

Summary
Actin polymerization in cells occurs via filament elongation at the barbed end. Proteins that cap the barbed end terminate this elongation. Heterodimeric capping protein (CP) is an abundant and ubiquitous protein that caps the barbed end. We find that the mouse homolog of the adaptor protein CARMIL (mCARMIL) binds CP with high affinity and decreases its affinity for the barbed end. Addition of mCARMIL to cell extracts increases the rate and extent of Arp2/3 or spectrin-actin seed-induced polymerization. In cells, GFP-mCARMIL concentrates in lamellipodia and increases the fraction of cells with large lamellipodia. Decreasing mCARMIL levels by siRNA transfection lowers theF-actin level and slows cell migration through a mechanism that includes decreased lamellipodia protrusion. This phenotype is reversed by full-length mCARMIL but not mCARMIL lacking the domain that binds CP. Thus, mCARMIL is a key regulator of CP and has profound effects on cell behavior.
Introduction
Actin polymerization, essential for cell migration, occurs primarily through elongation at the filament barbed end. The time and place of polymerization in a migrating cell is determined by local de novo nucleation of a filament with a free barbed end and/or by local severing or uncapping of an existing filament. Filament elongation then continues until the barbed end is capped by a capping protein. Often capping is rapid, thereby accounting for the very short filaments in the branched network of lamellipodia. Rapid capping gives the cell temporal and spatial control over where filaments elongate.
Because the barbed ends of most filaments in the cell are capped, the cytoplasmic pools of G-actin and profilin actin are maintained at a high level, rarely achieving steady state with free barbed ends. This large pool of monomeric actin allows rapid elongation by those barbed ends that become free. The in vitro movement of beads coated with proteins that activate the Arp2/3 complex depends on these pools of monomeric actin and thus requires a CP (Loisel et al., 1999).
Capping protein (CP), the nonmuscle homolog of capZ, is considered to be one of the key factors limiting the duration of filament elongation (DiNubile et al., 1995; Hug et al., 1995; Hopmann et al., 1996; Hopmann and Miller, 2003; Mejilano et al., 2004). CP is found in all eukaryotic cells, usually at high concentrations (1–3 μM), and it binds barbed ends with high affinity (KD 0.1–1 nM) (Cooper and Schafer, 2000; Wear et al., 2003). The off-rate of CP is slow (t1/2 ∼25 min), thus filaments in the lamellipodia may only rid themselves of CP by fully depolymerizing from their pointed end. Decreasing the concentration of CP in cells results in longer actin filaments in both Dictyostelium and mammalian cells (Hug et al., 1995; Mejilano et al., 2004). In the cell, not all filaments are short. Long actin filaments are characteristic of filopodia (Svitkina et al., 2003) and cells depleted of CP produce filopodia rather than lamellipodia (Mejilano et al., 2004). Thus, factors affecting filament length could determine the properties on actin-containing structures.
Regulation of barbed-end capping is emerging as a key mechanism of actin polymerization. Factors inhibit capping either by binding to the filament and protecting it from CP or by binding to CP and inhibiting its capping activity or by uncapping. Formins nucleate filaments and then transiently protect the new filament’s barbed end from capping (Moseley et al., 2003; Zigmond et al., 2003; Harris et al., 2004). VASP, an F-actin binding protein, inhibits capping by CP in vitro and increases filament length in vivo (Bear et al., 2002). Capping by Eps8, a newly discovered CP, is activated by binding to Abi1 (Disanza et al., 2004). Other F-actin binding proteins such as tropomyosin and members of the ADF family including coactosin can, by steric inhibition and/or by altering the filament structure, interfere with capping proteins (Rohrig et al., 1995; Nyakern-Meazza et al., 2002; Li et al., 2004).
Phosphoinositides bind and inhibit both gelsolin and CP; the proteins V-1/myotrophin and CKIP-1 bind and specifically inhibit CP (Taoka et al., 1994; Chellaiah and Hruska, 1996; Schafer et al., 1996; Yang et al., 1998; Allen, 2003; Taoka et al., 2003; Yin and Janmey, 2003; Canton et al., 2005). Phosphoinositides are the only molecules known to uncap capped filaments; no uncapping proteins have been reported thus far. Twin-filin binds CP but does not affect its capping activity; rather this binding is necessary for the localization and activity of twinfilin (Falck et al., 2004). The scaffolding protein CMS/CD2AP/CIN85 binds CP, but its effect on capping activity is unknown (Hutchings et al., 2003).
CARMIL, a protein isolated from Dictyostelium, binds CP as well as myosin type I and the Arp2/3 complex (Jung et al., 2001). Dictyostelium CARMIL activates Arp2/3, but its effect, and the effect of CARMIL isolated from Acanthamoeba, on CP activity are unknown (Remmert et al., 2003). Dictyostelium amoebae with mutations in CARMIL have defects of cell migration, endocytosis, and chemotaxis. The basis for these effects, given the multiple binding partners of Dictyostelium CARMIL, could include regulation of the distribution and/or activity of any of these binding partners.
To further explore the interaction between CARMIL and CP, we cloned and expressed fragments of human and mouse homologs of CARMIL as well as full-length mouse CARMIL (mCARMIL). We found that recombinant mammalian CARMIL binds CP with high affinity and inhibits its capping activity. Interestingly, these CARMILs also rapidly uncap filaments pre-capped by CP, a property not observed with either formins or V-1. In vivo, GFP-CARMIL localizes to the front of lamelli-podia. Overexpression of CARMIL enhances lamelli-podia formation, while decreasing CARMIL with siRNA dramatically decreases lamellipodia formation and slows cell migration. The phenotype of the siRNA can be rescued with full-length mCARMIL but not with mCARMIL lacking the CP binding domain.
Results
Identification of Mammalian CARMIL
Mammalian homologs of Dictyostelium CARMIL (Jung et al., 2001) were identified by BLAST searching of EST databases. Using human and mouse ESTs, we then BLAST searched the genome sequences and identified a CARMIL homolog on mouse chromosome 13 and human chromosome 6. The mouse protein, mCARMIL (accession number AY437876) predicted from cDNAs has 1374 amino acids with a calculated MW of 151.9 kDa (Figure S1 available with this article online). Northern blots of human tissue reveal a widely expressed ∼5.4 kb mRNA that is most abundant in kidney and other epithelial tissues (Figure 1A).
Figure 1.

Characterization of a Mammalian CARMIL
(A) Northern blot of human CARMIL in different tissues. Northern blotting was performed using a 12-lane MTN blot (Clontech) probed with a 250 bp 5′ fragment of the human cDNA. A 5.4 kb mRNA reactive band was revealed.
(B) Diagram of mCARMIL constructs used.
Leucine-rich regions are noted by black squares and regions that inhibit CP by open rectangles. GST- or MBP-fusion proteins expressed in E. coli were isolated and assayed (at concentrations up to 1 μM concentration) for their ability to inhibit capping by CP (8 nM). Fragments that inhibited capping by more than 50% were scored positive.
(C) The C-1 fragment of mCARMIL inhibits capping by CP. Fragments of mCARMIL were assayed for the ability to increase the rate of polymerization of 2 μM actin nucleated by seeds (0.2 nM) in the presence of CP (4 nM). GST-fusion proteins were tested at 80 nM (GST-C-1, filled blue circles) or 1 μM (GST-C-2, open black square; C-3, filled black squares; N, open pink triangle). Also shown are seeds alone (no CP, open red circles), seeds with CP and no CARMIL (open green circles), and G-actin alone without seeds (half-filled red squares). Pyrenylactin fluorescence is plotted versus time in s.
