

SUMMARY
Self-renewal is a hallmark of stem cells and cancer, but existence of a shared stemness program remains controversial. Here we construct a gene module map to systematically relate transcriptional programs in embryonic stem cells (ESCs), adult tissue stem cells, and human cancers. This map reveals two predominant gene modules that distinguish ESCs and adult tissue stem cells. The ESC-like transcriptional program is activated in diverse human epithelial cancers and strongly predicts metastasis and death. c-Myc, but not other oncogenes, is sufficient to reactivate the ESC-like program in normal and cancer cells. In primary human keratinocytes transformed by Ras and IκBα, c-Myc increases the fraction of tumor-initiating cells by 150-fold, enabling tumor formation and serial propagation with as few as 500 cells. c-Myc-enhanced tumor initiation is cell-autonomous and independent of genomic instability. Thus, activation of an ESC-like transcriptional program in differentiated adult cells may induce pathologic self-renewal characteristic of cancer stem cells.
INTRODUCTION
The stem cell model of tissue organization posits that a small number of progenitor cells maintains the self-renewal and organization of the tissue (reviewed by Beachy et al., 2004; Clarke and Fuller, 2006). The unique ability of ESCs and adult tissue stem cells to self-renew and give rise to multiple cell lineages have formed the basic definitions of “stemness”. Accumulating evidence suggests that cancers may have equivalent cancer stem cells, defined as the small fraction of cells that can initiate and perpetuate tumor growth and heterogeneity in serial transplantation experiments (Al-Hajj et al., 2003; Bonnet and Dick, 1997). If cancers are truly maintained by a small population of cancer stem cells, then the ability to identify, target, and eliminate cancer stem cells would be critical for cancer diagnosis and therapy. It has been theorized that a critical event in cancer initiation may be the inappropriate activation of the self-renewal machinery that is normally restricted to stem cells. However, it remains unclear whether activation of a normal stem cell program is a pervasive feature or required element in common human epithelial cancers.
Given the unique roles of normal stem cells in tissue maintenance, recent evidence has suggested a surprising plasticity of stem cell fates. Several types of adult tissue stem cells can be apparently reprogrammed into cell types of other lineages (Slack, 2007). In addition, introduction of four genes, c-Myc, Oct4, Sox2, and Klf4, into mouse or human fibroblasts was sufficient to reprogram them to confer pluripotency indistinguishable from authentic ESCs (Takahashi et al., 2007; Takahashi and Yamanaka, 2006). These findings raise the possibility that other types of stem cells, including cancer stem cells, may be experimentally created by the appropriate combination of genes.
Several studies have attempted to define a core transcriptional program of “stemness”, but the results have been controversial (Fortunel et al., 2003; Ivanova et al., 2002; Ramalho-Santos et al., 2002). Fortunel et al. compared the shared microarray expression profiles of ESCs, neural stem cells (NSCs), and hematopoietic stem cells (HSCs) across three independent studies, and they only found a single gene shared among the three studies. The inability to define a consensus stemness signature in a gene-by-gene analysis may suggest that different types of stem cells utilize distinct mechanisms to achieve self-renewal and pluripotency. Alternatively, the failure to identify a robust signature may be due to technical variations in stem cell isolation, degrees of cell purity, microarray platforms, or statistical analysis methods. Because intersection of gene-level analyses are more prone to noise and are inherently limited by the technically worse dataset under consideration (Bild and Febbo, 2005; Segal et al., 2004; Subramanian et al., 2005), we hypothesized that a higher-order, systems-level analysis may improve the organization and classification of stem cell transcriptional programs. Here, we use a method termed gene module map (Segal et al., 2004) to identify shared expression programs of ESCs and adult tissue stem cells, focusing on groups of genes, termed modules, that are supported by the consensus of multiple independent observations. We find that adult tissue stem cells can be separated into two large groups, one of which shares a core transcriptional program with ESCs. Further, the ESC-like transcriptional program is frequently activated in aggressive human epithelial cancers. c-Myc is uniquely able to activate the ESC-like transcriptional program in adult epithelial cells, which in the appropriate genetic context confers cardinal properties of human cancer stem cells in vivo.
