

Abstract
Ubiquitin carboxy terminal hydrolase-L1 (UCH-L1) belongs to the UCH proteases family that deubiquitinates ubiquitin-protein conjugates in the ubiquitin-proteasome system. Previous research showed that UCH-L1 was expressed in mouse retinal cells and testicular germ cells, and its function was associated with apoptosis. But it is still unclear whether UCH-L1 is concerned with apoptosis in tumor cells. In order to clarify the role of UCH-L1 in tumor cells, multi-drug resistance (MDR) human breast carcinoma cell line MCF7/Adr, that expresses relatively high UCH-L1, and its parental cell line MCF7, that expresses relatively low UCH-L1, were chosen for this study. We transfected pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA into MCF7 and MCF7/Adr cells, respectively. Using 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay, Western blot, Hoechst 33258 staining assay and flow cytometry, we found that over-expression of UCH-L1 in MCF7 cells induced apoptosis. On the other hand, silencing of UCH-L1 in MCF7/Adr cells led to the opposite effect. Moreover, to explore the mechanism underling these observations, we further investigated the expression of phospho-Akt and its downstream signal phospho-IκB-α and other signal molecules including Fas, Fas-L, Trail, DR4, DR5, Bax, cytochrome C, active caspase-3, phospho-p53, phospho-Mdm-2, Bcl-2, Bcl-xL, p21 and p27. The results indicated that the process of apoptosis triggered by UCH-L1 is, at least in part, probably through Phosphoinositide 3-kinase (PI3K)/ Akt signal pathway. Our findings suggest that modulating the ubiquitination and deubiquitination pathway could be a novel method for tumor therapy.
Keywords: ubiquitin carboxy terminal hydrolase-L1, MCF7, MCF7/Adr, apoptosis, PI3-kinase/Akt pathway
Introduction
Covalent conjugation of ubiquitin to proteins plays a crucial role in a wide variety of biological processes, including the cell cycle, cell proliferation, development, apoptosis, signal transduction, and membrane protein internalization (1). Ubiquitination is reversible, i.e. ubiquitin is recycled by proteolytic removal from its conjugating protein by deubiquitinating enzymes (DUBs), a family of proteases with exquisite specificity for ubiquitinated substrates. Deubiquitination is widely recognized as an important component of regulatory mechanisms in all ubiquitin-dependent pathways (2). Ubiquitin carboxy terminal hydrolase (UCH) is a subclass of DUBs, which catalyzes the hydrolysis of COOH-terminal ubiquityl esters and amides. It removes ubiquitin from ubiquitinated cellular proteins, thereby preventing them from targeted degradation via the proteasome pathway. One member of the UCH family is represented by UCH-L1, which is selectively expressed in the testis/ovary and brain (3-5). Previous work on UCH-L1 function in gracile axonal dystrophy (gad) mice with an exon deletion for UCH-L1 suggests that these mice are resistant to apoptotic stress in retinal cells and testicular germ cells (6,7). This observation is consistent with a recent report that the over-expression of UCH-L1 induces testicular germ cell apoptosis in UCH-L1 transgenic mice (8). Recently some studies showed that UCH-L1 was abnormally expressed in some tumors, and correlated with cancer cell differentiation, metastasis and multi-drug resistance (MDR) (9-12). But it is still unclear whether UCH-L1 is concerned with apoptosis in tumor cells. Since it is well known that human breast carcinoma cell line MCF7 expresses low UCH-L1, and its MDR counterpart MCF7/Adr expresses relatively high UCH-L1, to investigate the effect of UCH-L1 on apoptosis in breast cancer cells, we transfected pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA into MCF7 and MCF7/Adr cells, respectively. Results showed that UCH-L1 inhibited the growth of breast cancer cells and its action was dependent on the inducement of cell death, showing the typical characteristics of apoptosis but not necrosis. These results suggest that UCH-L1 plays an important role in tumor therapy via the inducement of apoptosis in breast cancer cells.
Phosphoinositide 3-kinase (PI3K) plays a central role in a diverse range of cellular responses including cell growth, survival and malignant transformation (13,14). Akt (also named protein kinase B) is a downstream signal of PI3K and a 60 KD serine/threonine kinase and is also a critical mediator of survival signals to protect cells from apoptosis (15,16). Activation of the PI3K/Akt pathway in response to cytokines leads to phosphorylation and activation of the nuclear factor NFκB p65/RelA subunit, which regulates expression of anti-apoptotic genes (17,18). To clarify the mechanism underling our observations in MCF7 and MCF7/Adr cells, the expression of Akt and phospho-IκB-α was further investigated and results demonstrated that UCH-L1 induced apoptosis in breast cancer cells, possibly through the PI3K/Akt pathway.
Materials and methods
Cell culture
The human mammary carcinoma cell lines, MCF7 and its MDR counterpart MCF7/Adr were cultured in RPMI-1640 (Gibco-BRL, Karlsruhe, Germany) containing 10% fetal bovine serum (FBS; PAA Laboratories, Linz, Austria), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. All cultures were discarded after 3 months and new cultures were obtained from frozen stocks.
Plasmid transfection
MCF7 cells were transfected with eukaryotic expression vectors pcDNA3.1-UCH-L1 or pcDNA3.1 empty loading control using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. In brief, 80% confluent cells grown in 100-mm dishes were washed twice with Opti-MEM (Gibco-BRL) medium before the addition of 25 μg of plasmid DNA and 60 μl of Lipofectamine 2000 in 3 ml of Opti-MEM media. After 8-h incubation at 37°C, 7 ml of medium containing 10% FBS was added. Incubations were continued at 37°C for another 28 h.
