

Abstract
Reelin controls neuronal positioning in the developing brain by binding to the two lipoprotein receptors, very-low-density lipoprotein receptor and apolipoprotein E receptor 2, to stimulate phosphorylation of Disabled-1 (Dab1) by the Fyn and Src tyrosine kinases. Crk and Crk-like (CrkL) have been proposed to interact with tyrosine phosphorylated Dab1 to mediate downstream events in the Reelin pathway. However, these adaptor proteins are widely expressed, and they fulfill essential functions during embryonic development. To address their specific roles in Reelin-mediated neuronal migration, we generated mutant mice, by Cre-loxP recombination, lacking Crk and CrkL in most neurons. These animals displayed the major anatomic features of reeler including, cerebellar hypofoliation, failure of Purkinje cell migration, absence of preplate splitting, impaired dendritic development, and disruption of layer formation in the hippocampus and cerebral cortex. However, proximal signaling involving tyrosine phosphorylation and turnover of Dab1 occurred normally in the mutant mouse brain and in primary cortical neurons treated with Reelin. In contrast, two downstream signaling events, Reelin-induced phosphorylation of C3G and Akt, were not observed in the absence of Crk and CrkL in mouse embryonic cortical neurons. These findings place C3G and Akt phosphorylation downstream of Crk and CrkL, which play essential overlapping functions in the Reelin signaling pathway.
Keywords: Crk, CrkL, Reelin, Dab1, neuronal migration, C3G, Akt
Introduction
Layer formation in the cerebral cortex, cerebellum, and hippocampus, which requires coordination of neurogenesis, neuronal migration, and cell positioning, is critically dependent on the Reelin pathway (for review, see Rice and Curran 2001; Ayala et al., 2007). The reeler mutation was first reported in mice (Falconer, 1951), but it was not until much later that the gene responsible for the widespread lamination defects characteristic of the reeler brain was identified (Miao et al., 1994; D'Arcangelo et al., 1995). The discovery of reelin triggered a rapid succession of studies, involving both spontaneous and genetically engineered mutant strains of mice, that elaborated many details of the Reelin signaling pathway. Reelin is an extracellular protein secreted by several neuronal populations, that binds to the very-low-density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (apoER2), to induce tyrosine phosphorylation of Disabled-1 (Dab1) by the Src family tyrosine kinases Fyn and Src (Keshvara et al., 2001; Arnaud et al., 2003a; Bock and Herz, 2003; Kuo et al., 2005). Subsequently, Dab1 undergoes ubiquitin-dependent degradation (Rice et al., 1998; Arnaud et al., 2003b; Bock et al., 2004). Tyrosine phosphorylated Dab1 associates with several adaptor proteins including Nck, Crk and Crk-like (CrkL) (Pramatarova et al., 2003; Ballif et al., 2004; Chen et al., 2004; Huang et al., 2004). Although tyrosine phosphorylation of Dab1 is a key event in Reelin signaling (Howell et al., 2000) and its biochemical interactions with Crk and CrkL have been demonstrated, the functional contributions of Crk and CrkL to Reelin signaling in vivo are unclear.
Crk and CrkL are expressed ubiquitously and they have been implicated in diverse biological processes including cell migration and adhesion (for review, see Feller, 2001). Crk family proteins relay signals from receptor tyrosine kinases, and nonreceptor tyrosine kinase-coupled receptors, to various downstream effectors by protein–protein interactions via their SH2 and SH3 domains. Crk-null embryos exhibit defects in cardiovascular and craniofacial development, but they die during embryonic development making analysis of brain structures difficult (Park et al., 2006). CrkL-null embryos exhibit lethal defects in cardiac and neural crest development (Guris et al., 2001), precluding analysis of mature brain structures. However, there is a failure of preganglionic neuronal migration in the spinal cord mice lacking CrkL, but this defect is distinct from that seen in reeler (Yip et al., 2007). To analyze the tissue-specific contributions of Crk and CrkL to Reelin signaling, while avoiding the confounding effect of embryonic lethality, we generated floxed alleles for Crk and CrkL and crossed them with a Nestin-Cre transgene (Knoepfler et al., 2002) to obtain conditional mutations in Crk and CrkL in mouse neurons. Double conditional knock-out mice (dcKO) exhibited strikingly similar anatomical defects to those observed in reeler mice. However, in contrast to reeler and in mice lacking both Reelin receptors, tyrosine phosphorylation of Dab1 was not reduced and it's steady-state level was not elevated in dcKO embryonic brain. These results provide compelling evidence that Crk and CrkL fulfill overlapping functions downstream of Dab1 in the Reelin pathway.
Materials and Methods
Construction of the targeting vector and generation of CrkL floxed mice.
Mouse embryonic stem (ES) cells carrying a floxed allele of CrkL were generated as described previously (Park et al., 2006). CrkL genomic DNA was obtained by screening CJ7/129SV CITB, Mouse BAC DNA pools (Invitrogen) using a 422-bp probe, CrkL-U1E4 by Southern blotting. CrkL-U1E4 contains 298 bp of the first exon of CrkL and the adjacent 124 bp of the 5′ upstream region, and was amplified using CrkL-U1E4F (5′-TTTGTTGCTGTGGCTATTGG-3′) and CrkL-U1E4R (5′-TCTTGCTTGGGAAGCCTTTA-3′) primers, and genomic DNA as a template. A 10.4 kb BstBI-ClaI genomic fragment containing the first exon of CrkL was isolated and subcloned into the ClaI site of the pBluescript II KS- vector. Then a 52 bp oligo containing a loxP and a 2.8 kb neomycin resistance–thymidine kinase (NeoTK) cassette flanked by loxP sites were sequentially inserted into the AatII and PacI restriction enzyme sites, respectively. The gene-targeting vector was linearized with XhoI and electroporated into 129/SvEv mouse ES cells (Specialty Media) according to the manufacturer's instruction. Genomic DNA was isolated from ES cells and digested with either XbaI or BsmI, and probed with CrkL-5U7 (Fig. 1 A, thick blue bar) and CrkL-3DS21 (Fig. 1 A, thick red bar), respectively. CrkL-5U7 was amplified using CrkL-5U7F (5′-GCATTGTCCTGACTGGGAGT-3′) and CrkL-5U7R (5′-TCAGTTTCTCGCATCATTGC-3′). CrkL-3DS21 was amplified using CrkL-3DS2F (5′-CAAACCTGGGATCCTTTCAA-3′) and CrkL-3DS1R (5′-CTTCCAACTCCAAGCTGAGG-3′).
Figure 1.

Targeted disruption of Crk and CrkL (dcKO) in mice. A, Schematic diagram of WT and homologous recombinant (floxed) alleles of CrkL. Filled triangles represent loxP sites. The relevant restriction sites XbaI (X), BstBI (BB), AatI (A), PacI (P), ClaI (C), and BsmI (B) are indicated. Colored bars indicate the locations of Southern probes (thick) and DNA fragments (thin) detected by the probes. B, Southern blot analysis of CrkL floxed mice. Genomic DNA from mouse tail was digested with XbaI and BsmI and hybridized with 5′- and 3′-external CrkL probes, respectively. C, Growth curves of dcKO mice and WT littermates. Body weight (in grams) of mice was measured two or three times a week, and data from females (27 dcKO and 35 WT are shown here) and males (data not shown) were analyzed separately, and revealed similar trends. Each point represents the mean ± SEM. D, Photograph of a 4-week-old dcKO mouse and a WT littermate. E, Image of perfused brains from a 4-week-old dcKO mouse and a wild-type littermate. Note the cerebellum is much smaller in dcKO than WT (see arrow). Scale bars: D, 1 cm; E, 0.5 cm.