RESULTS
Gene module map relating embryonic and adult tissue stem cells
To systematically relate gene expression programs among ESCs, adult stem cells, disease states, and key pathways that regulate such programs, we created a module map of stem cell genes (Figure 1A). We curated a compendium of mouse stem cell data, including 102 genome-wide expression profiles of mouse ESCs, NSCs, HSCs, retinal stem cells, neural crest stem cells, hair bulge stem cells, mammary stem cells, and various differentiated cells (Table S1). We also compiled over 3,000 gene sets, including signature genes that are differentially expressed between mouse stem cells and their differentiated counterparts, genes that are occupied by key stem cell transcription factors as defined by genome-scale chromatin immunoprecipitation (ChIP-chip or ChIP-PET), genes that are functionally regulated by key stem cell factors as defined by RNA interference, and genes that encode proteins with shared functions, such as those in Gene Ontology and KEGG pathways (Table S2). Gene module map enabled unbiased discovery of genes that are coordinately but perhaps subtly regulated in one or more types of stem cells as supported by multiple functional criteria and independent, reproducible observation (see Supplementary Text).
Figure 1. Gene Module Analysis of Stem Cells.

(A) Flow chart of the steps in our gene module analysis of stem cells.
(B) Mouse stem cell gene module map: a matrix of gene sets (rows) versus expression arrays (columns), where a red (or green) entry indicates that the individual genes within the gene set are significantly induced (or repressed) in the expression array more than expected by chance (FDR<0.05, p<0.01). The intensity of each entry corresponds to the average fold change of all of the genes within the gene set in the array. The arrays are labeled as differentiated cells or the type of stem cell. A subset of significant gene sets is shown; redundant gene sets were removed for clarity. Gene sets defined by gene ontology terms are listed in black; published gene sets defined by expression arrays of stem cells and differentiated cells are listed in purple; gene set defined by RNA interference experiments is listed in orange; gene set defined by genome-wide chromatin immunoprecipitation experiments is listed in blue. Note the clustering of two groups of stem cells, labeled “ESC-like” and “adult tissue stem”.
The module map of mouse stem cell genes revealed the existence of at least two distinct classes of adult tissue stem cells (Figure 1B). We found that the mouse ESCs from four independent studies, NSCs from three independent studies, fetal liver HSCs, and retinal stem cells are clustered together by the coordinate activation of a similar collection of gene sets (collectively termed “ESC-like”), demonstrating that ESCs and a subset of adult tissue stem cells share a robust transcriptional program. Interestingly, the ESC-like signature was not composed of any down-regulated gene sets, suggesting that the repressed genes are not shared but instead confer specificity among different types of stem cells. The ESC-like gene sets included several stem cell signatures previously defined in ESCs and NSCs, as well as select genes occupied by Oct4 and Nanog in ESCs by ChIP-PET and genes downregulated by RNAi of Oct4, Nanog, or Sox2 in ESCs. Gene signatures identified by one study were significantly recapitulated in independent studies by other groups, demonstrating the strength of gene module map in identifying shared expression patterns. The member genes that hit in each of the gene sets are grouped together to form the mouse ESC-like module (FDR<0.05, p<0.001; Table S3). The ESC-like module contains many transcriptional regulators, in particular several associated with pluripotency, including Sox2, c-Myc, Dnmt1, Cbx3, Hdac1, and Yy1. Oct4 and Nanog are not in the ESC-like module because they are specifically expressed in ESCs. This result implies that a portion of the transcriptional program mediated by these key ESC regulators, Oct4 and Nanog, can be regulated in other stem cells by alternate mechanisms.
In contrast, the majority of adult tissue stem cells, including bone marrow HSCs, neural crest stem cells, hair bulge stem cells, and mammary stem cells, clustered together by coordinate activation of a distinct collection of gene sets (collectively termed “adult tissue stem”) (Figure 1B). Bone marrow HSCs and hair bulge stem cells from three and two independent studies, respectively, each clustered together, demonstrating that the gene module map approach accurately clusters the stem cell transcriptional patterns. For instance, genes induced in hair bulge stem cells as defined by two studies, were each cross-validated by activation in each other’s arrays, and furthermore were induced in bone marrow HSCs, mammary stem cells, and neural crest stem cells relative to their differentiated counterparts. Interestingly, NSCs profiled by three independent studies clustered with ESCs whereas the NSCs from a fourth study clustered with the adult tissue stem cells; the reason for this discrepancy is at present unclear. The member genes that hit in each of these gene sets are grouped together to form the mouse adult tissue stem gene module (FDR<0.05, p<0.001, Table S4). Notably, the adult tissue stem module is populated by many transcriptional regulators of differentiation, such as Hox genes, GATA and forkhead family transcription factors, MLL family of histone methyltransferases, and lineage-specific regulators like Rest. Thus, the gene module map demonstrated two distinct classes of stem cells, which we term “ESC-like” and “adult tissue stem”.