RNA interference
MCF7/Adr cells were transfected with UCH-L1 siRNA duplex oligoribonucleotides (Sense: 5’-AGG ACU AAC UUC UUG UCC CUU CAG C-3’, Anti-sense: 5’-GCU GAA GGG ACA AGA AGU UAG UCC U-3’) or RNAi negative control duplexes (Invitrogen) using Lipofectamine 2000 following the manufacturer’s protocol. In brief, 50% confluent cells grown in 60-mm dishes were washed twice with Opti-MEM medium before the addition of 20 nM UCH-L1 siRNA duplex oligoribonucleotides or 20 nM RNAi negative control duplexes and 10 μl of Lipofectamine 2000 in 1 ml of Opti-MEM media. After 8-h incubation at 37°C, 3 ml of medium containing 10% FBS was added. Incubations were continued at 37°C for another 40 h.
Immunofluorescence analysis
Cells were seeded, incubated overnight on sterile cover glasses, and after transfetion, were washed with phosphate buffered saline (PBS) and fixed by cold acetone for 8 min at 4°C. The fixed cells were washed thrice with PBS and then incubated with anti-UCH-L1 (Chemicon, Temecula, CA, USA) antibody for 1 h at 37°C, followed by 45-min incubation at 37°C with TRITC-conjugated secondary antibody, finally the nuclei were stained with Hoechst 33258 (Sigma, St. Louis, MO, USA) for 5 min at room temperature. The labeled cells were analyzed by fluorescence microscopy (Zeiss, Oberkochen, Germany). To detect non-specific antibody binding, cells were incubated with isotype-match non-specific IgG (R&D, Minneapolis, MN, USA). The control samples did not show any significant cell labeling (data not shown).
Reverse transcription and quantitative real-time polymerase chain reaction
Total RNA of cells was extracted using Tripure isolation reagent (Invitrogen). The RNA samples were subjected to reverse transcription (RT) with 2 μg RNA, Oligo (dT)18, dNTP, and reaction buffer supplied with M-MLV reverse transcriptase (Promega, Madison, WI, USA). Quantitative real-time polymerase chain reaction (qRT-PCR) was then performed in 20 μl solution with 2 μg cDNA and 1 mM of each forward and reverse primer and 2X SYBR-Green mix (Takara, Shuzo, Kyoto, Japan). Changes in the mRNA expression level were calculated following normalization with glyceraldehyde-3-phosphatedehydrogenase (GAPDH). Relative gene expression was determined by the fluorescence intensity ratio of the target gene to GAPDH. The primers used in the real-time PCR reactions were designed based on information from the human genomic database. The following are the primers used for the specific amplification of GAPDH and UCH-L1: GAPDH forward primer: 5’-AAC GGA TTT GGT CGT ATT G-3’, and reverse primer: 5’-GGA AGA TGG TGA TGG GAT T-3’; UCH-L1 forward primer: 5’-GCC AAT GTC GGG TAG ATG-3’, and reverse primer: 5’-CAA AGT CCC TCC CAC AGA-3’.
Western blot
Cells were harvested and lysed in lysis buffer [10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Nonidet P (NP)-40, 1 mM ethylene diamine tetra-acetic acid (EDTA), 1 mM Na3VO4 and 1 mM phenylmethylsulfonyl fluoride (PMSF)]. Proteins (50 μg) were then subjected to standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For Western blot analysis, proteins were transferred to polyvinylidene fluoride (PVDF) membranes, blocked in 5% non-fat milk in 10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% (w/v) Tween-20 for 1 h. The membranes were first incubated with antibodies against ß-actin (Sigma), UCH-L1, PCNA (Cell Signaling Technology, Beverly, MA, USA), Fas (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Fas-L (Santa Cruz Biotechnology), Trail (Santa Cruz Biotechnology), DR4 (Santa Cruz Biotechnology), DR5 (Affinity Bioreagents, Golden, CO, USA), Bax (Santa Cruz Biotechnology), cytochrome C (Cell Signaling Technology), active caspase-3 (BioVision, Palo Alto, CA, USA), phospho-p53 (Cell Signaling Technology), phospho-Mdm-2 (Sigma), Bcl-2 (Santa Cruz Biotechnology), Bcl-xL (Santa Cruz Biotechnology), p21 (Sigma), p27 (Chemicon), phospho-IκB-α (Cell Signaling Technology), phospho-specific and total Akt (Cell Signaling Technology), respectively, overnight at 4°C, followed by 1-h incubation with the appropriate secondary antibody consisting of horseradish peroxidase (HRP). Detection by enzyme-linked chemiluminescence (ECL; Pierce, Rockford, USA) was performed according to the manufacturer’s protocol. Controls for protein loading were identified by ß-actin as the internal standard.
Cell growth curve
For the analysis of cell growth rate, cells were transfected and plated in 96-well plates at a density of 3×103 cells/well and viable cells were counted from day 1 to day 9. Every day, cells were stained with 20 μl sterile MTT dye (Sigma, 5 mg/ml) at 37°C for 4 h followed by removing the culture medium and mixing 150 μl of dimethylsulfoxide (DMSO) thoroughly for 10 min. Spectrometric absorbance at 490 nm was measured with a microplate reader. The results were obtained from three independent cultures and each was repeated three times.