Initially, Cre excision was performed in ES cells harboring the CrkL homologous recombinant allele and we obtained CrkL floxed ES cells without NeoTK. However, we failed to get chimeric mice from the ES cells. Therefore, we did not select for loss of NeoTK before injecting blastocysts to avoid the risk of genetic modifications occurring in ES cells during the process of Cre excision. Although selection markers such as neomycin and thymidine kinase were still present in the CrkL homologous recombinant allele, they were located in an intron following the first exon and were removed successfully by the Cre excision in the initial ES cell screen. Since the presence of the NeoTK selection markers in the intron following the first exon did not disturb expression of CrkL (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we considered the CrkL homologous recombinant allele as a CrkL floxed allele. C57BL/6 × 129 chimeras were obtained and one of the chimeras exhibited germline transmission, which was confirmed by PCR and Southern blot analysis. To genotype CrkL alleles by PCR, the following four primers were used: LGT1 (5′- AGGGTGAGGCGACTTCATAA-3′), LGT2 (5′-TGATTTACCAGGGCCTTCTG-3′), LGT3 (5′-GGAGAGGCTTTTTGCTTCCT-3′), and LGT4 (5′-CCACCCCACCTTCATTATTC-3′). The combination of LGT1 and LGT4 primers amplifies the wild-type allele with a 132 bp product. Combination of LGT2 and LGT4 primers amplifies the null allele with a 418 bp product. Combination of LGT3 and LGT4 primers amplifies the floxed allele with a 241 bp product. Nestin-Cre transgenic mice (Knoepfler et al., 2002) were purchased from Jackson laboratory, and they were bred with floxed Crk and CrkL mice. Mouse colonies were maintained on a mixed background of C57BL/6 and 129SvEv. When dcKO pups exhibited an obvious decrease in body weight, ∼3–4 weeks of age, they were provided with water gel (Napa Nectar, SE. Lab Group). All mouse studies were performed according to protocols approved by the Institutional Animal Care and Use Committee at the Children's Hospital of Philadelphia.
Histology and immunohistochemistry.
Embryos at different stages were dissected from dams and examined for normal heartbeat and blood flow. Embryos were fixed overnight in 4% paraformaldehyde at 4°C. Fixed embryos were embedded in paraffin and sectioned at a thickness of 5 μm. Every tenth section was stained with hematoxylin and eosin (H&E), and sections from similar anatomical planes were chosen for images and further immunohistochemical analyses. For immunohistochemical staining, sections were processed through deparaffinization and rehydration followed by antigen retrieval in 10 mm sodium citrate, pH 6.0, for 30 min at 95°C, as described previously (Romer et al., 2004). Slides were treated with 1.5–3% H2O2, blocked in PBS supplemented with 3.5% bovine serum albumin, and incubated with anti-Crk (BD PharMingen), anti-CrkL (Santa Cruz, C-20), anti-Dab1 (B3 from Jonathan Cooper, Fred Hutchinson Cancer Research Center, Seattle, WA), anti-Map2 (Sternberger, SMI-151), anti-Calbindin (Sigma, CB-955), and anti-BrdU (Serotec, MCA2060) overnight at room temperature. Immunoreactivity was detected using the Vectastain Elite ABC kit (Vector Laboratories) and the diaminobenzidine reagent set (Kirkegaard and Perry Laboratories) according to the manufacturers' instructions. Low magnification images of fixed tissue samples and sections were captured using an Olympus MVX10 macroscope with a DP71 digital color camera. High magnification images of sections were captured using a Nikon 90i microscope equipped with Roper EZ monochrome and DS-Fi1 color cameras. Images were imported into Adobe Photoshop CS for analysis.
Purkinje cells were counted in anti-Calbindin-stained slides from 4-week-old dcKO and wild-type (WT) littermate brains as described previously (Goldowitz et al., 1997) with some modifications. All Calbindin-positive cells with a distinctive circular shape were counted in every 20th 5 μm sagittal section starting from the midline throughout one-half of the cerebellum. In mutant brains, the percentages of misplaced Purkinje cells were calculated using counts from the first five sections, and their average values are presented. In WT brains, no misplaced Purkinje cells were observed.
BrdU birthdating analysis.
The thymidine analog, BrdU, was injected into pregnant female mice as described previously (Rice et al., 1998; Magdaleno et al., 2002). BrdU (Sigma) was injected at embryonic day (E) 11.5 by intraperitoneal injections at a concentration of 50 μg/g of body weight to label subplate neurons. At E17.5, embryos were fixed and processed as described above. BrdU-labeled cells were detected by immunohistochemical analysis.
Preparation of embryonic tissue extracts and primary cortical neurons.
Embryonic yolk sacs were used to prepare genomic DNA for genotyping. Cerebral cortical hemispheres were dissected at E15.5 to prepare total protein extract. For primary cortical neuron cultures, minced cortical hemispheres were digested with trypsin-EDTA, triturated, and plated onto Biocoat ploy-d-lysine-coated dishes (Becton Dickinson) in Neurobasal medium supplemented with B-27 (Invitrogen). After 5 d, neurons were treated with purified Reelin (Benhayon et al., 2003) or control medium (mock) for 15 min to observe stimulation of signaling molecules or for 3 h to investigate degradation of Dab1 at 37°C, and then lysed on ice.
Western blot analysis.
Lysates of E15.5 cortical hemispheres were prepared using 1% NP-40 lysis buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 25 mm NaF, 2 mm Na3VO4, 0.1% sodium deoxycholate, protease inhibitor mixture complete mini (Roche), and phosphatase inhibitor mixture PhosSTOP (Roche). After quantifying the protein content of each cell lysate using Bio-Rad protein assay kit, total cell lysates containing the same amount of protein were loaded, separated by SDS-PAGE, and transferred to nitrocellulose membranes (Invitrogen) (Keshvara et al., 2002). Membranes were then subjected to Western blot analysis using anti-Crk (BD PharMingen), anti-CrkL (Santa Cruz, s.c.-319), and anti-Dab1 (CT38 rabbit polyclonal), anti-C3G (Santa Cruz, s.c.-869), anti-phospho-Akt (serine 473; BioSource), anti-Akt (Cell Signaling), and anti-PLCγ1 (Santa Cruz Biotechnology) antibodies. Immunodetection was performed using the WestDura chemiluminescence kit (Pierce). For quantitative analyses of Western blotting results, x-ray films were scanned and densitometric analysis was performed using NIH ImageJ. To determine the quantitative effect of Reelin on protein phosphorylation of Dab1, C3G, and Akt, we calculated the ratio of phosphorylated protein to total protein in each case and normalized these values to mock-treated neuronal extracts. Quantitation of Dab1 degradation was determined by calculating the ratio of Dab1 to PLCγ1 with normalization to mock-treated neuronal extracts. These ratios are presented as fold changes comparing mock treatment to Reelin treatment in the text.