(D) Full-length CARMIL and C-1 have similar abilities to inhibit CP.
Actin (1 μM) was nucleated by seeds (0.1 nM) in the presence of CP (4 nM) and various concentrations of full-length His-tagged mCARMIL (red triangles) and GST-C-1 (green symbols, two different experiments indicated by circles and squares). The initial rate of elongation is plotted as a percentage of the rate observed with seeds alone (no CP) versus mCARMIL concentration.
(E) Sequence alignment of CARMIL C-1 regions. The amino acid sequences of C-1 from mouse (mCARMIL-AY: 437876), aligned with similar sequences from the databases of rat (rCARMIL, GI: 34876156), human (hCARMIL, GI: 15147715), mosquito (agCP4344, GI: 31210773), Drosophila (CG1399PB, GI: 22024065), and Dictyostelium (dCARMIL, GI: 14701866). Totally conserved residues (*); very conserved changes (two dots); conserved residues (one dot); and K991 and R993 (!) are noted. The comparable sequence for Acanthamoeba (Acn125) is shown separately because it diverges significantly in this region.
(F) R993 contributes to inhibition of capping activity. Seeds (0.1 nM) alone (filled black diamond) or with CP (3 nM) were incubated with varying concentrations of wt C-1 (blue squares), R993E-C-1 (red circles), or R993A-C-1 (green triangles) before addition of pyrenylactin and determination of the rate of polymerization. The data plotted are the initial rate of elongation, as a percentage of the rate observed with seeds and no CP, versus GST-C-1 concentration.
The C Terminus of mCARMIL Inhibits CP’s Capping Activity
His-tagged and GST-fusion proteins of full-length mCARMIL and various mCARMIL fragments (Figures 1B and 1C) were expressed, purified, and tested for effects on the capping activity of purified recombinant CP. Full-length His-tagged mCARMIL increased the rate of elongation from seeds in the presence of CP. It did not increase polymerization in the absence of seeds, indicating that it had no ability to nucleate filaments. A GST-fusion protein containing only the C terminus of mCARMIL, and the comparable fragment of human CARMIL, also reduced CP’s ability to inhibit barbed-end elongation (Figure 1B). Expression of truncated mCARMIL constructs localized the CP inhibition activity first to the C-terminal half of the molecule (resides 962–1374) and then to a 123 amino acid fragment (residues 962–1084) termed C-1 (red box in Figure 1B). Other fragments from the C-terminal half, i.e., C-2 and C-3, at concentrations >20 times that at which C-1 inhibited capping, showed no inhibition of capping (Figure 1C); at high concentrations (μM), C-2 had some barbed-end capping activity on its own (data not shown). C-1 constructs before and after cleavage of the GST tag showed similar levels of CP inhibition (data not shown). Dose-response curves of GST-C-1 and His-tagged full-length mCARMIL indicated that they have similar activity (Figure 1D). Neither full-length mCARMIL nor C-1 inhibited capping by gelsolin, indicating that the inhibition was specific for CP (data not shown).
The C-1 region is conserved in all metazoan homologs thus far examined and contains a number of highly conserved amino acids (Figure 1E). A basic sequence KXRXKRXK (aa 991–998) is conserved in mammals and insects. This region is less conserved in amoeboid versions of CARMIL. Mutations of R993 to A or E decreased the ability of C-1 to inhibit CP (Figure 1F). The mutation K991A also inhibited activity but not as severely as R993A (data not shown). Another conserved sequence, with 4 basic residues out of 8 (aa 1077–1084), is present at the C-terminal of C-1. However, a C-1 with the mutation K1077A inhibited capping as well as wild-type C-1 (data not shown).
C-1 Is a High-Affinity Inhibitor of CP
We determined the Ki of this C-terminal fragment (C-1) on CP by measuring the concentration dependence of GST-C-1 on capping activity (Figure 2A). The dose-response curve (using the affinity of CP for the barbed ends as 1 nM, see Figure 2B) defined the Ki of C-1 for CP as ∼1.5 nM (range 0.9 to 1.7). Interestingly the inhibition of CP by C-1 was not complete; rather the extent of inhibition plateaued at 80%–85% (range 70%–95%). Thus the GST-C1/CP complex may retain some affinity for the barbed end of the actin filament.
Figure 2.

C-1 Inhibits Capping by CP and Uncaps Barbed Ends Capped by CP; the CP/C-1 Complex Retains Some Capping Activity
(A) Dose response of GST-C-1 on inhibition of capping by several moderate concentrations of CP. Stabilized seeds (0.1 nM) were incubated with CP at 0.6 nM (squares), 2.2 nM (circles), or 3.8 nM (triangles) and with different concentrations of C-1 for 7 min before adding pyrenylactin (1 μM) and measuring the rate of elongation. The rate of elongation from seeds with 100 nM C-1 and no CP was set as 100%. See Figure 2B for description of smooth lines fitting curves
(B) Dose response of CP in a high concentration of mCARMIL. Stabilized seeds (0.1 nM) were incubated with different concentrations of CP or CP plus 200 nM C-1 for 7 min before addition of pyrenylactin and the rate of elongation determined. The elongation rate from seeds in the presence of 200 nM C-1 was set as 100%.
The smooth lines in (A) and (B) are drawn using consensus best fit equilibrium constants for this and several similar experiments using the same preparations.
Capping of ends by CP, KD = 1 nM.
Binding of CP to C-1, KD = 1.5 nM.
Capping of ends by CP/C-1 complex, KD = 15 nM. Thus the CP/C-1 complex retains capping activity but with an affinity 10-fold lower (range in different experiments: 10- to 50-fold) than that of free CP.
(C) GST-C1 rapidly uncaps barbed ends capped with CP. Polymerization of pyrenylactin (1.2 μM) nucleated by seeds in the absence or presence of 2 nM CP. After 600 s, PIP2 (35 μM), GST-C1 (200 nM), or GST-C1 and 28 nM Cytochalasin-D (CD) were added (arrow).
(D) Uncapping by 200 nM C-1 is maximal within ∼15 s. Stabilized seeds (0.6 nM) were incubated without (red dot) or with CP (4 nM) (blue squares) for 20 min before addition of C-1 (100 nM) for various times before dilution into pyrenylactin and determination of the initial rate of elongation. The earliest rate measurable after addition of C-1 was ∼15 s. The rate of elongation of a sample with C-1 present during the entire 20 min incubation with CP is also shown (green diamond).
To determine if the CP/C-1 complex retained some capping activity, we examined the concentration dependence of capping by CP with and without a constant high concentration (200 nM) of C-1 (Figure 2B). The data without C-1 gave an affinity of CP for the barbed end of 1 nM (range 0.3 to 1.5 nM). The data with C-1 could be fit by an equilibrium model assuming that the CP/C-1 complex had no capping activity, but this required the affinity of C-1 for CP to be 10-fold weaker than that determined above (Figure 2A). On the other hand, the data with C-1 were well fit by the model using the previously determined KD of C-1 for CP, 1.5 nM, if we assumed that the CP/C-1 complex caps the barbed end with an affinity about 15-fold lower than that of pure CP. In this experiment, binding of C-1 decreased the affinity of CP from 1 to 15 nM. Over a series of experiments with varying seed numbers and preincubation times, the CP/C-1 complex capping affinity was 10- to 50-fold weaker than that of CP alone.
mCARMIL Rapidly Uncaps Barbed Ends Capped by CP
We next asked whether C-1 could uncap ends previously capped by CP. CP, once bound, dissociates very slowly (koff = 5 × 10−4 s−1; t1/2 ∼25 min) (Wear et al., 2003). C-1 was added to a polymerization reaction in which the filament seeds were pre-capped by CP. Free barbed ends appeared rapidly, reaching their maximal level within 10 s, the shortest time we could assay (Figures 2C and 2D). This result suggests that C-1 binds CP on the barbed end and that this binding results in a large (≥100-fold) increase in CP’s off-rate (t1/2 from 1400 s to <10 s). CARMIL thus differs from two other inhibitors of CP, formin and V-1, that are not able to uncap capped filaments (Taoka et al., 2003) (M.H. and S.Z., unpublished data).