In order to validate the stem cell gene modules defined in mouse and identify the functional subset conserved in evolution, we applied our gene module map analysis to a compendium of human stem cell transcriptional profiles (Figure S1A). Similar to the mouse stem cell module map, we first curated a compendium of human stem cell data, including genome-wide expression profiles of human ESCs, embryonal carcinoma cells, and various differentiated cells (Table S1). We also compiled over 3,000 gene sets, including signature genes that are differentially expressed between human ESCs and their differentiated counterparts, genes that are occupied by key stem cell transcription factors as defined by genome-scale chromatin immunoprecipitation experiments, and genes that encode proteins with shared functions, such as those in Gene Ontology and KEGG pathways (Table S2). The module map of human stem cell genes revealed a robust human ESC-like gene module that is induced in two independent studies of human ESCs and embryonal carcinomas (member genes listed in Table S5, FDR<0.05, p<0.0001). Importantly, the human ESC-like module overlapped significantly with the mouse ESC-like module (p<10-83, hypergeometric distribution; Figure S1B), but not with the mouse adult tissue stem module. We defined the 335 genes shared between mouse and human as the core ESC-like gene module (Table S6).
An important caveat is that many stem cells were grown in culture, and one of the most consistent and expected responses of stem cells in culture is proliferation. Therefore, some of the concordance in expression programs in stem cells, in particular the ESC module, may be due to proliferation genes. We therefore determined the contribution of genes directly related to proliferation as well as exposure to serum in culture (Chang et al., 2004; Whitfield et al., 2002). A minority (31 and 5 percent) of the genes in the core ESC-like module and adult tissue stem module, respectively, have been previously shown to have periodic expression during the cell cycle or to be induced in response to serum. Notably, neither stem cell module contained any of the genes transcriptionally induced in the S and G2/M phases of the cell cycle (Whitfield et al., 2002). Moreover, removal of the gene sets associated with proliferation did not impact the clustering of the ESC-like stem cell arrays or the conservation between mouse and human ESC-like modules, demonstrating that the ESC-like module is not simply a signature of proliferation.
Widespread activation of the ESC module in human cancers and impact on cancer prognosis
Previous studies have demonstrated that cell-type specific transcripts from minority cell populations can be detected in tumor samples (Perou et al., 2000), suggesting that a specific cancer stem cell signature can be detected in expression profiles of bulk tumors (Liu et al., 2007). To investigate whether a stem cell expression signature is activated in human cancer, we examined the expression of genes comprising the core stem cell signatures in a “human cancer compendium” of publicly available expression profiles of a variety of human cancers and their corresponding normal tissues (Segal et al., 2004). The ESC-like gene module is significantly activated in various human cancers relative to corresponding normal tissue, and repressed in various normal tissues relative to cancer (Figure 2A, B). In contrast, the adult tissue stem gene module has an opposite pattern: activated in various normal tissues relative to cancer, and repressed in various human cancers relative to normal tissue. Thus, these results suggest that diverse human cancers exhibit an ESC-like expression pattern.