Results
Generation of mice with conditional mutations in Crk and CrkL
Generation of a floxed allele of Crk (Crk +/FL) was described previously (Park et al., 2006). A similar strategy was adopted to generate a floxed allele of CrkL (CrkL +/FL). As shown in Figure 1 A, the CrkL targeting construct contained the first exon of CrkL, flanked by two loxP sites, followed by a NeoTK gene and another loxP site. Southern hybridization analysis of mouse genomic DNA (Fig. 1 B) showed that both the wild-type and floxed alleles were present in CrkL +/FL mice, whereas the wild-type allele was absent in CrkLFL/FL mice. We also confirmed that homozygous floxed Crk or CrkL mice, or both together expressed normal levels of Crk and CrkL proteins by Western analysis (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The Nestin-Cre transgene (Knoepfler et al., 2002) was introduced into all three lines to ablate Crk and CrkL, individually and together, from the developing nervous system. Loss of Crk or CrkL alone in the neuronal progenitor cells did not result in any obvious neuroanatomic abnormalities in the cerebellum, hippocampus or cerebral cortex (supplemental Figs. 2, 3, available at www.jneurosci.org as supplemental material). However, both CrkFL/FL;Nestin-Cre and CrkLFL/FL;Nestin-Cre mice were ∼23% smaller than wild-type mice based on body weight. Growth retardation in these cKO mice may reflect impaired mitogenic signaling by insulin-like growth factor-I (IGF-I) since Crk family proteins have been demonstrated to be involved in IGF-I signaling (Beitner-Johnson et al., 1996; Butler et al., 1997). In addition, dcKO mice suffered severe growth retardation, with a significant (39–59%) decrease in body weight detected at 2–4 weeks of age, and they required water gel for survival (Fig. 1 C). The dcKO mice that survived were 15–29% smaller than wild-type mice (Fig. 1 D), and their brains were grossly abnormal in appearance, particularly the cerebellum (Fig. 1 E, arrow).
Defective cell migration and foliation in dcKO cerebellum
Immunostaining analysis of young adult mouse brain was carried out, using anti-Crk and anti-CrkL antibodies, to confirm neural-specific deletion of Crk and CrkL induced by Nestin-Cre. In normal mouse cerebellum, high levels of Crk expression were observed in the internal granule cell layer whereas weaker staining was observed in the molecular layer (Fig. 2 A,C). In contrast, Crk was completely absent in the dcKO cerebellum (Fig. 2 B,D). CrkL was expressed in normal cerebellum, with particularly strong staining in Purkinje cells compared with weaker staining in the granule cell and molecular layers (Fig. 2 E,G). In the dcKO cerebellum CrkL was dramatically reduced, although there was some weak residual immunoreactivity (Fig. 2 F,H). These results suggest that Crk and CrkL are expressed in the normal mouse cerebellum, but are significantly reduced in dcKO mice.
Figure 2.

Histological analysis of WT and dcKO cerebellum from 4-week-old mice. A–D, Crk immunostaining of low (A, B) and high (C, D) resolution images of WT (A, C) and dcKO (B, D). E–H, CrkL immunostaining of low (E, F) and high (G, H) resolution images of WT (E, G) and dcKO (F, H). I–L, H&E staining of low (I, J) and high (K, L) resolution images of WT (I, K) and dcKO (J, L). Ectopic granule cells are indicated as yellow arrows. Red arrow indicates boundary between the igl and ml layers is not clear. M–P, Calbindin immunostaining of low (M, N) and high (O, P) resolution images of WT (M, O) and dcKO (N, P). Double-headed arrows indicate thickness of the molecular layers. Black and blue arrows indicate Purkinje cells that failed to migrate and those that partially migrated, respectively. The red arrow indicates Purkinje cells that reached their final destination. ml, Molecular layer; igl, internal granule cell layer; e-gl, ectopic granule cell layer. Scale bars: A, E, I, M, 500 μm; C, G, K, O, 50 μm.
The dcKO cerebellum had a reduced structure, with poorly developed foliation and layer formation, compared with normal cerebellum (Fig. 2 I,J). Boundaries between layers were not clear and granule cells were often scattered, particularly in the subcortical region (Fig. 2 J, red arrow). In addition, several clusters of ectopic granule cells were observed outside of the internal granule cell layer (Fig. 2 J,L, yellow arrows), but not in normal cerebellum (Fig. 2 K). Purkinje cell migration, which fails to occur in reeler mice, was significantly reduced in the absence of Crk and CrkL. Purkinje cell bodies, identified by calbindin immunohistochemistry, align in a single layer underneath the molecular layer in normal cerebellum (Fig. 2 M), with their highly developed dendritic arbors extending into the molecular layer (Fig. 2 O). Only a small subset (∼29%) of Purkinje cells reached their final destination to extend shorter, less elaborate, dendrites into the reduced molecular layer of dcKO mice (Fig. 2 N,P, red arrow; also compare double-sided arrows in Fig. 2 O,P for the thickness of the molecular layer). The majority of Purkinje cells either failed to migrate (∼41%) (Fig. 2 N, black arrow), or migrated partially (∼31%) (Fig. 2 P, blue arrow), and their neuronal processes remained poorly developed and randomly oriented. These results indicate that Crk and CrkL play important roles in cerebellar development, particular in the migration and dendritic development of Purkinje cells. These defects are consistent, but less extensive, than those observed in reeler.
Disruption of hippocampus in dcKO mice
Strong expression of Crk was observed in both pyramidal and granule neurons of wild-type hippocampus, whereas CrkL was most prevalent in pyramidal cells (Fig. 3 A,E). In pyramidal neurons Crk and CrkL were mainly expressed in the cytoplasm, but not in the nucleus (Fig. 3 C,G). In the dcKO hippocampus Crk expression was completely missing (Fig. 3 B,D). Expression of CrkL was also decreased dramatically in the dcKO hippocampus (Fig. 3 F,H), but some cells, including pyramidal neurons in the CA3 region of dcKO mice, retained mild CrkL expression (Fig. 3 F, arrow). The reduced efficiency of CrkL deletion, compared with that of Crk, in the dcKO hippocampus may reflect the fact that the CrkL floxed allele carries three loxP sites, since the NeoTK gene is present, whereas only two loxP sites, flanking the first exon, are present in the Crk floxed allele. It is unclear why CrkL immunoreactivity was retained specifically in the dcKO CA3 region.
Figure 3.

Histological analysis of WT and dcKO hippocampus from 4-week-old mice. A–D, Crk immunostaining of low (A, B) and high (C, D for CA1) resolution images of WT (A, C) and dcKO (B, D). E–H, CrkL immunostaining of low (E, F) and high (G, H for CA1) resolution images of WT (E, G) and dcKO (F, H). Arrow in F indicates scattered CrkL expression in the CA3 region of dcKO mice. I–N, H&E staining of low (I, J) and high (K, L for CA1; M, N for dentate gyrus) resolution images of WT (I, K, M) and dcKO (J, L, N). O, P, Ki67 immunostaining with hematoxylin (H) counterstaining of WT (O) and dcKO (P). Proliferating subgranule cells in the dentate gyrus are indicated as yellow arrows (M–P). Q–T, Map2 immunostaining of low (Q, R) and high (S, T for CA1) resolution images of WT (Q, S) and dcKO (R, T). spb, Suprapyramidal blade; ipb, infrapyramidal blade; h, hilus; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; gl, granule cell layer; sgl, subgranular cell layer. Scale bars: A, E, I, 500 μm; O, Q, 200 μm; C, G, K, S, 50 μm.