C-1 Binds CP Directly and with High Affinity
To investigate how C-1 inhibits CP, we examined the ability of glutathione beads coated with GST-C-1 to pull down pure CP. CP pelleted with beads coated with GST-C-1 but not a GST-N-terminal fragment of mCARMIL or GST alone (Figure 3A). The C-1 mutant R993E, which does not inhibit capping, did not pull down CP. Thus, inhibition by C-1 involves direct binding to CP.
Figure 3.

C-1 Binds Directly to CP
(A) Pure CP is pelleted by GST-C-1; binding to CP is decreased by the R993E mutation. GST or mCARMIL fusion proteins, GST-N-mCARMIL, GST-C-1, or GST-R993E-C-1 (each ∼500 nM) were incubated with glutathionesepharose beads, washed, and then incubated with CP, a heterodimer of α1 and β2 (100 nM). After washing, the bound protein was eluted with SDS buffer, separated on 10% SDS-PAGE, and stained with silver.
(B and C) GST-C-1 binds to wt CP and the CPΔ/Δ mutant with similar affinity. The ability of glutathione-sepharose-coupled GST-C-1 to pull down pure CP was determined by loss of CP from the supernatant. The CP concentration was constant at 1 μM, and increasing amounts of beads, providing a total concentration of GST-C1 up to 5 μM, were added. (B) shows representative Tris-SDS-polyacrylamide gels (12% acrylamide) illustrating the GST-C1 concentration-dependent depletion of CP or CPΔ/Δ from the supernatant (S/N). Controls for GST-C-1, wt CP added, and wt CP pelleted with uncoupled beads are shown in lanes labeled a–c, and for CPΔ/Δ in lanes labeled d–f. (C) is a plot of the concentration of bound CP (open circles) or CPΔ/Δ (filled triangles) versus the concentration of immobilized GST-C-1. Values are the mean ± SEM, n = 3. The data were least squares fit to equation 1. The difference is not statistically significant (p > 0.7).
(D) CPΔ/Δ mutant competes with wt CP for GST-C-1. Actin polymerization was followed from pyrenylactin fluorescence (a.u.) as a function of time (s) after nucleation by seeds in the presence of wt CP (3 nM) in the absence or presence of GST-C-1 (250 or 125 nM), and in the absence or presence of various concentrations of CPΔ/Δ. CPΔ/Δ at high concentrations completely relieved the GSTC-1-mediated inhibition of CP’s capping activity. Actin was used at 2 μM (5.8% pyrene labeled).
We next asked if C-1 bound to the C-terminal regions of CP subunits (approximately the last 30 amino acids) that are primarily responsible for its actin-capping activity (Wear et al., 2003; Kim et al., 2004). A double-deletion mutant missing both of these C-terminal regions (CPΔ/Δ) does not bind actin (Wear et al., 2003; Kim et al., 2004). However, GST-C1 on beads was equally able to pull down wild-type (wt) CP and CPΔ/Δ (Figures 3B and 3C). Thus, C-1 does not bind to these C-terminal regions of CP. We confirmed this result with a functional assay. We added the CPΔ/Δ to a seeded actin assembly assay containing wt CP (at 3 nM) and a nearly saturating concentration of GST-C-1, 125 nM (Figure 3D). Increasing amounts of CPΔ/Δ relieved the GST-C-1-mediated inhibition of wt CP’s capping activity (green time courses, Figure 3D), indicating that CPΔ/Δ successfully competed with wt CP for binding to GST-C-1.
mCARMIL Binds CP and Enhances Actin Polymerization in a Cell Extract
Although pure mCARMIL and CP interact, it is important to determine if this interaction also occurs in cytoplasm. First, we found that GST-C-mCARMIL-coated beads could pull down CP from high-speed supernatant of lysed neutrophils (Figure 4A). Thus, C-1 can bind CP in cytoplasm. Second, immunoprecipitation of endogenous CP from HeLa cell extract pulled down endogenous CARMIL (Figure 4B). Thus, the CARMIL-CP complex exists in cytoplasmic extracts. We were unable to perform the converse reaction since none of our CARMIL antibodies work for immunoprecipitation.
Figure 4.
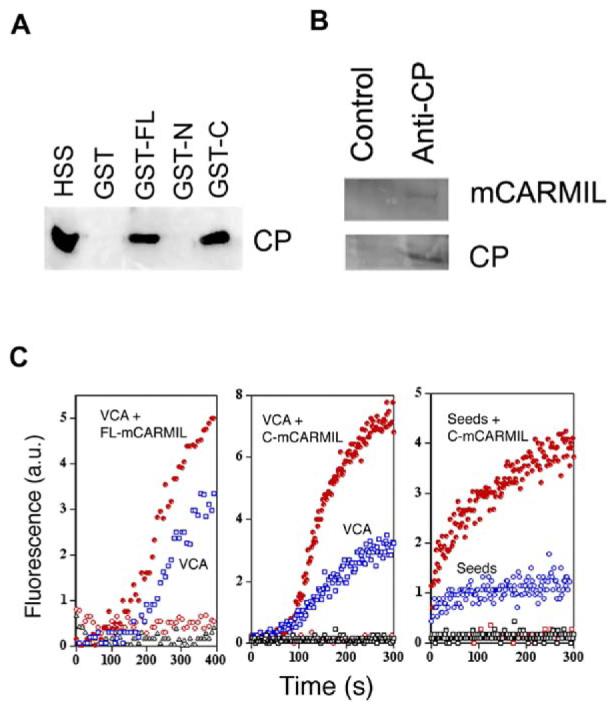
mCARMIL Binds and Inhibits CP in Cell Extracts
(A) GST-C-mCARMIL pulls down CP from cell extract. GST (∼2 mM) or mCARMIL fusion proteins, GST-FL mCARMIL(GST-FL), GST-N-mCARMIL (GST-N), or GST-C-mCARMIL (GST-C), were incubated with glutathione-sepharose beads, washed, and then incubated with high-speed supernatant of lysed neutrophil (HSS). After washing, bead bound CP was analyzed by Western blot with antibody against CP β chain.
(B) Endogenous mCARMIL and CP coimmunoprecipitate. High-speed extracts of Hela cells were incubated with rabbit anti-CP antibody (R26) or a control rabbit IgG. The immunoprecipitates were analyzed by Western blots probed with antibody against mCARMIL or CP.
(C) mCARMIL enhances barbed-end actin polymerization induced in neutrophil extract. GST-VCA (left [20 nM] and middle [10 nM] panels), seeds ([10 nM] right panel), or buffer (all panels) was added to cell extract containing 1 μM pyrenylactin in the absence (open black symbols) or presence of full-length mCARMIL (0.9 μM, left panel) or the C terminus of mCARMIL (0.5 μM, middle and right panels). Symbols: GST-VCA or seeds without mCARMIL (open blue symbols); with mCARMIL (filled red circles); mCARMIL without GST-VCA or seeds (red open circles). GST-VCA without mCARMIL (open blue squares). Polymerization was followed by the increase in pyrenylactin fluorescence.