Figure 2. Activation of ESC Module in Human Cancers.
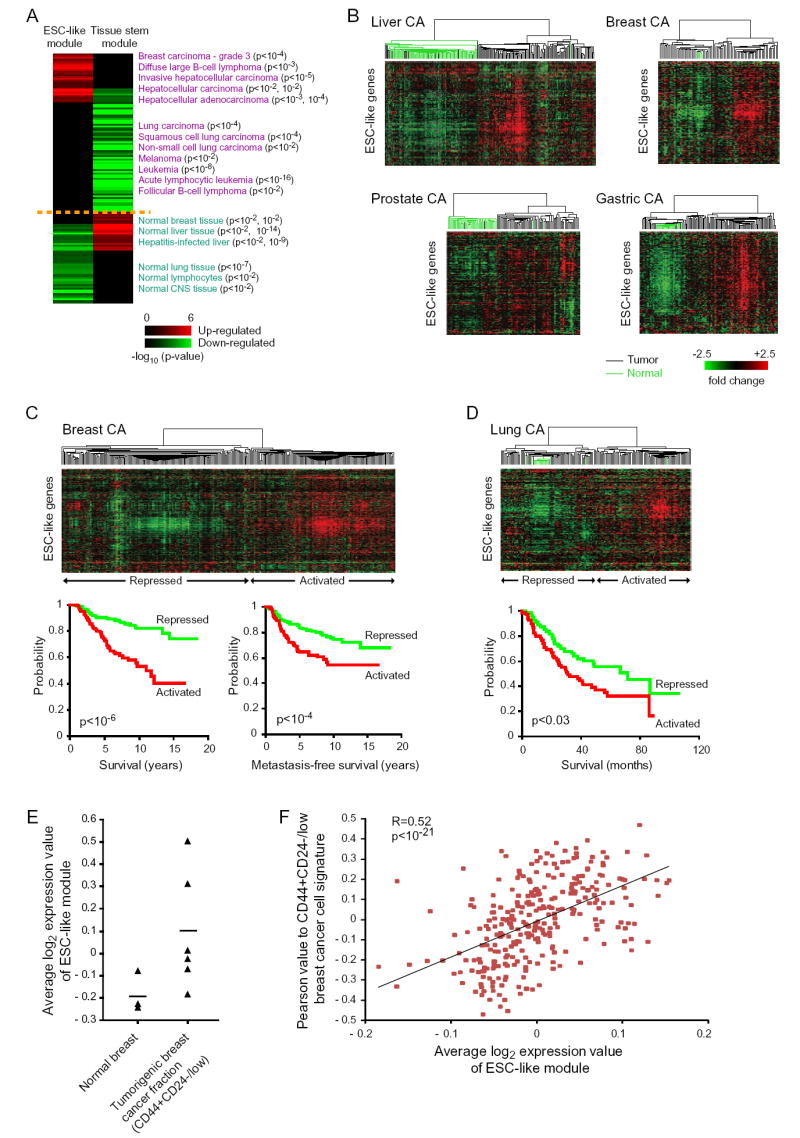
(A) Cancer gene module map: a matrix of stem cell gene modules (columns) and array clinical annotations (rows), where a red (or green) entry indicates that the arrays in which the corresponding module was significantly induced (or repressed) contained more arrays with a given annotation than would be expected by chance (FDR<0.05, p<0.01). The intensity of the entries corresponds to the significance, i.e. −log10 (p-value). A subset of significant annotations is shown; redundant annotations were removed for clarity.
(B-D) Expression patterns of ESC-like genes in primary human cancers and corresponding normal tissues are organized by hierarchical clustering. Note the coordinate regulation of the genes in the normal and cancer tissues. The dendrogram at the top of each data represent the similarities among the samples in their expression of ESC-like genes. Tumors are indicated by black branches, and normal tissues by green branches. Kaplan-Meier survival curves of tumors stratified into two classes based on expression of the ESC-like module are shown for data in (C) and (D).
(E) Average log2 expression value of all the genes in the ESC-like module in previously published expression arrays of normal breast and FACS-sorted CD44+CD24-/low tumorigenic breast cancer cells (Liu et al., 2007). A line indicating mean expression value among all samples is shown for each.
(F) Correlation of Pearson value to CD44+CD24-/low tumorigenic breast cancer cell signature with average log2 expression value of ESC-like module in the primary human breast cancers shown in (C).
To investigate whether the varying degrees of ESC-like module activation have clinical implications in human cancers, we examined the expression of the ESC-like genes in a consecutive series of 295 primary human stage I and II breast cancers (van de Vijver et al., 2002). The expression profiles were biphasic, allowing separation of these tumors into two classes: ESC-activated and ESC-repressed (Figure 2C). The primary breast cancers with the ESC-activated signature were significantly associated with poorer tumor differentiation (p<10-16; data not shown), consistent with cancers expressing a stem cell-like phenotype. In addition, these tumors with activated ESC signature were more likely to progress to metastasis and death (p< 10-4 and p<10-6, respectively; Figure 2C). Thus, the ESC-like signature is a powerful predictor of prognosis in human breast cancer. Multivariate analysis with patient age, tumor diameter, tumor grade, estrogen receptor status, lymph node status, vascular invasion status, mastectomy versus breast conserving therapy, use or nonuse of chemotherapy, use or nonuse of hormonal treatment, St. Gallen criteria, and National Institutes of Health consensus criteria, confirmed that the ESC-like signature is an independent prognostic factor of metastasis and death (hazard ratio=1.7, p<0.02 and hazard ratio=2.2, p<0.001, respectively). We also examined the expression of the ESC-like genes in 71 patients with stage I and II lung adenocarcinomas (Bhattacharjee et al., 2001). The expression profiles were biphasic, and the tumors with the ESC-activated signature were associated with significantly higher risk of death compared to tumors with the ESC-repressed signature (p<0.03; Figure 2D). These data demonstrate that the degree of ESC module activation is associated with poor differentiation and increased risk of metastasis and mortality in multiple tumor types.