In dcKO mice, pyramidal and granule neurons of the hippocampus were dispersed compared with the well developed, compact neuronal layers typical of a normal brain (compare Fig. 3 I,J). The dcKO pyramidal cell layer in the CA1 region was split and neurons were scattered with poor discrimination of the stratum oriens, stratum pyramidale, and stratum radiatum by H&E staining, compared with normal hippocampus (compare Fig. 3 K,L). Similarly, the dentate gyrus was diffuse in dcKO mice and the distinction between the suprapyramidal blade, hilus and infrapyramidal blade all but disappeared (compare Fig. 3 I,J). Proliferating subgranule cells, normally located adjacent to the hilar region in the dentate gyrus, stained with Ki67 (Fig. 3 M,O) were distributed throughout the dcKO dentate gyrus (Fig. 3 N,P, arrows).
In addition to disruptions in cell positioning, Map2 staining revealed defective development of dendrites in the dcKO hippocampus. In the normal hippocampus, the organization of cell bodies and dendrites is clearly segregated (Fig. 3 Q). Furthermore, dendrites located between the CA1 and dentate gyrus are well developed and arrayed in a radial orientation (Fig. 3 S). In contrast, cell bodies were scattered in the dcKO hippocampus and interspersed among the dendrites (Fig. 3 R), which were shorter than their wild-type counterparts and displayed abnormal orientation (Fig. 3 T). These results suggest that the defects observed in the dcKO hippocampus closely resemble those of reeler and that Crk and CrkL are required for layer formation and dendritic development in the hippocampus.
Defective lamination in dcKO cerebral cortex
The classic lamination defect associated with reeler, results from the failure of migrating neurons to split the preplate into the marginal and the subplate zones, causing disruption and apparent inversion of cortical layers (Rice and Curran, 1999). Crk and CrkL (chopbgem id = 401 and chopbgem id = 402 in http://www.chopbgem.org) are present in the ventricular zone before neuronal migration and expression persists in all cortical layers after migration is complete (Fig. 4 A,C). Expression of both Crk and CrkL was reduced dramatically in dcKO cortex with weak Crk immunoreactivity detected only in the pia and in a few cells distributed throughout the cortex (Fig. 4 B), although CrkL expression was high in a small number of scattered cells throughout the cortex and weak in the region beneath the pia (Fig. 4 D). Histology revealed that the overall organization of the cerebral cortex was abnormal in dcKO cortex (compare Fig. 4 E,F). There was no obvious laminar structure and cells appeared to be distributed randomly. The marginal zone, clearly visible in the normal cortex (Fig. 4 G, double-headed arrow), was populated with cell bodies in the dcKO cortex resembling the superplate structure described in reeler (Fig. 4 H). Map2 staining confirmed the presence of neuronal cell bodies below the pial surface in the dcKO cortex, but not in the normal cortex (Fig. 4 J,I, arrow). Apical dendrites of pyramidal neurons, stained with anti-Map2 antibody, that normally align in the radial direction most notably in layers III and IV (Fig. 4 K, arrows) were shorter and heterogeneous in orientation in the dcKO cortex (Fig. 4 L, black and blue arrows). To investigate cortical layer formation, we used anti-calbindin antibodies to identify neurons located predominantly in layers II and III, as well as interneurons dispersed throughout the cortex (Hof et al., 1999). In contrast to the predominant staining of superficial neurons in layers II/III of normal cortex (Fig. 4 M,O, double-headed arrow), calbindin immunoreactivity predominated in deeper layers of the dcKO cortex, suggesting layer inversion (Fig. 4 N,P). However, the scattered pattern of interneuron staining was unaffected in dcKO mice (Fig. 4 M,N, arrows). These findings parallel the situation in reeler mice, in which defective radial migration results in an apparent inversion of cortical layers whereas tangential migration of interneurons is unaffected (Hevner et al., 2004; Britanova et al., 2006).
Figure 4.

Histological analysis of WT and dcKO cerebral cortex from 4-week-old mice. A–D, Immunostaining with anti-Crk (A, B) and anti-CrkL (C, D). E–H, H&E staining. High-resolution images of E (WT) and F (dcKO) are shown in G and H, respectively. The marginal zone (mz) is indicated by a yellow double-sided arrow. I–L, Map2 immunostaining. Neuronal cell bodies under the marginal zone or under the pial surface are indicated by black arrows in I and J. High-resolution images of I and J are shown in K and L, respectively, and apical dendrites of pyramidal neurons are marked depending on their orientation (radial, black arrows; random, blue arrows). M–P, Calbindin immunostaining. Layer-specific predominant staining is indicated by double-sided arrows and staining of dispersed interneurons is indicated by black arrows. High-resolution images of M and N are shown in O and P, respectively. mz, Marginal zone; spp, superplate. Scale bars: A, C, E, I, M, 250 μm; G, K, O, 50 μm.
Failure of preplate splitting in dcKO embryonic cortex
During cortical development in reeler mice, the first wave of migrating neurons transits the intermediate zone, but fails to bypass subplate neurons. Although the leading edges of migrating neurons extend into the marginal zone the nucleus fails to translocate into the leading processes. This failure of nucleokinesis (also referred to as somal translocation) results in arrested migration below the preplate. Subsequent waves of migrating neurons fail to bypass their predecessors generating an apparently inverted, abnormal architectonic structure (for review, see Lambert de Rouvroit and Goffinet, 2001; Tissir and Goffinet, 2003). To investigate the origin of the cortical lamination defect in dcKO mice, we examined mouse embryonic cortices at E15.5, while neurons were undergoing radial migration. Crk was expressed in all the layers of embryonic cortex with particularly strong expression in the subplate and intermediate zone (Fig. 5 A), and it was dramatically decreased in the dcKO embryonic cortex, with only very weak expression in cells in the superficial layers (Fig. 5 B, double-headed arrow). CrkL was also expressed throughout the normal embryonic cortex. As shown in Figure 5 C, relatively strong expression of CrkL occurred in the intermediate zone and a superficial part of the cortical layer. However, CrkL expression decreased significantly in all the layers in dcKO embryonic cortex (Fig. 5 D). These immunohistochemical findings were confirmed by very low expression of Crk and CrkL in Western analysis of dcKO embryonic cortex (see Fig. 7 D). Therefore, our results demonstrate that Crk and CrkL had already been deleted significantly from migrating cortical neurons at E15.5.
Figure 5.

Reduced expression of Crk and CrkL in E15.5 dcKO cortex. Sagittal paraffin sections of embryonic cortex were immunostained with anti-Crk (A, B) and anti-CrkL (C, D) antibodies. Double-headed arrow in B indicates very weak expression of Crk in superficial layers. mz, Marginal zone; cp, cortical plate; sp, subplate; iz, intermediate zone; vz, ventricular zone. Scale bar, 100 μm.
Figure 7.