To examine the functional activity of mCARMIL in cell extracts, we induced actin polymerization from barbed ends in a neutrophil extract by adding the VCA fragment of N-WASP to activate the Arp2/3 complex (Figure 4C). Addition of full-length mCARMIL had no effect on its own, indicating that it did not stimulate nucleation but addition did enhance the rate and extent of polymerization induced by VCA. Addition of the C-terminal half of mCARMIL also increased the rate and extent of polymerization induced by VCA or by spectrin-actin seeds (Figure 4C, middle and right panels).
Increases in F-actin could have resulted from either increased nucleation or decreased capping. Increased nucleation was a consideration because Dictyostelium CARMIL can weakly activate the Arp2/3 complex (Jung et al., 2001). To discriminate between these alternatives, we determined whether the total number of barbed ends present changes: they would not increase if mCARMIL merely uncaps but would increase if mCARMIL stimulated de novo filament nucleation. To determine the total number of barbed ends, parallel samples of supernatant in the presence or absence of C-1 were stimulated with VCA and then diluted 50-fold into pyrenylactin containing PIP2, which uncaps barbed ends (Schafer et al., 1996). The rate of polymerization after dilution is thus proportional to the total number of barbed ends. C-1 had no effect on the total number of filaments alone or in the presence of VCA (Figure S2). On the other hand, when the samples were diluted into pyrenylactin without PIP2 (where, because the off-rate of pure CP is slow, the rate of polymerization is proportional to the number of free barbed ends), C-1 did increase the rate of polymerization (Figure S2). Thus, the stimulation of polymerization by C-1 is due to decreased capping not increased nucleation.
Inhibition of CP should slow the rate of capping and thereby increase the lengths of the actin filaments in the extract. We found this to be the case. Addition of C-1 increased the median length of actin filaments induced by VCA, measured by fluorescent phalloidin staining (Figure S3). Together these results show that C-1 enhances polymerization in cytoplasm by inhibiting capping, as it does with pure proteins.
Localization of GFP-mCARMIL in Human Glioblastoma Cells
CFP-tagged full-length mCARMIL (FL mC) expressed in glioblastoma cells was highly concentrated in lamelli-podia. When expressed in cells expressing YFP-tagged CP, the two colocalized (Figure 5A). GFP-mCARMIL co-localized with F-actin in lamellipodia but did not concentrate with F-actin in stress fibers (Figure 5B). When cells were lysed before fixation, the GFP-mCARMIL remained concentrated in the lamellipodia and colocalized with a lamellipodial marker cortactin (Figure 5C). In contrast, cytoplasmic label was lost upon lysis when GFP alone was transfected. This supports the conclusion that mCARMIL enrichment in lamellipodia is due to its specific association with the cytoskeleton but not due to a local variation in cell volume.
Figure 5.

Expressed mCARMIL in Cells
(A) CARMIL colocalizes with CP in lamellipodia. Cells stably expressing YFP-CP (β2 subunit) were cotransfected with CFP-mCARMIL. Cells were examined for each fluorophore by confocal microscopy. Shown are CFP-CARMIL (green), YFP-CP (red), and the merged image.
(B) Middle domain of mCARMIL mediates lamellipodia localization.
Glioblastoma cells were transfected with GFP-vector (vector), GFP full-length mCARMIL (FLwt), GFP-N terminus (N), GFP-middle to C terminus (MC), GFP-C-terminus, (C). Cells were fixed and stained with rhodamine phalloidin before viewing in the confocal microscope.
(C) Full-length and middle domain of mCARMIL colocalize with cortactin in lysed cells. Glioblastoma cells were cotransfected with pDsRed2-cortactin and GFP-vector (vector), or GFP-full length mCARMIL or GFP-tagged mCARMIL fragments. The cells were extracted with 1% Triton X-100 in PEM buffer (100 mM PIPES, [pH 6.9], 1 mM MgCl2, 1 mM EGTA) containing 4% PEG, MW 35,000, for 5 min followed by fixation with 4% paraformaldehyde before viewing in the confocal microscope.
To determine the region of mCARMIL responsible for its localization, we expressed GFP fused to different fragments of the protein. The distribution of a GFP-tagged fragment containing both the middle and C-terminal of mCARMIL (MC) was similar to that of full-length mCARMIL; however, neither the C terminus alone, the N terminus alone, nor GFP itself localized to lamellipodia (Figures 5B and 5C; see also Figure 7A).
Figure 7.

Full-Length CARMIL, but Not ΔC-1 CARMIL, Rescues CARMIL siRNA
(A) Morphology. Cells expressing human CARMIL si-RNA had a retracted morphology with few lamellipodia and stress fibers (first column); coexpression of human CARMIL siRNA with full-length GFP-tagged mouse CARMIL resulted in formation of lamellipodia (second column) that were larger than those in control cells expressing GFP alone (third column). Expression of ΔC-1 CARMIL in control cells resulted in cells that appeared retracted and had few lamellipodia or stress fibers (fourth column). When small lamelli-podia were present, the GFP-ΔC-1 mCARMIL localized at the front with the F-actin (fifth column, arrows). In each panel cells expressing the indicated GFP-fusion constructs (top row) were stained by phalloidin (middle row) and photographed by confocal microscopy. Merged images are shown in a bottom row.
(B–D) Migration speed and persistence. Time-lapse movies were acquired as in Figure 6C for untransfected cells (UT, n = 35), cells transfected with CARMIL siRNA alone (n = 30), with CARMIL siRNA and full-length mCARMIL (+FL, n = 39), or with CARMIL siRNA ΔC-1 mCARMIL (+ΔC-1, n = 34). Other cells were transfected with control siRNA alone (n = 30), with control siRNA and full-length mCARMIL (+FL, n = 34), or with control siRNA and ΔC-1 mCARMIL (+ΔC-1, n = 34).
(B) Display of representative paths of untransfected cells, cells transfected with CARMIL siRNA plus full-length mCARMIL, and cells transfected with control siRNA plus ΔC-1 mCARMIL. The traced cells were selected on the basis of their rate being near median for the population.
(C) Instantaneous speed was calculated as in Figure 6C. While the reduction in cell speed caused by mCARMIL siRNA was reversed by FL mCARMIL (p < 0.0001), it was not reversed by ΔC-1-mCARMIL. Rather, expression of ΔC-1-mCARMIL decreased the migration rate of cells transfected with control siRNA cells (p < 0.0001). Cells transfected with control siRNA moved at a rate similar to that of untransfected cells. Data are expressed as mean ± SEM.
(D) Inhibition of directional persistence. Quantitation of directional persistence was determined (as in Figure 6E) for the same sets of cells as described in (A). Expression of ΔC-1 CARMIL did not restore the loss of directional persistence caused by mCARMIL siRNA but rather resulted in a loss of directional persistence in cells treated with control siRNA (p < 0.001). Control siRNA cells had a directional persistence similar to that of untransfected cells. Data show the ratio ± SEM, The n values for UT = 34; CARMIL siRNA alone, 30; CARMIL siRNA + FL, 33; CARMIL siRNA + ΔC-1, 34; control siRNA alone, 30; control siRNA + FL, 34; control siRNA + ΔC-1, 32.