Hoechst 33258 staining
Cells were seeded, incubated overnight on sterile cover glasses, and after transfetion, were fixed by methanol for 5 min at room temperature. The fixed cells were washed twice with PBS and then incubated with Hoechst 33258 for 5 min at room temperature and observed under fluorescence microscopy using a 4’,6-diamidino-2-phenylindole (DAPI) filter. Fragmented or condensed nuclei were scored as apoptotic.
Flow cytometry analysis
For induction of cell apoptosis and detection of cell cycle, cells were scraped from the bottom of the dish and pooled with the floating cells and centrifuged at 2,000 rpm for 5 min. Cells were washed with PBS before fixation in citric acid. After the fixation, cells were sent to be analyzed for apoptotic ratio and cell cycle by flow cytometry by propidium iodide (PI) staining of nuclei, which is based on the principle that after DNA fragmentation permeabilized cells exhibit a reduced chromatin stainability and accessibility to fluorochromes in the Institute of Cell Biology (Shanghai, P.R. China).
Statistical analysis
Statistics were conducted by SPSS software. The results are presented with mean ± standard errors (SEM). ANOVA and Student’s t-test analysis followed by Dunnett’s multiple comparison tests were used to compare mean values. A probability value of P<0.05 was defined as statistical significance.
Results
PcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA caused specific and effective up- or down-regulation of UCH-L1 expression, respectively
To investigate the role of UCH-L1 in breast cancer cells, firstly we transfected pcDNA3.1-UCH-L1 plasmid in MCF7 cells that express UCH-L1 in a relatively low level, and secondly transfected UCH-L1 siRNA in MCF7/Adr cells that express UCH-L1 in a relatively high level. The transfection efficiencies of individual transfectants were initially evaluated using immunofluorescence analysis. As shown in Fig. 1A, the fluorescence intensity which represented UCH-L1 expression level in UCH-L1 transfectants and UCH-L1 siRNA transfectants displayed a significant promotion or reduction when compared with their negative controls. In addition, qRT-PCR and Western blot analysis showed the responsive changes in corresponding transfectants (Fig. 1B and C).
Figure 1.
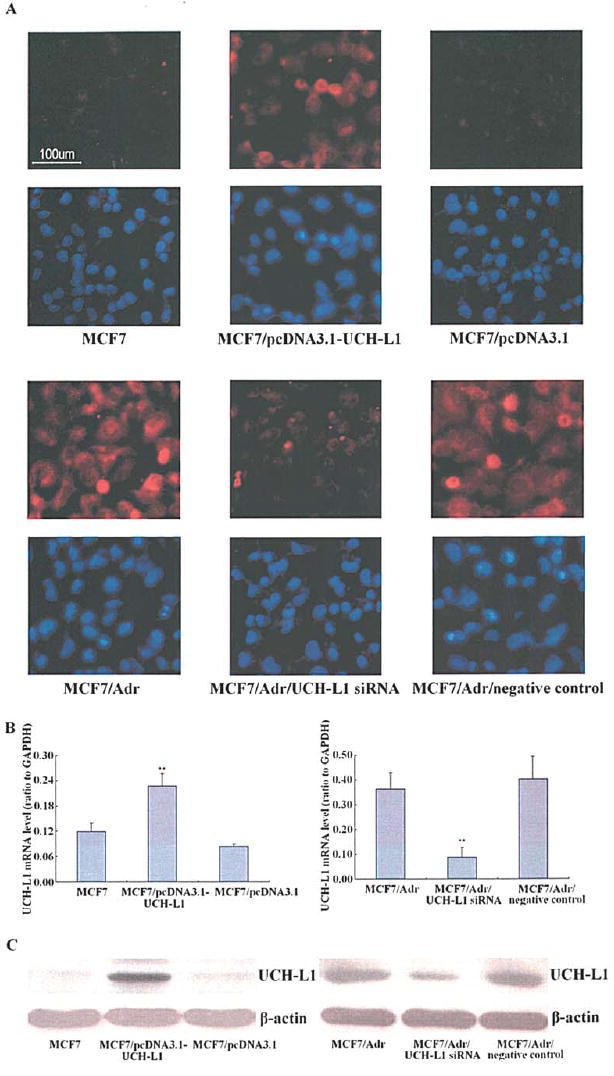
Production of UCH-L1 mRNA and protein in MCF7, MCF7/Adr cells were up- or down-regulated after transfecting with pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA, respectively. (A) Transfection efficiencies identified by immunofluorescence, the nuclei were stained with Hoechst 33258. All fields are representative of multiple fields observed in three independent experiments. (B) UCH-L1 mRNA assessed by quantitative real-time polymerase chain reaction (qRT-PCR), GAPDH served as the internal standard. Each bar indicates the mean of normalized UCH-L1 mRNA ± SEM of three independent experiments. **P<0.05 vs. control cells. (C) UCH-L1 protein assessed by Western blot, ß-actin served as control for sample loading. Results are representative of three similar experiments.