Reelin signaling pathway in E15.5 dcKO cortex and primary cortical neurons. A–C, Immunostaining of mouse E15.5 cortex of WT, dcKO, and reeler with anti-Dab1 antibody. Black vertical bars indicate layers of strong Dab1 immunoreactivity. Neuronal cell bodies located between the ventricular and intermediate zones are indicated as black arrows for comparison of their Dab1 immunoreactivity. Scale bar, 100 μm. D, Western blot analysis of E15.5 WT and dcKO cerebral cortical hemispheres with anti-Dab1, anti-Crk, and anti-CrkL antibodies. Anti-ShpII was used as a loading control. The ratio of Dab1 to ShpII was calculated and normalized to the ratio in WT cortical extract. E, F, Immunoprecipitation (IP) of extracts from cerebral cortical hemispheres from E15.5 dcKO (E) or reeler (F), and their WT embryos with anti-Dab1 antibody followed by blotting with anti-phosphotyrosine (pY-Dab1, 4G10) and anti-Dab1 antibodies. Both the ratio of Dab1 protein itself and the ratio of tyrosine phosphorylated Dab1 to total Dab1 protein were calculated and normalized to the ratio in WT cortical extract. G, H, Mouse embryonic cortical neurons were prepared from WT and dcKO embryos, cultured, and stimulated with either mock or purified Reelin for 15 min (G) to see stimulation of signaling molecules or for 3 h (H) to detect degradation of Dab1. Total protein extract was subjected to IP with anti-Dab1 and anti-C3G antibodies to detect tyrosine phosphorylation of Dab1 and C3G, respectively. Total protein extract was also subjected to blotting with antibodies against phospho-Akt (serine 473), Akt, Crk, CrkL, and phospholipase Cγ1 (PLCr1) as a loading control. The arrow in G indicates location of tyrosine phosphorylated C3G. I, Schematic diagram of the role of Crk and CrkL in the Reelin signaling pathway. We suggest the Reelin pathway consists of two subroutines, specific subroutine (Subroutine S) and general subroutine (Subroutine G). Molecules in Subroutine S include the pathway-specific molecules such as Reelin, Reelin receptors VLDLR and ApoER2, and Dab1, while Subroutine G consists of common molecules such as Crk, CrkL, Akt, and Rap1. We suggest that the two subroutines operate separately, but are coupled to each other by the interaction of tyrosine phosphorylated Dab1 (pY-Dab1) with Crk/CrkL. PI3K, Phosphatidylinositol 3-kinase; Rap1, repressor activator protein; rl, reeler.
Histology revealed a close similarity of the dcKO embryonic cerebral cortex to that of reeler (Fig. 6 A–C). In contrast to the cell-sparse marginal zone, clearly identifiable subplate, and laminar cortical plate characteristic of the normal embryonic cortex, it was difficult to distinguish distinct layers in both the dcKO and reeler cortices. In these cases, cells had infiltrated the marginal zone, the subplate was absent and a superplate structure had formed near the pial surface. Cortical neurons were not organized radially and there were several cell-free rifts in the cortical plate (Fig. 6 B,C, arrows). In contrast, the arrangement of cells in the ventricular zone was similar in wild-type, dcKO, and reeler cortices (Fig. 6 A–C). Map2 immunostaining confirmed the similarities between dcKO and reeler brains. In wild-type E15.5 embryos the cortical plate was filled with neurons and the marginal zone exhibited a high level of Map2. The subplate was readily distinguished by Map2 staining (Fig. 6 D, arrow). Although the superplate showed high Map2 immunoreactivity in dcKO and reeler mice, there was no clear-cut boundary such as the subplate between the cortical plate and the intermediate zone (Fig. 6 E,F). In addition, some neurons infiltrated the marginal zone forming heterotopic cell clusters (Fig. 6 E, arrow). To investigate the disappearance of subplate neurons both in dcKO and reeler mice, we used calbindin immunostaining. As reported previously (Del Río et al., 2000), calbindin-positive neurons are well separated into the subplate and the marginal zone of normal E15.5 cortex (Fig. 6 G). In contrast, calbindin-stained neurons were dispersed throughout the superplate and cortical plate of dcKO and reeler mice (Figs. 6 H,I, double-sided arrows), indicating that the preplate failed to split into the marginal zone and the subplate in both cases. It should be also noted that there are some differences between dcKO and reeler cortices. Map2 staining in the cortical plate appears to be more homogeneous and broad in reeler brain (Fig. 6 F). Calbindin staining in reeler cortex is weaker than in dcKO cortex (Fig. 6 I). Nevertheless, the general organization of cortical layers between dcKO and reeler mice is very similar.
Figure 6.
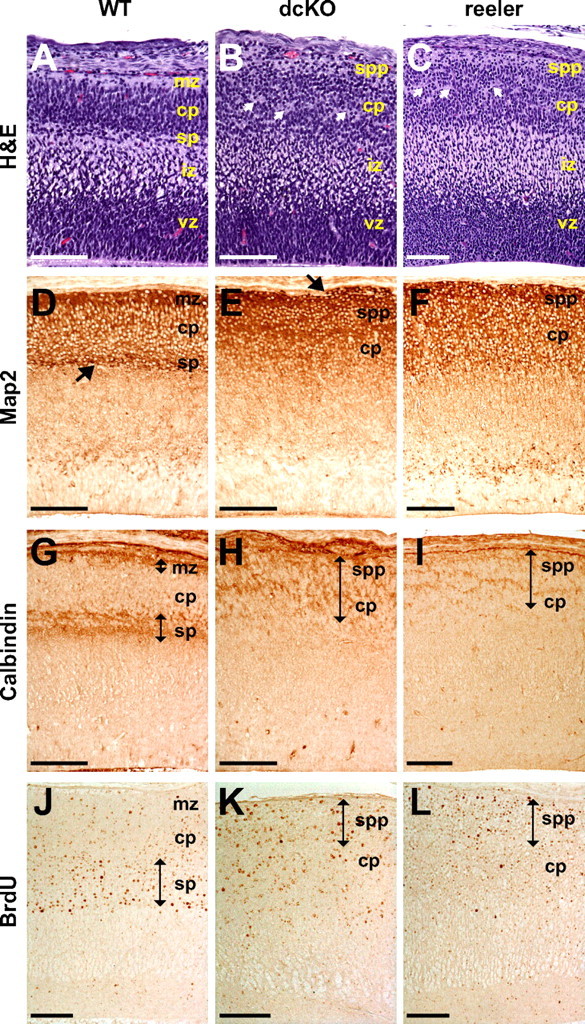
Histological analysis of E15.5 dcKO cortex. A–I, Every tenth sagittal paraffin section of whole embryos was stained with H&E (A–C), and representative images of the cortex were taken from the sections. Matching sections were processed for immunostaining. White arrows in B and C indicate aberrant cell-free rifts. Black arrow in D indicates subplate stained with anti-Map2 antibody, while black arrow in E indicates a heterotopia formed in the marginal zone of dcKO cortex. Black double-sided arrows in G–I indicate layers of strong calbindin immunoreactivity. J–L, Birthdating analysis. BrdU was administered to pregnant females at E11.5 to label subplate neurons. Locations of the cell populations were analyzed at E17.5. Black double-sided arrows in J–L indicate layers of strong BrdU immunoreactivity. mz, Marginal zone; cp, cortical plate; sp, subplate; iz, intermediate zone; vz, ventricular zone; spp, superplate. Scale bar, 100 μm.
Similar results were obtained using BrdU-injected pregnant female mice carrying E11.5 embryos. As expected, normal embryos collected at E17.5 contained strongly BrdU-positive cells in the subplate (Fig. 6 J, double-sided arrow), and some cells in the cortical plate showed a lower level of BrdU immunoreactivity as a consequence of labeling progenitor cells that continued to divide. In dcKO and reeler mice, however, cells in the superplate exhibited the highest level of BrdU staining (Fig. 6 K,L, double-sided arrow), probably because later born neurons could not bypass the earlier born neurons. Together, these results demonstrate that the preplate failed to spilt into the marginal zone and the subplate in embryos lacking Crk and CrkL. Furthermore, subplate neurons were intermingled with cortical plate neurons in a superficial superplate structure. Thus, Crk and CrkL are essential for layer formation in the developing mouse cortex and their absence results in defects similar to those observed in reeler.