We also noted in these experiments that expression of full-length mCARMIL altered cell morphology by increasing the fraction of cells with large lamellipodia (Figures 5A and 7B). While 11% (11/104) of control cells had lamellipodia covering >10% of the cell perimeter, 80% (86/108) of the cells expressing GFP-mCARMIL had such lamellipodia. These data suggest that mCARMIL positively regulates lamellipodial protrusion. To further investigate this idea, we inhibited endogenous CARMIL in human glioblastoma cells using siRNA.
CARMIL siRNA Decreased Cell Migration and Lamellipodial Protrusion
Human glioblastoma cells were treated with siRNA for human CARMIL (CARMIL siRNA). The efficiency of transfection was ∼90%, and the total concentration of CARMIL was decreased by greater than 50% relative to cells treated with siRNA for CARMIL (control siRNA) from a different species (mouse) (Figure 6A). Expression of CARMIL siRNA caused a decrease in the total F-actin per cell (Figure 6A). This is consistent with an increase in CP activity lowering the fraction of free barbed ends and therefore the total F-actin level. Overall cell morphology and actin organization also changed dramatically in CARMIL siRNA-expressing cells. Cells looked partially retracted and had few lamellipodia (see below). Prominent stress fibers also disappeared, leaving only occasional short actin bundles scattered in the cytoplasm.
Figure 6.

Expression of mCARMIL siRNA Decreased F Actin and Cell Migration
(A) Depleting CARMIL decreases F-actin levels. Human glioblastoma cells transfected with siRNA for human CARMIL or control siRNA were cultured for 5 days. Cells were then lysed and examined for CARMIL content via a Western blot using Arp3 as a loading control (left panel). Other cells were fixed, permeabilized, stained with TRITC-phalloidin, and photographed using wide-field epifluorescence. The phalloidin staining (F-actin/cell) was quantified from the photos using ImageJ software. Data are expressed (right panel) as mean ± SEM (N for CARMIL siRNA = 79, for control siRNA = 105). Student’s t test showed the difference was significant, p < 0.001.
(B) Expression of CARMIL siRNA slowed cell migration in a wound-healing assay. A wound was scratched in confluent cultures of human glioblastoma cells transfected 4 days earlier with CARMIL siRNA or control siRNA. Shown are representative sections of the wound from CARMIL siRNA and control siRNA photographed 6 min and 22 hr after wounding.
(C–E) CARMIL siRNA slowed individual cell migration. Time lapse movies were acquired at 5 min intervals for 6 hr with 4x objective. (C) Inhibition of cell speed. Instantaneous cell speed was calculated from time lapse sequences using MetaMorph imaging software. Data are expressed as mean ± SEM, n = 34 (control siRNA) or 35 (CARMIL siRNA), p < 0.0001. (D and E) Inhibition of directional persistence. (D) Display of representative paths of cells selected on the basis of their rate being near median for the population. (E) Quantification of directional persistence. The ratio of total distance moved/net displacement was determined for each cell; this value is inversely related to the persistence in direction of movement. The percentage of cells within a range of values is shown in a histogram (higher value = the less persistence). The insert shows mean value of this ratio ± SEM, n = 34 (control siRNA) or 35 (CARMIL siRNA); p < 0.0001.
(F—I) Kymograph analysis. Individual frames and kymographs from time lapse movies of control siRNA and CARMIL siRNA cells recorded every 3 s for 10 min with a 100× objective. The line in individual frames of movies was drawn perpendicular to the tangent of the cell edge at the point of furthest protrusion; this line was used to generate kymograph using MetaMorph software.
Box and whisker plots for velocity (G), distance (how far a lamellipodium protrudes during a single protrusion/withdrawal cycle) (H), and frequency of individual protrusion events (I) from kymograph analysis. Dot shows mean; middle line of box shows median; top and bottom of box indicates 75th and 25th quartile, and whiskers indicate extent of 10th and 90th percentiles, respectively. Data for control siRNA are derived from 228 events of protrusion in 25 cells, for CARMIL siRNA, from 342 events of protrusion in 32 cells. The differences between CARMIL siRNA and control siRNA for velocity, protrusion distance, and frequency were each significant at p < 0.0001.
Depletion of CARMIL with siRNA decreased various measures of motility in glioblastoma cells. Cell spreading was slowed: after 3 hr, only 35% ± 5% (n = 264) of CARMIL siRNA-treated cells were spread (unspread cells were defined as round cells with not even a small portion of extended cytoplasm) while 86% ± 1% (n = 194) of control cells (transfected with control siRNA) were spread. Cell migration in a wound-healing assay was decreased: control cells completely filled the wound space within 22 hr, whereas a significant gap remained between wound edges with CARMIL siRNA cells at the same time point (Figure 6B). The inhibition of cell migration was also detected by following individual cell motility (Figures 6C–6E). Compared to cells treated with control siRNA, cells treated with CARMIL siRNA moved 50% more slowly (Figure 6C), and the direction of movement showed less persistence (Figures 6D and 6E).
To determine to what extent the decrease in rate of cell migration was due to impairment of lamellipodia protrusion, we monitored lamellipodia movements through a 100x lens (Figure 6F). CARMIL siRNA caused a dramatic inhibition of the protrusive behavior of many cells. Normal lamellipodia characteristic of control cells (Figure 6F, top) were rarely formed. Instead, these cells rapidly extended small, phase dark bleb-like structures at their margin (Figure 6F, bottom). A fraction of the CARMIL siRNA cells had a milder phenotype and extended recognizable lamellipodia (Figure 6F, middle). However, different parameters of protrusive behavior, such as the rate, distance, and frequency of protrusion, were significantly impaired in these residual lamelli-podia (Figures 6G–6I).
Expression of Full-Length mCARMIL but Not mCARMIL Lacking the C-1 Domain Rescued the CARMIL siRNA Phenotype
Morphologically, expression of full-length mCARMIL reversed the loss of lamellipodia seen with CARMIL siRNA. Cells cotransfected with mCARMIL again had large lamellipodia (Figure 7A). This ruled out the possibility that the effects of CARMIL siRNA were nonspecific. Interestingly, expression of a construct lacking the C-1 region, ΔC-1mCARMIL, did not restore lamelli-podia. Rather expression of ΔC-1mCARMIL even in control cells resulted in a partially retracted morphology and reduced lamellipodia and stress fibers (Figure 7A), and in severe cases, they showed blebbing at the edges. The fraction of cells expressing ΔC-1mCARMIL that had recognizable lamellipodia was 5-fold smaller than that of cells expressing GFP alone. Thus, the morphology of cells expressing ΔC-1mCARMIL was similar to that of cells with CARMIL siRNA. In the few cells that had lamellipodia, the GFP ΔC-1mCARMIL was localized at the front of the lamellipodia, consistent with the localization of mCARMIL being determined by its middle region and indicating that its localization did not depend on binding to CP (Figure 7A, 5th column).
To determine whether the migration defects of the CARMIL siRNA cells depended on the loss of CARMIL’s ability to bind CP versus some other function mediated by CARMIL, we examined the ability of full-length mCARMIL and ΔC-1mCARMIL to rescue. To monitor rescue quantitatively, we examined the migration of individual cells. Expression of FL mCARMIL restored the rate of migration and the directional persistence to CARMIL siRNA-transfected cells. However, expression of ΔC-1mCARMIL restored neither the rate of migration nor the directional persistence (Figures 7B–7D). Indeed, expression of ΔC-1mCARMIL in control siRNA cells decreased the rate of migration and directional persistence to levels similar to those of CARMIL siRNA cells.