Elevated expression of UCH-L1 in MCF7 cells resulted in growth suppression and inducement of apoptosis, whereas knockdown of UCH-L1 in MCF7/Adr cells led to the opposite effects
As UCH-L1 has been reported to be associated with apoptosis in some types of cells, it is essential to understand whether modulation of UCH-L1 expression has any effects on breast cancer cell growth and apoptosis. UCH-L1 over-expressing MCF7 cells and UCH-L1 silencing MCF7/Adr cells were therefore examined for growth capacity using MTT assay. As shown in Fig. 2A, elevated UCH-L1 expression in MCF7 cells suppressed cell growth, while knockdown of UCH-L1 in MCF7/Adr cells led to the opposite effect when compared with negative controls. To further confirm that the effect of UCH-L1 on cell growth was controlled by affecting proliferation or apoptosis or both pathways, we measured cell cycle and PCNA protein level which is associated with cell proliferation and DNA synthesis during S phase of cell cycle in the cells, and found that the UCH-L1 over-expressing MCF7 cells and UCH-L1 silencing MCF7/Adr cells did not show significant difference between their corresponding negative controls (Fig. 2B and C). Subsequently, we measured possible apoptotic morphology change and stained with Hoechst 33258. Fluorescence microscope revealed that as compared with negative controls, condensed bright apoptotic nuclei were readily observed in UCH-L1 over-expressing MCF7 cells, while after UCH-L1 siRNA transfection, the apoptotic nuclei were hardly observed in MCF7/Adr cells (Fig. 2D). To obtain a quantitative characterization of the apoptotic effect, we examined the above cells for apoptotic change by measuring DNA degradation using flow cytometry. Compared with their negative controls, up-regulation of UCH-L1 expression in MCF7 cells markedly increased the fraction of apoptotic cells, while down-regulation of UCH-L1 expression in MCF7/Adr cells led to the opposite effect (Fig. 2E).
Figure 2.
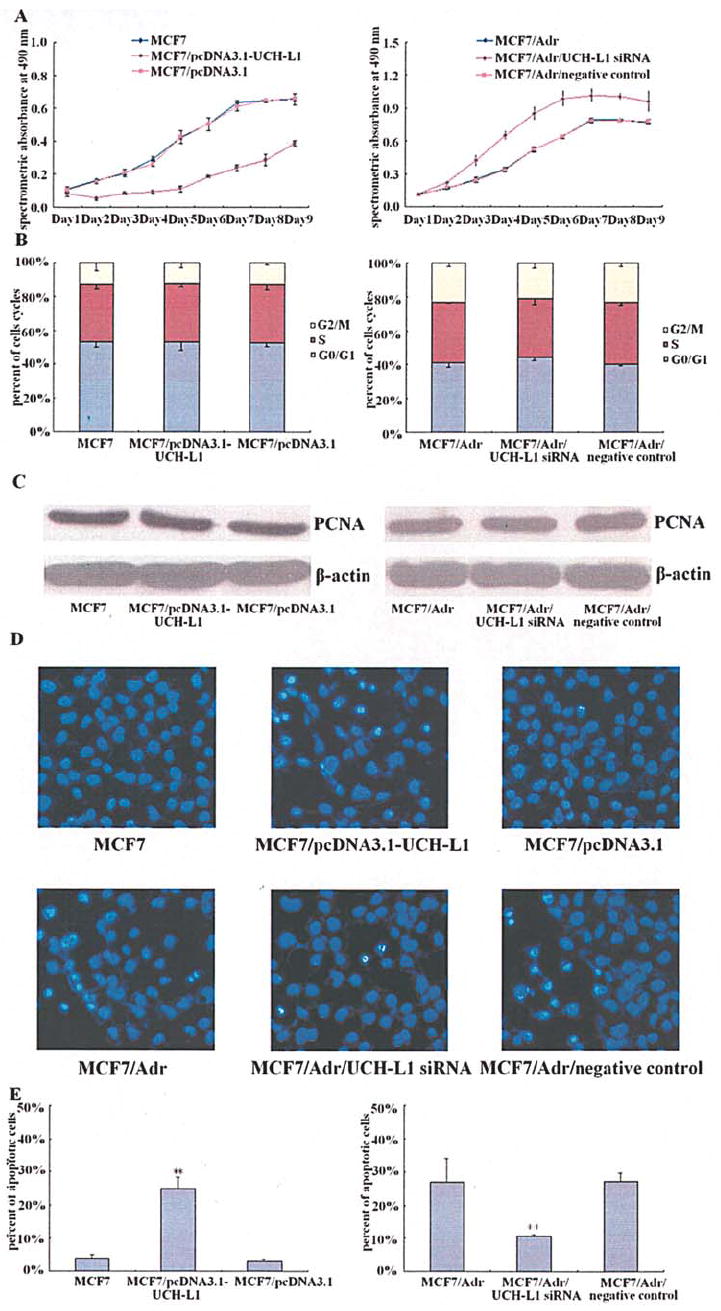
Effects of UCH-L1 on the growth and apoptosis in MCF7 and MCF7/Adr cells. MCF7 and MCF7/Adr cells were transfected with pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA, respectively. (A) Cells growth curve measured by 3-(4,5-dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide (MTT) assay. Each point indicates the mean of spectrometric absorbance ± SEM of three independent experiments. (B) Cells cycle analyzed by flow cytometry. Each bar indicates the distribution of cells cycle. (C) PCNA protein assessed by Western blot, ß-actin served as control for sample loading. Results are representative of three similar experiments. (D) Cells morphological change detected by Hoechst 33258 staining. All fields are representative of multiple fields observed in three independent experiments. (E) Cells apoptotic ratio analyzed by flow cytometry. Each bar indicates the mean of cell apoptosis ± SEM of three independent experiments. **P<0.05 vs. control cells.