Normal Dab1 turnover and tyrosine phosphorylation in the absence of Crk and CrkL
Dab1 accumulates in a hypophosphorylated form in the absence of Reelin signaling. In fact, displaced neurons can be identified, based on higher levels of Dab1 expression, in mice lacking Reelin, the VLDLR and ApoER2 receptors, and the Fyn and Src tyrosine kinases (Rice et al., 1998; Howell et al., 1999; Trommsdorff et al., 1999; Arnaud et al., 2003a; Bock et al., 2004). These findings led to the hypothesis that Dab1 is not functional in the absence of tyrosine phosphorylation and that it undergoes proteasome-mediated degradation after activation. The results described above imply that Crk and CrkL provide essential functions in the Reelin pathway, but they do not distinguish whether this effect occurs downstream of Dab1 or through some influence on Dab1 function. To address this question, we examined the status of Dab1 in the developing brain of dcKO mice.
In the normal E15.5 cortex, Dab1 is widely expressed in most neuronal populations and progenitors in the marginal zone, cortical plate, and the superficial portion of the intermediate zone (Fig. 7 A, vertical bar). Lower levels of Dab1 immunostaining are seen in the ventricular zone, while reduced levels are observed in the subplate and the proximal region of the intermediate zone (Fig. 7 A). In reeler mice, increased Dab1 staining was observed throughout the superplate, cortical plate and superficial region of the intermediate zone (Fig. 7 C). Although the distribution of Dab1-positive cells in the dcKO mice, throughout the superplate, cortical plate and superficial region of the intermediate zone, was rather similar to that in reeler, the intensity of Dab1 immunoreactivity was not noticeably increased compared with wild-type (Fig. 7 B, vertical bar). The intensity of Dab1 immunoreactivity in reeler was much higher than that in wild-type and dcKO cortices (Fig. 7 C, vertical bar). Many isolated neuronal cell bodies with very strong Dab1 were observed between the ventricular and intermediate zones of reeler, but not wild-type or dcKO mice (Fig. 7 A–C, arrows). The region showing the highest level of Dab1 immunoreactivity extended throughout the intermediate zone in reeler, but not in wild-type or dcKO mice (Fig. 7 A–C, vertical bars). Dab1 levels in ventricular zone cells were similar among the three genotypes, suggesting that Dab1 expression was elevated specifically in the superficial layers of reeler, but not in the dcKO cortex. Therefore, these results suggest that, in contrast to reeler, displaced neurons in dcKO mice express normal levels of Dab1. We also examined Reelin expression in the developing cortex of dcKO mice. Although the failure of preplate splitting, into the marginal zone and the subplate, resulted in redistribution of Reelin, the level of protein expression was similar to that in normal cortex (data not shown).
For a quantitative comparison of Dab1 levels in developing mouse brain, we performed Western blot analysis using total protein extracts prepared from E15.5 cortices. As shown in Figure 7 D, Crk and CrkL proteins were almost completely absent in the dcKO embryonic cortex, whereas the level of Dab1 was similar to that in wild-type mouse brain (the normalized ratios of Dab1 to ShpII in WT and dcKO were both 1.0). In contrast, Dab1 was present at a much higher level (13.5-fold) in E15.5 reeler cortex (Fig. 7 F). The overall level of tyrosine phosphorylation of Dab1 in dcKO embryonic cortex was similar to that in wild-type cortex (Fig. 7 E). In the case of reeler, the stoichiometry of Dab1 tyrosine phosphorylation was one tenth of that observed in wild-type brain (Fig. 7 F). These results demonstrate that Crk and CrkL do not affect the activation or degradation of Dab1 in vivo.
To confirm whether Dab1 is activated and degraded normally in the absence of Crk and CrkL upon Reelin stimulation of neurons, we isolated primary cortical neurons from dcKO mouse embryos. When cultured cortical neurons were stimulated with purified Reelin (Benhayon et al., 2003), Dab1 tyrosine phosphorylation increased 2.5-fold in wild-type and 2.4-fold in dcKO neurons (Fig. 7 G). Furthermore, continuous treatment with purified Reelin for 3 h resulted in a reduction in Dab1 levels of 0.5-fold in dcKO and 0.6-fold in normal cortical neurons (Fig. 7 H) although the basal level of Dab1 was lower in dcKO neurons. These results support the findings, obtained from analysis of dcKO cortical extracts, indicating that proximal Reelin signaling, including Dab1 tyrosine phosphorylation and proteolytic degradation, occur normally in dcKO mice, and they suggest that Crk and CrkL function downstream of Dab1.
Reelin has been proposed to induce association of phosphatidylinositol 3-kinase (PI3 kinase) with Dab1 and promote Akt phosphorylation based on biochemical and pharmacological experiments (Beffert et al., 2002; Bock et al., 2003; Jossin and Goffinet, 2007). However, the widespread role of PI3 kinase in brain development has made it difficult to test this model genetically. We investigated whether serine phosphorylation of Akt depends on the presence of Crk and CrkL. In normal neurons, we observed a low and variable level of Reelin-induced Akt phosphorylation (1.4-fold), whereas it went down by 0.6-fold in dcKO neurons treated with Reelin (Fig. 7 G). Thus, although Dab1 is phosphorylated on tyrosine and undergoes proteolytic degradation, Akt is not activated in dcKO neurons. This finding places Akt activation downstream of Crk and CrkL. Similarly, stimulation of neurons with Reelin causes increased C3G phosphorylation on tyrosine (4.3-fold); however, this fails to occur in dcKO neurons (Fig. 7 G). These data place C3G downstream of Crk and CrkL in the Reelin pathway.
Discussion
The genetic studies reported here demonstrate that the adaptor proteins Crk and CrkL fulfill essential functions in the Reelin pathway, downstream of Dab1, to control neuronal positioning in the developing brain. Mutation of either gene alone did not compromise Reelin signaling, indicating that Crk and CrkL have overlapping functions in the Reelin signaling. It is likely that both Crk and CrkL have additional functions in neurons, independently of the Reelin pathway, since the single and double cKO mice were smaller and had smaller brains. However, the primary role of these ubiquitous adaptor proteins in brain development appears to be the regulation of cell positioning in response to Reelin signaling. Both Crk and CrkL bind to tyrosine phosphorylated Dab1 following Reelin stimulation. In the absence of Crk and CrkL, Dab1 is phosphorylated on tyrosine and it is degraded at the same rate as in normal cells. This implies that Crk and CrkL provide the next key step in the Reelin pathway after phosphorylation of Dab1.
The cerebral cortex, cerebellum and hippocampus of dcKO mice exhibited profound defects in neuronal migration and positioning, very similar to those described in reeler. The dcKO cerebellum was smaller, hypofoliated and most Purkinje cells failed to migrate to the Purkinje cell plate. However, disruption of the cerebellum was less extensive than that reported in reeler mice, as ∼29% of Purkinje cells still reached the Purkinje cell layer. In contrast, only 5% of Purkinje cells are correctly positioned in the reeler cerebellum (Goldowitz et al., 1997). One interpretation of this result is that nestinCRE may cause gene excision just before migration so there may be sufficient residual Crk and CrkL in some dcKO Purkinje cells to support normal migration. Alternatively, other proteins may contribute to Reelin signaling downstream of Dab1 in some Purkinje cells. For example, it has been suggested that Nckβ can also bind to tyrosine phosphorylated Dab1 (Pramatarova et al., 2003).