Both the movement of cells expressing ΔC-1mCARMIL and their morphology were similar to those of CARMIL siRNA cells. Thus, the expression of ΔC-1mCARMIL had a dominant-negative phenotype. Furthermore, these data indicate that CARMIL function in both lamellipodial formation and cell migration depends on its ability to bind CP.
Discussion
A growing body of evidence demonstrates that capping of actin filament barbed ends is critical for actin-dependent mechanisms of cell motility. However, there is little knowledge regarding local regulation of capping and targeting of CPs. Capping can be inhibited by proteins such as formins and VASP that act by protecting filament barbed ends, while others such as V-1 act by sequestering CP. Phosphoinositides can uncap actin filaments in vitro, but whether they also uncap in vivo is difficult to test since the phosphoinositides contribute to activation of Arp2/3 and thus de novo actin nucleation.
CARMIL from Dictyostelium was shown previously to bind CP (Jung et al., 2001). However, these studies have not addressed the functional effects of CARMIL binding to CP. We report here that mammalian CARMIL binds CP with high affinity (w1 nM) and significantly decreases CP’s affinity for barbed ends (by 10- to 100-fold). Moreover, mCARMIL also rapidly uncaps CP-capped filaments, increasing the CP off-rate by about 100-fold. This is only protein that is able to uncap filaments capped with CP.
While binding of Dictyostelium CARMIL to CP was thought to occur at the N terminus, binding of CP by Acanthamoeba CARMIL (Remmert et al., 2003) and by mCARMIL localizes to the C terminus. We narrowed down the CP binding domain of mCARMIL to a 100 amino acid portion, C-1, that inhibits capping as well as full-length mCARMIL. Similar activities of full-length CARMIL and C-1 suggest that CARMIL is not an auto-regulated molecule but could be regulated by additional factors. C-1 contains a stretch of basic residues highly conserved between mammals and insects. Substitutions within this region of a highly conserved Arg993 and Lys991 decreased binding affinity. The comparable region of amoeboid CARMILs retains the key Arg at a position homologous to 993, but surrounding amino acids are poorly conserved (Figure 2B). For example, the Lys991 is converted to Ala in Dictyostelium and Pro in Acanthamoeba. Perhaps this substitution contributes to the lower affinity of Acanthamoeba CARMIL for CP (KD = 400 nM versus 1.5 nM for mCARMIL) (Remmert et al., 2003). This decrease in affinity may not result in a different function in vivo since we observed that the R993E mutant, which has a decreased affinity for CP, could still rescue CARMIL siRNA cells.
The region of CP that binds to C-1 does not include the C-terminal regions required for binding to the barbed end of a filament (Wear et al., 2003). Thus, C-1 does not compete with barbed ends for this critical surface on CP. This eliminates a key potential mode of inhibition. While mCARMIL dramatically decreases the affinity of CP for the barbed end, the mCARMIL-CP complex retains some ability to cap the barbed end. Energy square analysis (also called microreversibility or detailed balance; see Supplemental Data) indicates that C-1′s affinity for CP bound to a barbed end is 10-to 30-fold lower than for free CP. Thus, C-1 binding to free CP must include some surface or conformation that is not available when CP is bound to a barbed end. This raises the possibility that uncapping is a two-step process where C-1 binds first in a manner that weakly inhibits CP and then, when one of the C-terminal regions required for capping releases (each tail has an off-rate constant of 0.2/s), C-1 acquires access to an additional binding site that now sterically inhibits rebinding of the C-terminal region of CP to the barbed end. The two-step uncapping hypothesis suggests that C-1 is flexible and able to progressively spread over the surface of CP. Indeed, circular dichroism analysis of C-1 indicated that the peptide had little secondary structure and that no new structural peaks emerged upon mixing CP with C-1 (Figure S5).
Given the in vivo concentrations of mCARMIL (estimated from Western blots to be between 0.1 and 1 μM) and its affinity for CP (KD ∼1 nM), much of the mCARMIL in the cytosol is calculated to be bound to CP. This is supported by the coimmunoprecipitation of mCARMIL with CP and from previous experiments showing that Acanthamoeba CARMIL copurifies with CP. Furthermore, the two proteins colocalize at the front of lamellipodia. The localization of CARMIL in lamelli-podia required a distinct domain localized in the middle of the CARMIL sequence. Localization did not appear to depend on binding to CP. Rather, CARMIL may recruit CP to the front of the lamellipodia since CP mislocalizes in cells treated with CARMIL siRNA (not shown). However, because the overall lamellar morphology in these cells is so disturbed, it is difficult to know if mislocalization of CP is directly related to CARMIL depletion.
mCARMIL interacts with CP in cytoplasm, and this interaction is essential for lamellipodial protrusion and cell translocation. Dictyostelium CARMIL null mutants (Jung et al., 2001) have shown decreased migration, although the nature of motility defects remains unclear. Dictyostelium CARMIL, in addition to binding CP, binds the SH3 domain of type 1 myosins and binds and activates nucleation by the Arp2/3 complex. mCARMIL has regions of amino acid similarity to the major domains of Dictyostelium CARMIL, but we have detected no activation of the Arp2/3 complex by mCARMIL, either with pure Arp2/3 or in cell lysates. Yet, mCARMIL clearly affects lamellipodial behavior. Overexpression of CARMIL induced large lamellipodia, while depletion, using either siRNA or expression of CARMIL lacking the C-1 domain, led to a decrease in or complete abrogation of lamellipodial protrusion. The decrease in lamellipodial protrusion presumably led to both the decreased rate of migration and the decreased persistence of translocation. The similar phenotype of siRNA cells and expression of CARMIL lacking the C-1 domain demonstrate that the ability of CARMIL to bind CP is key to its cellular function.
The CARMIL-CP complex can still cap barbed ends, although with a lower affinity than pure CP (×15 nM versus 1 nM). However, given the concentrations of both CARMIL and CP in cells, the concentration of the complex (∼0.5 μM) should be sufficient to cap barbed ends. It would be surprising if the ability to uncap a filament were not utilized by the cell. Indeed, the decrease in F-actin observed with siRNA treatment and the morphology of cells treated with siRNA or ΔC-1 CARMIL are reminiscent of that seen in cytochalasin-treated cells. This suggests that a loss of CARMIL’s ability to bind CP results in an increase in capping activity. Future studies will be required to determine if additional players regulate the ability of CARMIL to function as an uncapper in vivo.
Experimental Procedures
See Supplemental Experimental Procedures for additional details.
Constructs, Northern, and Recombinant Protein
Blast analysis of the NCBI databases revealed the mouse and human homologs of Dictyostelium CARMIL. Two overlapping mouse cDNA clones (BC01229 and UI-M-BH0-aiy-g-05-0-UI) were obtained from the Research Genetics collection at Invitrogen and sequenced to give a full-length cDNA sequence (5010 bp) (GenBank accession number AY437876). Using this cDNA, mouse genomic database searches indicated that the mCARMIL gene is located on chromosome 13 and contains 35 introns. The predicted mCARMIL protein consists of 1374 amino acids with a calculated MW of 151.9 kDa (Figure S1, for full sequence). The human cDNA clone # COL00695, with homology to C-terminal CARMIL, was a gift from Dr. Hiroko Hata (University of Tokyo, Japan). The human gene is located on chromosome 6. The clone for pDsRed-cortactin was the gift of Dr S. Kojima of Northwestern University.