Regulation of pro-apoptotic, anti-apoptotic and cyclin-dependent kinase inhibitory proteins levels by UCH-L1 in MCF7 cells
To obtain clues to the mechanism by which elevated expression of UCH-L1 induces apoptosis in MCF7 cells, lysates from UCH-L1 over-expressing MCF7 cells and negative controls were prepared and Western blot was carried out. Results of multiple Western blot analysis showed that pro-apoptotic factors, Fas, Fas-L, Trail, DR4, DR5, Bax, cytochrome C, active caspase-3 and phospho-p53 levels were remarkably higher (Fig. 3A), anti-apoptotic factors, phospho-Mdm-2, Bcl-2 and Bcl-xL levels were significantly lower (Fig. 3B), cyclin-dependent kinase inhibitor p21 level was elevated, while p27 level was reduced (Fig. 3C) in UCH-L1 transfectants than in MCF7 cells and their empty loading control transfectants.
Figure 3.
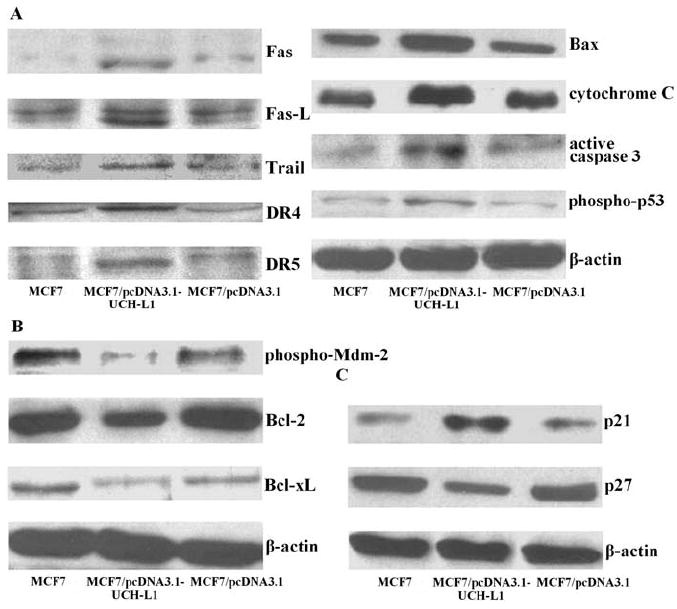
Production of pro-apoptotic, anti-apoptotic and cyclin-dependent kinase inhibitory proteins in MCF7 cells transfected with pcDNA3.1-UCH-L1 plasmid. (A) Pro-apoptotic factors, Fas, Fas-L, Trail, DR4, DR5, Bax, cytochrome C, active caspase-3 and phospho-p53 proteins assessed by Western blot. (B) Anti-apoptotic factors, phospho-Mdm-2, Bcl-2 and Bcl-xL proteins assessed by Western blot. (C) Cyclin-dependent kinase inhibitors, p21 and p27 proteins assessed by Western blot. ß-actin served as control for sample loading. Results are representative of three similar experiments.
Effects of UCH-L1 siRNA on pro-apoptotic, anti-apoptotic and cyclin-dependent kinase inhibitory protein levels in MCF7/Adr cells
To further confirm the role of UCH-L1 in breast cancer cell apoptosis, we used MCF7/Adr derived cells in which over-expression of UCH-L1 was suppressed by transfection of UCH-L1 siRNA. Western blot analysis revealed that protein levels of pro-apoptotic factors, Fas, Fas-L, Bax, cytochrome C, active caspase-3 and phospho-p53 were notably lower, while Trail, DR4 and DR5 showed no changes (Fig. 4A), anti-apoptotic factors, phospho-Mdm-2, Bcl-2 and Bcl-xL were significantly higher (Fig. 4B), cyclin-dependent kinase inhibitor p21 was decreased, while p27 was increased (Fig. 4C) in UCH-L1 silencing transfectants than in their negative controls.
Figure 4.
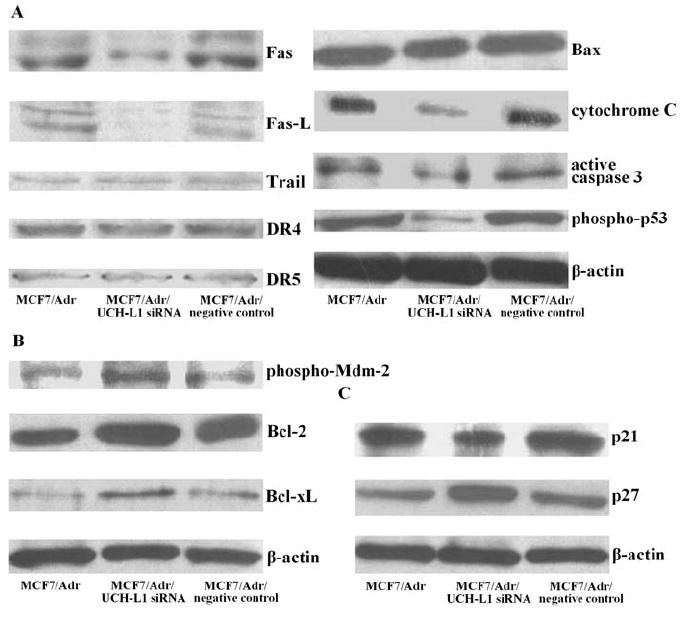
Production of pro-apoptotic, anti-apoptotic and cyclin-dependent kinase inhibitory proteins in MCF7/Adr cells transfected with UCH-L1 siRNA. (A) Pro-apoptotic factors, Fas, Fas-L, Trail, DR4, DR5, Bax, cytochrome C, active caspase-3 and phospho-p53 proteins assessed by Western blot. (B) Anti-apoptotic factors, phospho-Mdm-2, Bcl-2 and Bcl-xL proteins assessed by Western blot. (C) Cyclin-dependent kinase inhibitors, p21 and p27 proteins assessed by Western blot. ß-actin served as control for sample loading. Results are representative of three similar experiments.