The hippocampus of dcKO mice closely resembles that of reeler. Pyramidal neurons failed to complete migration and they separated into several layers in a classic reeler manner. Granule neurons of the dentate gyrus were distributed diffusely throughout the hilar region and the two blades were not distinct. The preplate of the cerebral cortex failed to split into the marginal zone and subplate, leading to formation of the superplate. These defects are hallmarks of the reeler phenotype and they have been described in mice lacking functional Reelin, the two Reelin receptors (VLDLR and ApoER2), Dab1, or the tyrosine kinases Fyn and Src (D'Arcangelo et al., 1995; Trommsdorff et al., 1999; Sheldon et al., 1997; Howell et al., 1997; Arnaud et al., 2003a). In addition, the poor dendritic development in the cerebellar molecular layer, the hippocampal CA1 region, and the cerebral cortex of dcKO mice is reminiscent of the defective dendritic development reported in reeler (Niu et al., 2004). This is consistent with a recent report from Matsuki et al. (2008), indicating that Reelin-induced dendritogenesis is reduced in the presence of inhibitory RNAs directed against Crk and CrkL in cultured hippocampal neurons. Together, our data provide clear genetic evidence supporting the contention that Crk and CrkL play critical, overlapping roles in Reelin signaling in vivo.
Crk and CrkL were first implicated in the Reelin pathway when they were identified as binding partners of tyrosine phosphorylated Dab1, the active form of Dab1 (Ballif et al., 2004; Chen et al., 2004; Huang et al., 2004). Although it was assumed that tyrosine phosphorylation of Dab1 by Fyn/Src recruited Crk and CrkL to the complex, overexpression of Crk promotes phosphorylation of Dab1 in 293T cells (Huang et al., 2004). This suggested that Crk and CrkL may regulate tyrosine phosphorylation of Dab1. Previous reports demonstrate that Reelin induces degradation of Dab1 (Rice et al., 1998; Arnaud et al., 2003b; Bock et al., 2004). Blocking Dab1 degradation, by targeting expression of its E3 ubiquitin ligase Cullin 5 by in utero electroporation of shRNA, results in excess migration of cortical neurons (Feng et al., 2007). However, we did not detect any significant change in the level of Dab1 protein or its degree of tyrosine phosphorylation in the absence of Crk and CrkL in the embryonic mouse cortex and primary cortical neurons. This indicates that the two adaptor proteins do not affect the modification or degradation of Dab1, rather they are recruited to Dab1 only when it is activated.
The highly specific function we have identified for Crk and CrkL during brain development is remarkable and it has important implications for our understanding of signal transduction pathways. v-Crk was initially identified as a viral oncoprotein and it was shown to induce a remarkable increase in tyrosine phosphorylation in transformed cells despite the lack of a protein kinase domain (Mayer et al., 1988). The cellular counterparts of v-Crk, Crk and CrkL, were identified later as adaptor proteins containing SH2 domain and two SH3 domains and suggested to function in signal transduction cascades involving tyrosine kinases. Subsequently, Crk family proteins have been shown to interact with many phosphotyrosine-containing proteins and proline-rich domain-containing proteins through their SH2 and SH3 domains, respectively. Like other SH2/SH3-containing adaptor proteins, they receive signals from tyrosine kinase receptors as well as tyrosine kinase-coupled membrane proteins, which they transduce to elicit downstream biological responses to changes in the extracellular environment. Crk family proteins are expressed ubiquitously and they have been proposed to participate in a variety of biological processes, including regulation of cell morphology, cell migration, proliferation, and differentiation (Feller, 2001). Therefore, it was generally accepted that they were involved in multiple signaling pathways. Complete loss of function of both Crk and CrkL was expected to cause cell death or a dramatic loss of cell function. Therefore, our observation of a highly specific phenotype in dcKO mice, demonstrating a specific function for these SH2/SH3 adaptor proteins in the Reelin pathway during brain development was quite surprising. Although neurons are misplaced in reeler and dcKO mice, and laminar organization is severely disrupted, the brain still functions remarkably well. In many cases, axons reach their appropriate targets, albeit by an indirect route, and there is not a massive loss of neuronal populations. The general implication of these findings is that proteins and signaling events thought to be generic may be used for very particular roles in specific biological contexts. Thus, common adaptor proteins, such as Crk and CrkL may function as part of a subroutine in numerous specific signaling pathways (Fig. 7 I). The upstream of the specific component of the pathway [Specific (S) subroutine] comprises molecules whose expression is anatomically and temporally restricted in the developing brain, whereas the downstream molecules, including Crk and CrkL, may comprise general signaling proteins [General (G) subroutine]. Reelin (D'Arcangelo et al., 1995; Rice et al., 1998; chopbgem id = 404), ApoER2 (Kim et al., 1996), and Dab1 (Howell et al., 1997; Rice et al., 1998) (chopbgem id = 1) are expressed primarily in a limited number of tissues in the developing and adult mouse brain. However, Crk and CrkL are broadly expressed in most of the tissues during development and in the adult (de Jong et al., 1995) (chopbgem id = 401; chopbgem id = 402). Other candidate molecules that might function downstream of Dab1 and Crk/CrkL, including C3G and Rap1 (Ballif et al., 2004), are also broadly expressed (Tanaka et al., 1994; Kim et al., 1990) (chopbgem id = 2184, chopbgem id = 3231). Thus, biological specificity is achieved through an integration of highly specific molecular interactions with very general cellular processes.
One of the key questions for the future concerns the mechanism whereby Crk and CrkL affect cell positioning in response to Reelin stimulation. The identification of downstream processes and molecules is likely to be challenging as many proteins have been reported to interact with these general adaptor molecules. GTPases, such as Rap1 and Rac, may be prime targets because of their participation in other signaling pathways involving Crk and CrkL. Additional possibilities include PI3 kinase-Akt and Lis1 which have also been implicated in Reelin signaling (Beffert et al., 2002; Assadi et al., 2003; Jossin and Goffinet, 2007). Our finding that Reelin-induced phosphorylation of C3G and Akt is blocked in dcKO neurons suggest the C3G-Rap1 pathway and the PI3 kinase-Akt pathway both depend on the interaction of Crk/CrkL with Dab1 in Reelin signaling (Fig. 7 I). Therefore, our dcKO mice provide a very useful resource for assessing the roles of such candidate molecules in Reelin signaling in the developing and adult brain. In addition, dcKO mice can be used to investigate the many proposed functions of Crk and CrkL in a whole animal context by inducible or tissue-specific gene ablation. Alternative approaches are more challenging due to overlapping functions of Crk and CrkL, as we found that presence of a single allele of either Crk or CrkL prevented most of the defects we describe here in the Reelin pathway.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant 5R37NS036558. We thank Dr. Jessica M. Y. Ng for critical reading of this manuscript.