All of the human cancer expression array data examined above were from bulk tumor tissue. However, multiple studies have demonstrated that only a small fraction of the cells in a cancer have the unique ability to initiate and perpetuate tumor growth and heterogeneity based on transplantation in immunodeficient mice (Al-Hajj et al., 2003; Bonnet and Dick, 1997). In order to determine whether the ESC-like gene expression program may be expressed in the tumorigenic cancer stem cell subpopulation, we first examined the expression of the ESC-like gene module in the CD44+CD24-/low subpopulation of breast cancer cells that are uniquely enriched for cancer stem cells (Liu et al., 2007). Liu et al. previously defined a gene expression signature based on the profiles of these enriched tumorigenic breast cancer cell populations relative to normal breast tissues. This 184-gene signature has an overlap of only four genes with our 335-gene core ESC-like gene module. However, despite the fact that the flow-sorted populations are only partially enriched for cancer stem cells, of the CD44+CD24-/low populations in six independent breast cancers, two had significantly up-regulated the ESC-like gene module relative to normal breast tissue (Figure 2E), suggesting that the ESC-like signature may be activated in cancer stem cell populations. We also examined the co-regulation of these two signatures in an independent set of primary human breast cancers (van de Vijver et al., 2002). Strikingly, expression of the ESC-like signature is significantly correlated with expression of the cancer stem cell enriched CD44+CD24-/low subpopulation signature in these primary human breast cancers (R=0.52, p<10-21; Figure 2F). Thus, these results suggest that the ESC-like signature may be activated in the tumorigenic fraction of cancer cells.
c-Myc activates the ESC module in adult epithelial cells
To gain further insight into the mechanisms by which the ESC-like module may be directly regulated, we performed a computational screen of cis-regulatory motifs that are enriched among the ESC-like module genes. We analyzed all 903 single, double, and three-way combinations of cis-regulatory motifs in TRANSFAC or identified by evolutionary conservation (Sinha et al., 2008). The top motifs or motif combinations significantly enriched among the ESC-like module genes are listed in Table S7. Notably, the cis-regulatory motifs that had the most occurrences at the top of the enriched list included that of c-Myc (binding motifs of MYC, MAX, and the MYC/MAX dimer) and HNF1 (binding motifs of HNF1, HNF1:SP1 pair, and HNF1:KROX pair). In combination with previous data showing that c-Myc is necessary and sufficient to maintain mouse ESC self-renewal (Cartwright et al., 2005) and is one of four genes sufficient to reprogram mouse and human fibroblasts to confer pluripotency indistinguishable from authentic ESCs (Takahashi et al., 2007; Takahashi and Yamanaka, 2006), this unbiased cis-regulatory motif analysis suggested that c-Myc may be a dominant regulator of the ESC-like program.
Based on this result, we next examined gene expression profiles of c-Myc as well as four additional oncogenic pathways (Bild et al., 2006). Intriguingly, we found that c-Myc, but not other oncogenic proteins such as Src, β-catenin, E2F3, and Ras, can coordinately induce expression of the ESC-like module in primary human mammary epithelial cells (Figure 3A, 3B). c-Myc also substantially inactivated the adult tissue stem gene module (Figure 3C). This result suggested that c-Myc may act as a switch between these apparently mutually exclusive transcriptional programs. We similarly observed induction of the ESC-like module by c-Myc in primary human fibroblasts and a nontransformed breast epithelial cell line (Figure S2A, S2B; Adler et al., 2006; Coller et al., 2000). In addition, 88 of the 335 (26%) ESC-like module genes have been previously identified as c-Myc targets (http://www.myc-cancer-gene.org/). To extend this result in vivo, we examined in detail the transcriptional response following induction of c-Myc in the epidermis by topical application of tamoxifen to K14-Myc-ER transgenic mice (Frye et al., 2003) and in pancreatic beta cells by systemic administration of tamoxifen in pIns-Myc-ER transgenic mice (Lawlor et al., 2006). Consistent with results in cultured cells, Myc-ER activation coordinately induced the ESC-like module (Figure 3D, 3E, S2C). In addition, Myc-ER activation silenced the adult tissue stem module (Figure 3F). Previous studies had documented that c-Myc induction in epidermis blocks hair follicular stem cell function (Arnold and Watt, 2001; Waikel et al., 2001). Our finding of the silencing of the adult tissue stem module (characteristic of hair bulge stem cells) therefore anticipates the previously observed biological phenotype and validates the gene module approach. In addition, these findings suggest that an unanticipated consequence of c-Myc induction is the reactivation of the ESC-like module in adult epithelia.
Figure 3. c-Myc Activates ESC Module in Epithelial Cells.