Northern blots using the human 12-lane MTN blot (Clontech) were probed with a 250 bp 5# fragment of the human cDNA clone COL00695 32P-labeled with Randam Primed DNA Labeling Kit (Amersham Pharmacia Biotech); equal loading was determined by human β-actin.
Recombinant Proteins
DNA fragments for full-length and various fragments of mCARMIL were amplified by PCR and cloned into pGEX-5X-1 or pMAL-c2X, and the resulting constructs were fully sequenced to assure that they were in correct reading frame. Fusion proteins were expressed in E. coli and isolated via glutathione-sepharose beads or amylose Resin (BioLabs). Purity of isolated proteins is shown in Figure S4.
His-Tagged mCARMIL Expression in Sf9 Cells
Recombinant baculovirus expressing N-terminal His6-tagged full-length mCARMIL was constructed using Bac-To-Bac Baculovirus Expression Systems (Life Tech).
Generation of mCARMIL Antibody
MBP-M-mCARMIL (595–959) was used to immunize rabbits and antibody was affinity-purified with GST-M-mCARMIL
Preparation of C-1 Mutants
The site-directed mutations replacing Arg993 with glutamic acid or alanine (R993E and R993A mutants) and Lys991 and Lys1077 with alanine (K991A and K1077A mutants) were performed using the QuikChange site-directed mutagenesis kit (Stratagene). The presence of the desired mutations was confirmed by DNA sequencing.
CP Immunoprecipatation
Protein A beads bound with affinity-purified rabbit antibodies prepared against the unique C-terminal region of the chicken CP β2 isoform (R25) or with control IgG were incubated with high-speed supernatant from HeLa cells overnight at 4°C. Immunocomplexes were washed and bound proteins were analyzed by Western blots.
CP and CPμ/μ Binding to GST-C-1 Beads by Supernatant Depletion
One micromole CP or CPΔ/Δ was incubated with increasing concentrations of GST-C1 bound to glutathione Sepharose-4B beads (Pharmacia) for 10 min at 25°C in IP buffer. The beads were pelleted and aliquots of supernatant were electrophoresed on SDS gels. The amount of CP was calculated by densitometry of gels stained with Coomassie blue using NIH Image software. Bound CP was plotted versus the total bead-coupled ligand concentration and least squares fit to equation 1, using Kaleidagraph v3.6 software (Synergy Software, Reading, Pennsylvania).
Isolation of CP and CP Mutant
CP was isolated from E. coli expressing recombinant mouse CP (α1β2) (Palmgren et al., 2001). The CP double C-terminal deletion mutant, CPΔ/Δ (missing the C-terminal 28 amino acids from the α subunit and the C-terminal 34 amino acids from the β subunit), was purified from a bacterial expression system with chicken cDNAs as described (Wear et al., 2003).
Actin
Actin was isolated from rabbit muscle and gel-filtered and a fraction was labeled with pyrene. Actin was stored as calcium ATP-actin but converted to magnesium ATP-actin immediately before use. Assays used between 5% and 30% pyrenylactin; varying the fraction labeled had no effect on the results.
Spectrin-Actin Seeds
Spectrin-actin seeds (seeds) were isolated from human red blood cells and stored in ethylene glycol at −20°C (Casella et al., 1986; Zigmond, 1998). The number of barbed ends was determined from the rate of polymerization monitored by the increase in pyrenylactin fluorescence Ex370/Em410) (Cano et al., 1991).
To stabilize seeds for extended incubation in IP buffer, the seeds were preincubated in 0.35 μM actin and 0.35 μM phalloidin in IP buffer for 30 min at 25°C.
Uncapping Assays
The uncapping assay was a modification of the seeded assembly assay and performed essentially as described (Schafer et al., 1996). Uncapping is quantified from the change in the rate of actin polymerization, calculated from the slope of the pyrene fluorescence versus time, following addition of either PIP2 or GST-C-1.
Neutrophil Extract
Extracts of rabbit peritoneal exudate neutrophils were obtained as described (Zigmond et al., 1997).
Culture and Transfection of Human Glioblastoma Cells
Human glioblastoma cells (SNB-19 cells) (a gift from Marc Symons, North Shore-Long Island Jewish Research Institute, New York) were transiently transfected with GFP-mCARMIL constructs (cloned into the pEGFP-C2 vector [Clontech]) using lipofectamine 2000 (Invitrogen).
Coexpression of CFP-mCARMIL and YFP-CP was achieved by first establishing SNB-19 glioblastoma cells that stably express pEYFP-CPβ2 by selecting pEYFP-CPβ2 transfected cells using G418 at 500 μg/ml for 2 weeks and then transfecting these cells with pECFP-FL mCARMIL.
siRNAs
A specific siRNA for human CARMIL coding region (5′-AAGAAA TAGGGAAGGTGGAAC-3′) (hCARMIL siRNA) and a control siRNA nucleotide (5′-AAATTTACAGGACTTCAGTCA-3′) from the mouse CARMIL coding region were obtained from Dharmacon Research (Lafayette, Colorado) and Cy3-labeled using a Silencer siRNA labeling kit (Ambion). Cells were transfected using Lipofectamine 2000 (Invitrogen). Effects of transfection were analyzed 3 to 5 days posttransfection. The efficiency of transfection was ∼90% and the decrease in CARMIL, as assayed by Western blots, was ≥50%.
Cell Spreading and Lamellipodia Assays
hCARMIL siRNA and control siRNA transfected glioblastoma cells (72 hr posttransfection) were observed with a 20! objective and classified as round (bright sphere in phase contrast) or spread (having a thin rim or extended cytoplasmic processes). Lamellipodia presence was evaluated by a “blind” observer on cells co-stained with phalloidin.
Wounding Assay
siRNA-treated and control siRNA-transfected glioblastoma cells (72 hr posttransfection) were plated in 12-well tissue culture plate and grown to near confluency before a line of cells was removed by scraping with a pipet tip. After washing, fresh culture medium was added and cells were monitored at various times up to 22 hr.
Cell Tracking and Kymography
Cells transfected with CARMIL siRNA and control siRNA were mixed and plated at low density into a 35 mm dish. Timelapse sequences of motile cells were acquired 60 hr posttransfection using an inverted microscope (Eclipse TE2000-U, Nikon) and MetaMorph imaging software (Universal Imaging Corp). During imaging, cell cultures were maintained at 35°C by heating the microscope stage and/or objective lens. Cell paths were generated using Track Object tool in MetaMorph. After filming, cells containing CARMIL siRNA were identified by their Cy3 fluorescence under the microscope.
Supplementary Material
Acknowledgments
The work was supported by NIH grants AI 19883 to S.H.Z., GM070898 to T.M.S., and GM 38542 to J.A.C. We thank Michael Joyce for excellent technical assistance, D. Schafer for anti-CP antibody, M.M. Chou for human Northern blot, S. EauClaire for preliminary experiments of CP/C-1 binding, and J. Hammer for many helpful conversations.
Accession Numbers
The mouse CARMIL sequence reported in this paper has been deposited in GenBank under accession number AY437876.