PI3K/Akt pathway is possibly involved in the process of cell apoptosis induced by over-expression of UCH-L1 in MCF7 or suppressed by UCH-L1 siRNA in MCF7/Adr cells
PI3K/Akt pathway is one of the main signal pathways involved in breast cancer cell growth (19). To investigate the function role of the PI3K/Akt pathway in the process of cell apoptosis, we examined the expression of phosphospecific-Akt and total Akt in UCH-L1 over-expressing MCF7 cells and UCH-L1 silencing MCF7/Adr cells, respectively. As a result, we found that elevated UCH-L1 expression in MCF7 cells blocked phospho-Akt expression, while knockdown of UCH-L1 in MCF7/Adr cells led to the opposite effect when compared with their corresponding controls (Fig. 5A). We further examined the downstream signal of Akt and noted that NFκB is held in the cytoplasm as an inactive complex with inhibitor IκB and remains in an inactivate state without IκB phosphorylation. We further evaluated the phosphorylation of IκB with phospho-IκB-specific antibody. As shown in Fig. 5B, Western blot analysis showed the responsive changes in corresponding transfectants.
Figure 5.
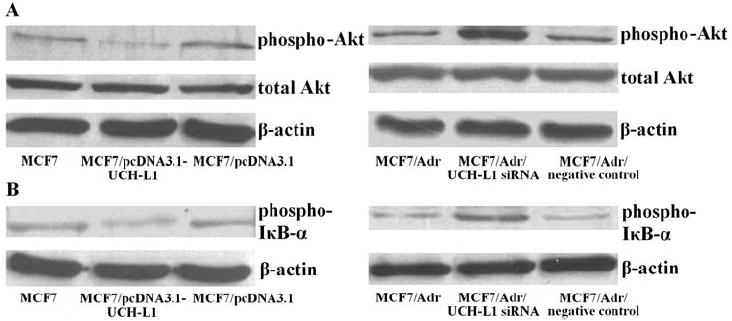
Expression of phospho-Akt, total Akt and phospho-IκB in MCF7 and MCF7/Adr cells transfected with pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA, respectively. (A) Phospho-Akt and total Akt proteins assessed by Western blot. (B) Phospho-IκB protein assessed by Western blot. ß-actin served as control for sample loading. Results are representative of three similar experiments.
Discussion
Ubiquitin is a 76-amino acid protein that, as its name implies, is ubiquitously distributed and highly conserved throughout eukaryotic organisms. The most widely accepted consequence of ubiquitination is proteasomal degradation. Ubiquitin level is controlled by the balance of ubiquitinating enzymes and DUBs. DUBs are subdivided into UCHs and ubiquitin-specific processing proteases (USPs). UCH-L1, a member of UCHs family, has a dual function: a hydrolase activity that removes small COOH-terminal ubiquitin to generate a ubiquitin monomer and a dimerization-dependent ubiquitin ligase activity (20).
UCH-L1 over-expression has been observed in several forms of cancers. In breast cancer, high UCH-L1 mRNA level was significantly associated with high histological grade, negative estrogen receptor (ER) status and progesterone receptor (PR) status, and indeed, had a poorer prognosis than those with low UCH-L1 mRNA level (9). To investigate the role of UCH-L1 in breast cancer, we chose two close associated mammary carcinoma cell lines, MCF7 and MCF7/Adr. MCF7/Adr possesses MDR and metastasis phenotype, and expresses much higher UCH-L1 than MCF7 (12,21,22). It is also well known that MCF7 is an ER positive and PR positive cell line, while MCF7/Adr is an ER negative and PR negative cell line. Furthermore, our previous data indicated that during the development of drug resistance to adriamycin in MCF7 cells, the promoted expression of MDR1 gene coincided with up-regulation of UCH-L1 and increase of invasion abilities in vitro. But the detailed role of UCH-L1 is unclear in these breast cancer cell lines that exhibit different characteristics. Therefore, we transfected pcDNA3.1-UCH-L1 plasmid and UCH-L1 siRNA into MCF7 and MCF7/Adr cells, respectively, then investigated whether up-regulation of UCH-L1 expression in MCF7 cells and down-regulation of UCH-L1 expression in MCF7/Adr cells had different effects on breast cancer cell growth and apoptosis. Our results revealed that MCF7/Adr cells exhibited much stronger growth capacity and higher apoptosis percentage in comparison with MCF7 cells. In addition, over-expression of UCH-L1 suppressed cell growth and its action was dependent on the inducement of apoptosis in MCF7 cells, because cell cycle and PCNA protein level in UCH-L1 over-expressing MCF7 cells showed no significant difference between negative controls. In contrast, knockdown of UCH-L1 in MCF7/Adr cells led to the opposite effects. Our results clearly demonstrated that UCH-L1 had a significant effect on inducing apoptosis in breast cancer cells in vitro.