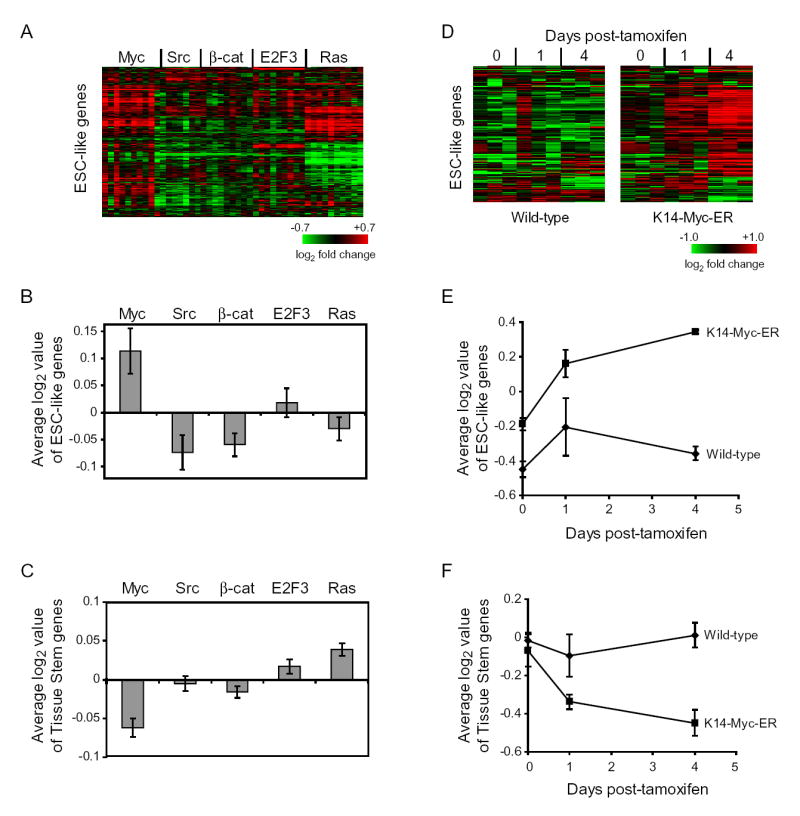
(A) Expression of ESC-like module genes (excluding the proliferation genes) in previously published expression arrays of primary human mammary epithelial cells transduced with indicated oncogenes relative to those cells transduced with GFP (Bild et al., 2006). Each row represents an individual gene within the ESC-like module and each column is an expression array. The level of expression of each gene in each sample relative to the mean level of expression of that gene in the cells transduced with GFP is represented using a red-green color scale. (B) Average log2 expression values of all of the genes in the ESC-like gene module are expressed as mean ± SEM. (C) Average log2 expression values of all of the genes in the adult tissue stem gene module are expressed as mean ± SEM.
(D) Expression of ESC-like module genes (excluding the proliferation genes) in previous published expression arrays of c-Myc induction in the epidermis of K14-Myc-ER transgenic mice, one and four days after topical application of tamoxifen (Frye et al., 2003). (E) Average log2 expression values of all of the genes in the ESC-like gene module are expressed as mean ± SEM. (F) Average log2 expression values of all of the genes in the adult tissue stem gene module are expressed as mean ± SEM.
c-Myc activates the ESC module in human cancer and induces epithelial tumor initiating cells
To determine the consequences of c-Myc-induced ESC module activation in human cancers, we turned to a genetically defined, in vivo model of human epithelial cancer. Primary human keratinocytes transduced with oncogenic Ras and a stabilized mutant of the NF-κB repressor IκBα, that are then grafted onto immunodeficient scid mice, produce well-differentiated squamous cell carcinomas (SCCs) that are phenotypically indistinguishable from authentic human lesions (Dajee et al., 2003). We retrovirally transduced primary human keratinocytes with oncogenic Ras and IκBα, plus either GFP, E2F3, or c-Myc. E2F3 serves as a control oncogene because it promotes proliferation but does not activate the ESC-like module (Figure 3A, 3B). Injection of 5 × 105 transduced keratinocytes of each genetic combination led to gross tumor formation with 100% efficiency, but with marked differences in gene expression and histology. First, microarray analysis showed that c-Myc, but not GFP or E2F3, led to the strong and coordinate activation of the ESC-like module in the generated SCCs, indicating that c- Myc expression is sufficient to activate the ESC-like module in human epithelial tumors in vivo (Figure 4A). For instance, c-Myc induced SOX2, a key transcriptional regulator in embryonic stem cells. In addition to activation of specific genes, ESCs are also characterized by silencing lineage-specific developmental regulators, frequently marked by “bivalent” chromatin domains bearing both histone H3 K4 and K27 methylation. Of the 87 genes previously described within the “bivalent” chromatin domains (Bernstein et al., 2006), 49 were present on our expression arrays; 26 of these 49 genes significantly changed in expression and all but two were repressed in the c-Myc-Ras-IκBα tumors relative to the GFP-Ras-IκBα tumors (Figure 4B). Thus, c-Myc expression led to transcriptional silencing of canonical ESC bivalent genes, such as HOX genes. Strikingly, the c-Myc-Ras-IκBα tumors also specifically activated a gene expression signature of the cancer stem cell as previously defined by the CD44+CD24-/low population (Figure 4C; Liu et al., 2007), suggesting an increased cancer stem cell population in the c-Myc-Ras-IκBα tumors. Thus, these three independent gene signatures—the ESC-like module, ESC bivalent genes, and a signature of CD44+CD24/low cells enriched for cancer stem cells—suggest a reprogramming of the cancer cells to a state resembling ESCs and potentially cancer stem cells.
