]
- 12.Wu R, Wang P. Adenosine A2A receptor activation: another potential therapy for trauma and hemorrhagic shock. Crit Care Med. 2006;34:1273–1275. doi: 10.1097/01.CCM.0000208437.93725.18. [DOI] [PubMed] [Google Scholar]
- 13.Morrell ED, Tsai BM, Crisostomo PR, Hammoud ZT, Meldrum DR. Experimental therapies for hypoxia-induced pulmonary hypertension during acute lung injury. Shock. 2006;25:214–226. doi: 10.1097/01.shk.0000191380.44972.46. [DOI] [PubMed] [Google Scholar]
- 14.Raju R, Bland KI, Chaudry IH. Estrogen: a novel therapeutic adjunct for the treatment of trauma-hemorrhage-induced immunological alterations. Mol Med. 2008;14:213–221. doi: 10.2119/2008-00001.Raju. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, Chaudry IH. G protein-coupled receptor 30-dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. 2007;170:1210–1218. doi: 10.2353/ajpath.2007.060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higuchi S, Wu R, Zhou M, Ravikumar TS, Wang P. Downregulation of hepatic cytochrome P-450 isoforms and PPAR-gamma: their role in hepatic injury and proinflammatory responses in a double-hit model of hemorrhage and sepsis. J Surg Res. 2007;137:46–52. doi: 10.1016/j.jss.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 17.Lowry SF, Calvano SE. Challenges for modeling and interpreting the complex biology of severe injury and inflammation. J Leukoc Biol. 2008;83:553–557. doi: 10.1189/jlb.0607377. [DOI] [PubMed] [Google Scholar]
- 18.Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Mechanism of estrogen-mediated attenuation of hepatic injury following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. J Leukoc Biol. 2007;82:1019–1026. doi: 10.1189/jlb.0607355. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hildebrand F, Hubbard WJ, Choudhry MA, Frink M, Pape HC, Kunkel SL, Chaudry IH. Kupffer cells and their mediators: the culprits in producing distant organ damage after trauma-hemorrhage. Am J Pathol. 2006;169:784–794. doi: 10.2353/ajpath.2006.060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider CP, Nickel EA, Samy TS, Schwacha MG, Cioffi WG, Bland KI, Chaudry IH. The aromatase inhibitor, 4-hydroxyandrostenedione, restores immune responses following trauma-hemorrhage in males and decreases mortality from subsequent sepsis. Shock. 2000;14:347–353. doi: 10.1097/00024382-200014030-00019. [DOI] [PubMed] [Google Scholar]
- 22.Redl H, Schlag G, Paul E, Davies J. Plasma glutathione S-transferase as an early marker of posttraumatic hepatic injury in non-human primates. Shock. 1995;3:395–397. [PubMed] [Google Scholar]
- 23.Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- 24.Yokoyama Y, Toth B, Kitchens WC, Schwacha MG, Bland KI, Chaudry IH. Role of thromboxane in producing portal hypertension following trauma-hemorrhage. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1293–G1299. doi: 10.1152/ajpgi.00268.2003. [DOI] [PubMed] [Google Scholar]
- 25.Ayala A, Perrin MM, Ertel W, Chaudry IH. Differential effects of hemorrhage on Kupffer cells: decreased antigen presentation despite increased inflammatory cytokine (IL-1, IL-6 and TNF) release. Cytokine. 1992;4:66–75. doi: 10.1016/1043-4666(92)90039-t. [DOI] [PubMed] [Google Scholar]
- 26.Catania RA, Schwacha MG, Cioffi WG, Bland KI, Chaudry IH. Does uninjured skin release proinflammatory cytokines following trauma and hemorrhage? Arch Surg. 1999;134:368–373. doi: 10.1001/archsurg.134.4.368. [DOI] [PubMed] [Google Scholar]
- 27.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 28.Ma Y, Wang P, Kuebler JF, Chaudry IH, Messina JL. Hemorrhage induces the rapid development of hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2003;284:G107–G115. doi: 10.1152/ajpgi.00217.2002. [DOI] [PubMed] [Google Scholar]
- 29.Ma Y, Toth B, Keeton AB, Holland LT, Chaudry IH, Messina JL. Mechanisms of hemorrhage-induced hepatic insulin resistance: role of tumor necrosis factor-alpha. Endocrinology. 2004;145:5168–5176. doi: 10.1210/en.2004-0524. [DOI] [PubMed] [Google Scholar]
- 30.Lyles MM, Gilbert HF. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry. 1991;30:613–619. doi: 10.1021/bi00217a004. [DOI] [PubMed] [Google Scholar]
- 31.Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 32.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 33.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 34.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong D, Ko B, Baumeister P, Swenson S, Costa F, Markland F, Stiles C, Patterson JB, Bates SE, Lee AS. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–5791. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 37.Schroder M, Kohno K. Recent advances in understanding the unfolded protein response. Antioxid Redox Signal. 2007;9:2241–2244. doi: 10.1089/ars.2007.1877. [DOI] [PubMed] [Google Scholar]
- 38.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 39.Datta R, Waheed A, Shah GN, Sly WS. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci U S A. 2007;104:19989–19994. doi: 10.1073/pnas.0708725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rissanen A, Sivenius J, Jolkkonen J. Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res. 2006;1087:60–66. doi: 10.1016/j.brainres.2006.02.095. [DOI] [PubMed] [Google Scholar]
- 41.Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- 42.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 43.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frink M, Hsieh YC, Thobe BM, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. TLR4 regulates Kupffer cell chemokine production, systemic inflammation and lung neutrophil infiltration following trauma-hemorrhage. Mol Immunol. 2007;44:2625–2630. doi: 10.1016/j.molimm.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh YC, Frink M, Thobe BM, Hsu JT, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. 17Beta-estradiol downregulates Kupffer cell TLR4-dependent p38 MAPK pathway and normalizes inflammatory cytokine production following trauma-hemorrhage. Mol Immunol. 2007;44:2165–2172. doi: 10.1016/j.molimm.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kan WH, Hsieh CH, Schwacha MG, Choudhry MA, Raju R, Bland KI, Chaudry IH. Flutamide protects against trauma-hemorrhage-induced liver injury via attenuation of the inflammatory response, oxidative stress, and apopotosis. J Appl Physiol. 2008;105:595–602. doi: 10.1152/japplphysiol.00012.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimizu T, Yu HP, Suzuki T, Szalay L, Hsieh YC, Choudhry MA, Bland KI, Chaudry IH. The role of estrogen receptor subtypes in ameliorating hepatic injury following trauma-hemorrhage. J Hepatol. 2007;46:1047–1054. doi: 10.1016/j.jhep.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iizaka T, Tsuji M, Oyamada H, Morio Y, Oguchi K. Interaction between caspase-8 activation and endoplasmic reticulum stress in glycochenodeoxycholic acid-induced apoptotic HepG2 cells. Toxicology. 2007;241:146–156. doi: 10.1016/j.tox.2007.08.095. [DOI] [PubMed] [Google Scholar]
- 49.Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7:520–532. doi: 10.1016/j.cmet.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tamaki N, Hatano E, Taura K, Tada M, Kodama Y, Nitta T, Iwaisako K, Seo S, Nakajima A, Ikai I, Uemoto S. CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am J Physiol Gastrointest Liver Physiol. 2008;294:G498–G505. doi: 10.1152/ajpgi.00482.2007. [DOI] [PubMed] [Google Scholar]