Although it is the first time that the effect of UCH-L1 on inducing apoptosis in cancer cells is reported, previous research has linked inhibition of UCH-L1 function with suppression of apoptosis in certain types of cells in vivo (6,7). Apoptosis is an extremely fine-tuned regulatory network consisting of pro- and anti-apoptotic factors. It has shown that ubiquitination is one of the major mechanisms for the regulation of key proteins of the cell apoptotic machinery, such as p53 and the molecules of Bcl-2 family. Ubquitin-dependent degradation of pro-apoptotic Bcl-2 family members, Bid, Bax and Bak, has been reported to promote cell survival (23,24). Moreover, recent studies suggest that through regulation of monoubiquitin level, UCH-L1 plays a role in balancing the expression of pro- and anti-apoptotic proteins (7,25). In our present study, we investigated the alteration of pro-apoptotic and anti-apoptotic proteins levels in UCH-L1 over-expressing MCF7 cells and UCH-L1 silencing MCF7/Adr cells. The results indicated that probably the extrinsic (Fas, Fas-L, Trail, DR4 and DR5) and intrinsic (cytochrome C) apoptotic pathways, the activation of p53 (phospho-Mdm-2 and phospho-p53) and the Bcl-2 family members (pro-apoptotic member Bax, anti-apoptotic members Bcl-2 and Bcl-xL) combined with each other, participating in the process of apoptosis induced by UCH-L1 in MCF7 cells. Whereas, Trail/DR4 and Trail/DR5 pathways were not involved in the apoptosis suppression conducted by UCH-L1 siRNA in MCF7/Adr cells.
Progression through the cell cycle is controlled by the activity of cyclin-dependent kinases (CDKs), which in turn is regulated by cyclins, CDK-inhibitory proteins (CKIs), and changes in the phosphorylation status of the CDKs. The activity of cyclin/CDK complexes is negatively regulated by the CKIs, which are grouped into two structurally related families. Of these, p21 and p27 have been most extensively studied. p21 acts downstream of numerous signaling pathways, and can be induced by p53, transforming growth factor-ß (TGF-ß), and other anti-mitogenic stimuli (26,27). In tissue culture systems, p21 is up-regulated in proliferating cells and appears to play a role in governing progression through the cell cycle (28). p27 is constitutively expressed in many cells, and may play an important role in governing the growth factor restriction point by ensuring CDK activity suppression during G0 and early G1 phase. In addition, p27 has been implicated as a mediator of growth arrest due to TGF-ß, cAMP, and other extra-cellular factors, and can be regulated by UCH-L1 (29-31). In order to further confirm our findings that the role of UCH-L1 on growth suppression was dependent on the inducement of apoptosis in breast cancer cells, we measured p21 and p27 levels in UCH-L1 over-expressing MCF7 cells and UCH-L1 silencing MCF7/Adr cells. The results revealed that p21 and p27 changed contrarily in both cell types, thus, we deduced that this might be the cause of the unchanged cell cycle in these cells when compared with their corresponding negative controls.
In addition, we demonstrated that elevated UCH-L1 expression in MCF7 cells blocked phospho-Akt and phospho-IκB-α expression, while knockdown of UCH-L1 in MCF7/Adr cells led to the opposite effect. The PI3K-Akt pathway acts as a survival (anti-apoptotic) signal and plays a key role in the regulation of apoptotic change in breast cancer cells. Akt is a downstream mediator of PI3K and the growth factor receptor (GFR) stimulation of Akt has been shown to be dependent on PI3K activity. Akt can exert its anti-apoptotic effects in several different ways, such as negatively regulating pro-apoptotic factors, Bad and pro-caspase-9, stimulating the NFκB survival pathway and suppressing p53 pro-apoptotic signaling by phosphorylation of the oncogene Mdm-2 (17,18,32,33). Activation of cytoplasmic NFκB requires the degradation of an inhibitor protein, IκB, which traps NFκB in the cytoplasm. In response to signals, IκB is phosphorylated on two Ser residues in its NH2-terminal regulatory domain, Ser32 and Ser36, and degraded (34). Our data suggest that PI3K/Akt pathway is possibly involved in apoptosis in breast cancer cells and blocked by UCH-L1.
In summary, the present data provide evidence for the first time that UCH-L1 induces apoptosis in cultured breast cancer cells in vitro and the apoptotic effect is possibly through the PI3K/Akt pathway. Further studies are needed to elucidate the mechanism of UCH-L1 induced apoptosis in detail.
Acknowledgments
This research was supported by Funds of the Shanghai Board of Health (No. 044082), Funds of the Shanghai Scientific Association (No. 05ZR14023) and Funds of the National Institutes of Health/National Cancer Institute (No. CA 66077 and No. CA109371). We thank members in our laboratory for helpful discussion.