Figure 4. c-Myc Induces ESC-like State in Cancer.
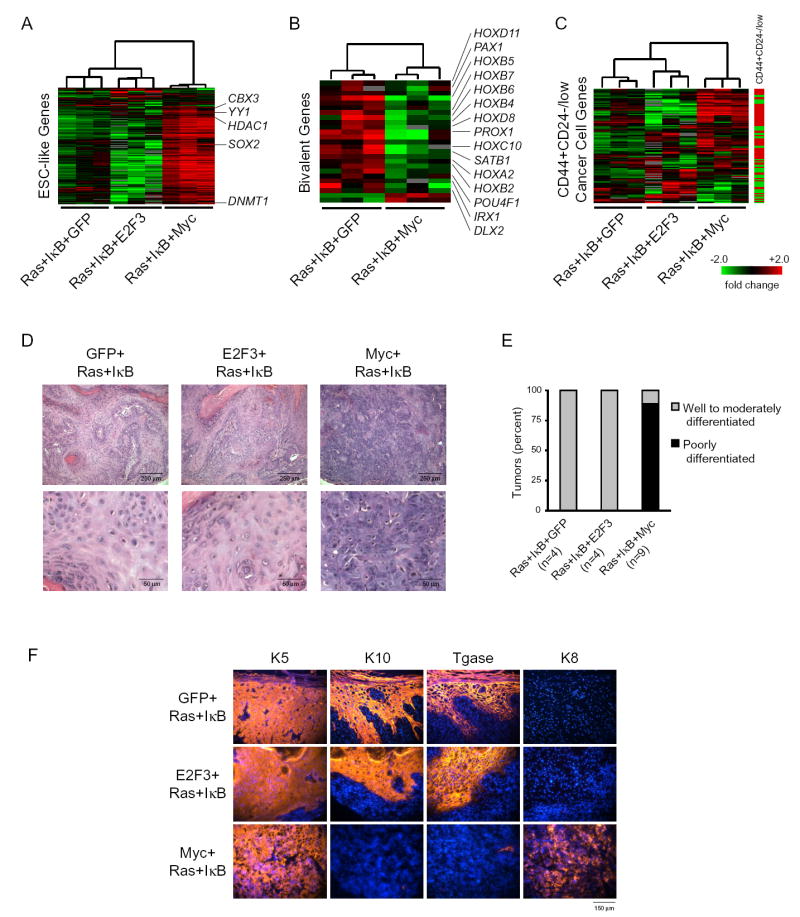
Subcutaneous scid mouse tumors derived from transduced primary human keratinocytes expressing the indicated proteins:
(A-C) Each row represents an individual gene and each column is an expression array of a subcutaneous tumor. The level of expression of each gene in each sample was normalized relative to its average expression in all the samples and is represented using a red-green color scale.
(A) RNA expression of genes in the ESC-like module.
(B) RNA expression of genes with bivalent chromatin domains that are repressed in ESCs (Bernstein et al., 2006).
(C) RNA expression of genes in a signature of CD44+CD24-/low cancer cell subpopulation enriched for cancer stem cells (Liu et al., 2007). The CD44+CD24-/low signature is composed of genes that are induced and repressed which is shown on the bar to the right (red is induced; green is repressed).
(D) Histology of subcutaneous tumors.
(E) Grades of subcutaneous tumors.