References
- Allen P. Actin filament uncapping localizes to ruffling lamellae and rocketing vesicles. Nat. Cell Biol. 2003;5:972–979. doi: 10.1038/ncb1059. [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6. [DOI] [PubMed] [Google Scholar]
- Cano ML, Lauffenburger DA, Zigmond SH. Kinetic analysis of F-actin depolymerization in polymorphonuclear leukocyte lysates indicates that chemoattractant stimulation increases actin filament number without altering the filament length distribution. J. Cell Biol. 1991;115:677–687. doi: 10.1083/jcb.115.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canton DA, Olsten MEK, Kim K, Doherty-Kirby A, Lajoie G, Cooper JA, Litchfield DW. The pleckstrin homology domain-containing protein CKIP-1 is involved in regulation of cell morphology and the actin cytoskeleton and interaction with actin capping protein. Mol. Cell. Biol. 2005;25:3519–3534. doi: 10.1128/MCB.25.9.3519-3534.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella JF, Maack DJ, Lin S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. J. Biol. Chem. 1986;261:10915–10921. [PubMed] [Google Scholar]
- Chellaiah M, Hruska K. Osteopontin stimulates gelsolin-associated phosphoinositide levels and phosphatidylinositol triphosphate-hydroxyl kinase. Mol. Biol. Cell. 1996;7:743–753. doi: 10.1091/mbc.7.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Schafer DA. Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 2000;12:97–103. doi: 10.1016/s0955-0674(99)00062-9. [DOI] [PubMed] [Google Scholar]
- DiNubile MJ, Cassimeris L, Joyce M, Zigmond SH. Actin filament barbed-end capping activity in neutrophil lysates: the role of capping protein-beta 2. Mol. Biol. Cell. 1995;6:1659–1671. doi: 10.1091/mbc.6.12.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disanza A, Carlier MF, Stradal TE, Didry D, Frittoli E, Confalornieri S, Croce A, Wehland J, Di Fiore PP, Scita G. Eps8 controls actin-based motility by capping the barbed ends of actin filaments. Nat. Cell Biol. 2004;6:1180–1188. doi: 10.1038/ncb1199. [DOI] [PubMed] [Google Scholar]
- Falck S, Paavilainen VO, Wear MA, Grossmann JG, Cooper JA, Lappalainen P. Biological role and structural mechanism of twinfilin-capping protein interaction. EMBO J. 2004;23:3010–3019. doi: 10.1038/sj.emboj.7600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E, Li F, Higgs H. The mouse formin, FRLa, slows actin filament barbed end elongation, competes with capping protein, accelerates polymerization from monomers, and severs filaments. J. Biol. Chem. 2004 doi: 10.1074/jbc.M312718200. [DOI] [PubMed] [Google Scholar]
- Hopmann R, Miller KG. A balance of capping protein and profilin functions is required to regulate actin polymerization in Drosophila bristle. Mol. Biol. Cell. 2003;14:118–128. doi: 10.1091/mbc.E02-05-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopmann R, Cooper JA, Miller KG. Actin organization, bristle morphology, and viability are affected by actin capping protein mutations in Drosophila. J. Cell Biol. 1996;133:1293–1305. doi: 10.1083/jcb.133.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug C, Jay PY, Reddy I, McNally JG, Bridgman PC, Elson EL, Cooper JA. Capping protein levels influence actin assembly and cell motility in dictyostelium. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Hutchings NJ, Clarkson N, Chalkley R, Barclay AN, Brown MH. Linking the T cell surface protein CD2 to the actin-capping protein CAPZ via CMS and CIN85. J. Biol. Chem. 2003;278:22396–22403. doi: 10.1074/jbc.M302540200. [DOI] [PubMed] [Google Scholar]
- Jung G, Remmert K, Wu X, Volosky JM, Hammer JA., 3rd. The Dictyostelium CARMIL protein links capping protein and the Arp2/3 complex to type I myosins through their SH3 domains. J. Cell Biol. 2001;153:1479–1497. doi: 10.1083/jcb.153.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Yamashita A, Wear M, Maeda Y, Cooper J. Capping protein binding to actin in yeast: Biochemical mechanism and physiological relevance. J. Cell Biol. 2004;164:567–580. doi: 10.1083/jcb.200308061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu X, Lou Z, Xin D, Wu H, Liu Y, Rao Z. Crystal structure of human coactosin-like protein at 1.9A resolution. Protein Sci. 2004;13:2845–2851. doi: 10.1110/ps.04937304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Mejilano MR, Kojima S.-i., Applewhite DA, Gertler FB, Svitkina TM, Borisy GG. Lamelliodial versus filopodial mode of actin nanomachinery: pivotal role of the filament barbed end. Cell. 2004;118:363–373. doi: 10.1016/j.cell.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Moseley J, Sagot I, Manning A, Xu Y, Eck M, Pellman D, Goode B. A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell. 2003;15:896–907. doi: 10.1091/mbc.E03-08-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyakern-Meazza M, Narayan K, Schutt CE, Lindberg U. Tropomyosin and gelsolin cooperate in controlling the microfilament system. J. Biol. Chem. 2002;277:28774–28779. doi: 10.1074/jbc.M203360200. [DOI] [PubMed] [Google Scholar]
- Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P. Interactions with PIP2, ADP-actin monomers, and capping protein regulate the activity and localization of yeast twinfilin. J. Cell Biol. 2001;155:251–260. doi: 10.1083/jcb.200106157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert K, Olszewski TE, Bowers MB, Dimitrova M, Ginsburg A, Hammer JA., III. CARMIL is a bonefide capping protein interactant. J. Biol. Chem. 2003;279:3068–3077. doi: 10.1074/jbc.M308829200. [DOI] [PubMed] [Google Scholar]
- Rohrig U, Gerisch G, Morozova L, Schleicher M, Wegner A. Coactosin interferes with the capping of actin filaments. FEBS Lett. 1995;374:284–286. doi: 10.1016/0014-5793(95)01130-7. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Jennings PB, Cooper JA. Dynamics of capping protein and actin assembly in vitro: uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 2003;160:409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka M, Ichimura T, Wakamiya-Tsuruta A, Kubota Y, Araki T, Obinata T, Isobe T. V-1, a protein expressed transiently during murine cerebellar development, regulates actin polymerization via interaction with capping protein. J. Biol. Chem. 2003;278:5864–5870. doi: 10.1074/jbc.M211509200. [DOI] [PubMed] [Google Scholar]
- Taoka M, Isobe T, Okuyama T, Watanabe M, Kondo H, Yamakawa Y, Ozawa F, Hisinuma F, Kubota M, Minegishi A, et al. Murine cerebellar neurons express a novel gene encoding a protein related to cell cycle control and cell fate determination proteins. J. Biol. Chem. 1994;269:9946–9951. [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maeda Y, Cooper JA. How capping protein binds the barbed end of the actin filament. Curr. Biol. 2003;13:1531–1537. doi: 10.1016/s0960-9822(03)00559-1. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nanduri S, Sen S, Qin J. The structural basis of ankyrin-like repeat function as revealed by the solution structure of myotrophin. Structure. 1998;6:619–626. doi: 10.1016/s0969-2126(98)00063-x. [DOI] [PubMed] [Google Scholar]
- Yin H, Janmey P. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Actin cytoskeleton: the Arp2/3 complex gets to the point. Curr. Biol. 1998;8:R654–R657. doi: 10.1016/s0960-9822(07)00415-0. [DOI] [PubMed] [Google Scholar]
- Zigmond SH, Joyce M, Borleis J, Bokoch GM, Devreotes PN. Regulation of actin polymerization in cell-free systems by GTPgammaS and Cdc42. J. Cell Biol. 1997;138:363–374. doi: 10.1083/jcb.138.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond SH, Evangelista M, Boone C, Yang C, Dar AC, Sicheri F, Forkey F, Pring M. Formin leaky cap allows elongation in the presence of capping proteins. Curr. Biol. 2003;13:1820–1823. doi: 10.1016/j.cub.2003.09.057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.