References
- Arnaud L, Ballif BA, Förster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol. 2003a;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol Cell Biol. 2003b;23:9293–9302. doi: 10.1128/MCB.23.24.9293-9302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D'Arcangelo G, Clark GD. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35:270–276. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14:606–610. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Blakesley VA, Shen-Orr Z, Jimenez M, Stannard B, Wang LM, Pierce J, LeRoith D. The proto-oncogene product c-Crk associates with insulin receptor substrate-1 and 4PS. Modulation by insulin growth factor-I (IGF) and enhanced IGF-I signaling. J Biol Chem. 1996;271:9287–9290. doi: 10.1074/jbc.271.16.9287. [DOI] [PubMed] [Google Scholar]
- Benhayon D, Magdaleno S, Curran T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Brain Res Mol Brain Res. 2003;112:33–45. doi: 10.1016/s0169-328x(03)00032-9. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, Liu P, Förster E, May P, Goffinet AM, Herz J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J Biol Chem. 2003;278:38772–38779. doi: 10.1074/jbc.M306416200. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, May P, Bergner O, Herz J. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J Biol Chem. 2004;279:33471–33479. doi: 10.1074/jbc.M401770200. [DOI] [PubMed] [Google Scholar]
- Britanova O, Alifragis P, Junek S, Jones K, Gruss P, Tarabykin V. A novel mode of tangential migration of cortical projection neurons. Dev Biol. 2006;298:299–311. doi: 10.1016/j.ydbio.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Butler AA, Blakesley VA, Koval A, deJong R, Groffen J, LeRoith D. In vivo regulation of CrkII and CrkL proto-oncogenes in the uterus by insulin-like growth factor-I. Differential effects on tyrosine phosphorylation and association with paxillin. J Biol Chem. 1997;272:27660–27664. doi: 10.1074/jbc.272.44.27660. [DOI] [PubMed] [Google Scholar]
- Chen K, Ochalski PG, Tran TS, Sahir N, Schubert M, Pramatarova A, Howell BW. Interaction between Dab1 and CrkII is promoted by Reelin signaling. J Cell Sci. 2004;117:4527–4536. doi: 10.1242/jcs.01320. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- de Jong R, Haataja L, Voncken JW, Heisterkamp N, Groffen J. Tyrosine phosphorylation of murine Crkl. Oncogene. 1995;11:1469–1474. [PubMed] [Google Scholar]
- Del Río JA, Martínez A, Auladell C, Soriano E. Developmental history of the subplate and developing white matter in the murine neocortex. Neuronal organization and relationship with the main afferent systems at embryonic and perinatal stages. Cereb Cortex. 2000;10:784–801. doi: 10.1093/cercor/10.8.784. [DOI] [PubMed] [Google Scholar]
- Falconer DS. 2 new mutants, trembler and reeler, with neurological actions in the house mouse (mus-musculus l) J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldowitz D, Cushing RC, Laywell E, D'Arcangelo G, Sheldon M, Sweet HO, Davisson M, Steindler D, Curran T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of reelin. J Neurosci. 1997;17:8767–8777. doi: 10.1523/JNEUROSCI.17-22-08767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–298. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Daza RA, Englund C, Kohtz J, Fink A. Postnatal shifts of interneuron position in the neocortex of normal and reeler mice: evidence for inward radial migration. Neuroscience. 2004;124:605–618. doi: 10.1016/j.neuroscience.2003.11.033. [DOI] [PubMed] [Google Scholar]
- Hof PR, Glezer II, Condé F, Flagg RA, Rubin MB, Nimchinsky EA, Vogt Weisenhorn DM. Cellular distribution of the calcium-binding proteins parvalbumin, calbindin, and calretinin in the neocortex of mammals: phylogenetic and developmental patterns. J Chem Neuroanat. 1999;16:77–116. doi: 10.1016/s0891-0618(98)00065-9. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Huang Y, Magdaleno S, Hopkins R, Slaughter C, Curran T, Keshvara L. Tyrosine phosphorylated Disabled 1 recruits Crk family adapter proteins. Biochem Biophys Res Commun. 2004;318:204–212. doi: 10.1016/j.bbrc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 2007;27:7113–7124. doi: 10.1128/MCB.00928-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshvara L, Benhayon D, Magdaleno S, Curran T. Identification of reelin-induced sites of tyrosyl phosphorylation on disabled 1. J Biol Chem. 2001;276:16008–16014. doi: 10.1074/jbc.M101422200. [DOI] [PubMed] [Google Scholar]
- Keshvara L, Magdaleno S, Benhayon D, Curran T. Cyclin-dependent kinase 5 phosphorylates disabled 1 independently of Reelin signaling. J Neurosci. 2002;22:4869–4877. doi: 10.1523/JNEUROSCI.22-12-04869.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Iijima H, Goto K, Sakai J, Ishii H, Kim HJ, Suzuki H, Kondo H, Saeki S, Yamamoto T. Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J Biol Chem. 1996;271:8373–8380. doi: 10.1074/jbc.271.14.8373. [DOI] [PubMed] [Google Scholar]
- Kim S, Mizoguchi A, Kikuchi A, Takai Y. Tissue and subcellular distributions of the smg-21/rap1/Krev-1 proteins which are partly distinct from those of c-ras p21s. Mol Cell Biol. 1990;10:2645–2652. doi: 10.1128/mcb.10.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo G, Arnaud L, Kronstad-O'Brien P, Cooper JA. Absence of Fyn and Src causes a reeler-like phenotype. J Neurosci. 2005;25:8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM. Neuronal migration. Mech Dev. 2001;105:47–56. doi: 10.1016/s0925-4773(01)00396-3. [DOI] [PubMed] [Google Scholar]
- Magdaleno S, Keshvara L, Curran T. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron. 2002;33:573–586. doi: 10.1016/s0896-6273(02)00582-2. [DOI] [PubMed] [Google Scholar]
- Matsuki T, Pramatarova A, Howell BW. Reduction of Crk and CrkL expression blocks reelin-induced dendritogenesis. J Cell Sci. 2008;121:1869–1875. doi: 10.1242/jcs.027334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- Miao GG, Smeyne RJ, D'Arcangelo G, Copeland NG, Jenkins NA, Morgan JI, Curran T. Isolation of an allele of reeler by insertional mutagenesis. Proc Natl Acad Sci U S A. 1994;91:11050–11054. doi: 10.1073/pnas.91.23.11050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D'Arcangelo G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41:71–84. doi: 10.1016/s0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- Park TJ, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol. 2006;26:6272–6282. doi: 10.1128/MCB.00472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A, Ochalski PG, Chen K, Gropman A, Myers S, Min KT, Howell BW. Nck beta interacts with tyrosine-phosphorylated disabled 1 and redistributes in Reelin-stimulated neurons. Mol Cell Biol. 2003;23:7210–7221. doi: 10.1128/MCB.23.20.7210-7221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Curran T. Mutant mice with scrambled brains: understanding the signaling pathways that control cell positioning in the CNS. Genes Dev. 1999;13:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D'Arcangelo G, Nakajima K, Goldowitz D, Curran T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Morishita T, Hashimoto Y, Hattori S, Nakamura S, Shibuya M, Matuoka K, Takenawa T, Kurata T, Nagashima K, et al. C3G, a guanine nucleotide-releasing protein expressed ubiquitously, binds to the Src homology 3 domains of CRK and GRB2/ASH proteins. Proc Natl Acad Sci U S A. 1994;91:3443–3447. doi: 10.1073/pnas.91.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Yip YP, Kronstadt-O'Brien P, Capriotti C, Cooper JA, Yip JW. Migration of sympathetic preganglionic neurons in the spinal cord is regulated by Reelin-dependent Dab1 tyrosine phosphorylation and CrkL. J Comp Neurol. 2007;502:635–643. doi: 10.1002/cne.21318. [DOI] [PubMed] [Google Scholar]