Abbreviations
- UCH-L1
ubiquitin carboxy terminal hydrolase-L1
- MDR
multi-drug resistance
- PI3K
phosphoinositide 3-kinase
- DUBs
deubiquitinating enzymes
- gad
gracile axonal dystrophy
- USPs
ubiquitin-specific processing proteases
- ER
estrogen receptor
- PR
progesterone receptor
- CDKs
CKIs, cyclin-dependent kinases
- CDK
inhibitory proteins
- TGF-ß
transforming growth factor-ß
- GFR
growth factor receptor
References
- 1.Hershko A, Ciechanover A, Varshavsky A. Basic medical research award. The ubiquitin system. Nat Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 2.Nijman SM, Luna-Vargas MP, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Kwon J, Wang YL, Setsuie R, et al. Developmental regulation of ubiquitin C-terminal hydrolase isozyme expression during spermatogenesis in mice. Biol Reprod. 2004;71:515–521. doi: 10.1095/biolreprod.104.027565. [DOI] [PubMed] [Google Scholar]
- 4.Sekiguchi S, Yoshikawa Y, Tanaka S, et al. Immunohistochemical analysis of protein gene product 9.5, a ubiquitin carboxyl-terminal hydrolase, during placental and embryonic development in the mouse. Exp Anim. 2003;52:365–369. doi: 10.1538/expanim.52.365. [DOI] [PubMed] [Google Scholar]
- 5.Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246:670–673. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- 6.Harada T, Harada C, Wang YL, et al. Role of ubiquitin carboxy terminal hydrolase-L1 in neural cell apoptosis induced by ischemic retinal injury in vivo. Am J Pathol. 2004;164:59–64. doi: 10.1016/S0002-9440(10)63096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon J, Wang YL, Setsuie R, et al. Two closely related ubiquitin C-terminal hydrolase isozymes function as reciprocal modulators of germ cell apoptosis in cryptorchid testis. Am J Pathol. 2004;165:1367–1374. doi: 10.1016/S0002-9440(10)63394-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang YL, Liu W, Sun YJ, et al. Overexpression of ubiquitin carboxyl-terminal hydrolase L1 arrests spermatogenesis in transgenic mice. Mol Reprod Dev. 2006;73:40–49. doi: 10.1002/mrd.20364. [DOI] [PubMed] [Google Scholar]
- 9.Miyoshi Y, Nakayama S, Torikoshi Y, et al. High expression of ubiquitin carboxy-terminal hydrolase-L1 and -L3 mRNA predicts early recurrence in patients with invasive breast cancer. Cancer Sci. 2006;97:523–529. doi: 10.1111/j.1349-7006.2006.00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamazaki T, Hibi K, Takase T, et al. PGP9.5 as a marker for invasive colorectal cancer. Clin Cancer Res. 2002;8:192–195. [PubMed] [Google Scholar]
- 11.Tezel E, Hibi K, Nagasaka T, Nakao A. PGP9.5 as a prognostic factor in pancreatic cancer. Clin Cancer Res. 2000;6:4764–4767. [PubMed] [Google Scholar]
- 12.Yang JM. Emerging roles of deubiquitinating enzymes in human cancer. Acta Pharmacol Sin. 2007;28:1325–1330. doi: 10.1111/j.1745-7254.2007.00687.x. [DOI] [PubMed] [Google Scholar]
- 13.Toker A. Protein kinases as mediators of phosphoinositide 3-kinase signaling. Mol Pharmacol. 2000;57:652–658. [PubMed] [Google Scholar]
- 14.Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 15.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 16.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 17.Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 19.Isakoff SJ, Engelman JA, Irie HY, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 21.Li QQ, Wang WJ, Xu JD, et al. Involvement of CD147 in regulation of multidrug resistance to P-gp substrate drugs and in vitro invasion in breast cancer cells. Cancer Sci. 2007;98:1064–1069. doi: 10.1111/j.1349-7006.2007.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li QQ, Wang WJ, Xu JD, et al. Up-regulation of CD147 and matrix metalloproteinase-2, -9 induced by P-glycoprotein substrates in multidrug resistant breast cancer cells. Cancer Sci. 2007;98:1767–1774. doi: 10.1111/j.1349-7006.2007.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Yu X. Regulation of apoptosis: the ubiquitous way. FASEB J. 2003;17:790–799. doi: 10.1096/fj.02-0654rev. [DOI] [PubMed] [Google Scholar]
- 24.Maki CG, Howley PM. Ubiquitination of p53 and p21 are differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osaka H, Wang YL, Takada K, et al. Ubiquitin carboxy terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12:1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 26.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esposito F, Cuccovillo F, Vanoni M, et al. Redox-mediated regulation of p21Waf1/Cip1 expression involves a post-transcriptional mechanism and activation of the mitogen-activated protein kinase pathway. Eur J Biochem. 1997;245:730–737. doi: 10.1111/j.1432-1033.1997.00730.x. [DOI] [PubMed] [Google Scholar]
- 28.Macleod KF, Sherry N, Hannon G, et al. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 29.Polyak K, Kato JY, Solomon MJ, et al. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 30.Kato JY, Matsuoka M, Polyak K, Massagué J, Sherr CJ. Cyclin AMP-induced G1 phase arrest mediated by an inhibitor (p27kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79:487–496. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 31.Caballero OL, Resto V, Patturajan M, et al. Interaction and colocalization of PGP9.5 with JAB1 and P27 (Kip1) Oncogene. 2002;21:3003–3010. doi: 10.1038/sj.onc.1205390. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 33.Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–467. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- 34.Brockman JA, Scherer DC, McKinsey TA, et al. Coupling of a signal response domain in IκBα to multiple pathways for NFκB activation. Mol Cell Biol. 1995;15:2809–2818. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]