(F) Subcutaneous tumor tissue: protein expression of keratinocyte differentiation markers keratin 5 (K5), keratin 10 (K10), transglutaminase 1 (Tgase), and keratin 8 (K8), all in orange. Hoechst 33342 nuclear staining in blue.
c-Myc-transduced tumors were also strikingly different in histological appearance (Figure 4D). Despite having a high frequency of mitotic figures, the E2F3-Ras-IκBα tumors, like control GFP-Ras-IκBα tumors, were well-differentiated squamous cell carcinomas, characterized by mitotically active cells adhering to basement membranes that stratify to give rise to eosinophilic cells with decreased nuclear to cytoplasmic ratios and keratin whorls (Figure 4D and S3A; data not shown). In contrast, the c-Myc-Ras-IκBα tumors formed poorly differentiated tumors, composed of mostly basophilic cells with nuclear pleomorphism and atypia with high nuclear to cytoplasmic ratios. Moreover, c-Myc-transduced tumors showed absence of stratification and tissue polarity. Grading of the tumors on histologic sections by a dermatopathologist unaware of the genotypes confirmed the poor differentiation of the tumors that overexpressed c-Myc (Figure 4E). Immunofluorescence staining demonstrated that the c-Myc-Ras-IκBα tumors lost expression of differentiated epidermal markers, including transglutaminase 1 and keratin 10 (Figure 4F), demonstrating that c-Myc is sufficient to inhibit differentiation of the keratinocyte-derived carcinoma. The c-Myc-Ras-IκBα tumors retained keratins 5 and 14, which are expressed by the basal cell compartment of the skin epidermis that contains the epidermal stem cells, demonstrating that they still express the undifferentiated markers of the keratinocyte lineage (Figure 4F; data not shown). In addition, the c-Myc-Ras-IκBα tumors activated expression of keratin 8, which is a marker of simple epithelia, suggesting that c-Myc induces de-differentiation (Figure 4F). The same histological results were seen with tumors derived from transduced keratinocytes that were placed on devitalized dermis and grafted onto scid mice (data not shown).
The ability of c-Myc to induce an ESC-like transcriptional program and tumor de-differentiation raised the possibility that c-Myc may promote the formation of epithelial cancer stem cells. Therefore, we performed a limiting dilution analysis to determine whether c-Myc was sufficient to increase the fraction of epithelial tumor-initiating cells (Table 1). Transduced keratinocytes were diluted in a ten-fold series from 5 × 105 to 50 cells prior to subcutaneous injection into scid mice. With 5×105 injected cells, co-expression of c-Myc or E2F3 produced more rapidly growing tumors than cells transduced with GFP-Ras-IκBα confirming the expected role of c-Myc and E2F3 in tumor mass proliferation (Figure 5A). With 5×104 injected cells, both E2F3- and GFP-transduced tumors grew much more slowly and formed tumors at substantially lower efficiency compared to c-Myc-transduced tumors (Figure 5B). Strikingly, all 16 injections of 5 × 103 c-Myc-Ras-IκBα cells formed tumors, whereas none of the injections of 5 × 103 GFP-Ras-IκBα or E2F3-Ras-IκBα cells formed a tumor (Figure 5C). In fact, as few as 500 c-Myc-Ras-IκBα cells, were sufficient to form a tumor in three of 17 injections (Figure 5D). All of the c-Myc-Ras-IκBα tumors formed were histologically identical to those formed with 5 × 105 cells (Figure S3B). Based on a Poisson distribution (Fazekas de St, 1982), we estimate that c-Myc increased the frequency of successful tumor initiation events from 1/330,000 to 1/2,200 cells, a 150-fold increase in efficiency. We next performed serial re-transplantation experiments from three c-Myc-Ras-IκBα tumors derived from initial injection of as few as 500 cells, as well as from two GFP-Ras-IκBα tumors. Tumors were excised at six weeks after initial injection and digested into a single cell suspension, then re-injected into new recipient scid mice. Neither of the two injections from the GFP-Ras-IκBα tumors formed secondary tumors. In contrast, all three injections from the c-Myc-Ras-IκBα tumors formed secondary tumors with identical histology to the original tumor (Figure S3C) and identical growth kinetics, providing further evidence for self-renewal capacity of experimentally created tumor initiating cells. Thus, c-Myc is sufficient to induce a dramatic increase in the cancer stem cell population.
Table 1.
Tumorigenicity Is Highly Enriched by c-Myc
| # cells per injection
|
|||||
|---|---|---|---|---|---|
| Genotype | 5 × 105 | 5 × 104 | 5 × 103 | 5 × 102 | 5 × 101 |
| GFP+Ras+IκB | 3/3 | 1/7 | 0/10 | 0/10 | 0/10 |
| E2F3+Ras+IκB | 3/3 | 2/5 | 0/12 | 0/12 | 0/10 |
| c-Myc+Ras+IκB | 3/3 | 7/7 | 16/16 | 3/17 | 0/17 |
Number of tumors formed over number of subcutaneous injections performed is shown.
Figure 5. c-Myc Increases Fraction of Tumor-Initiating Cells.
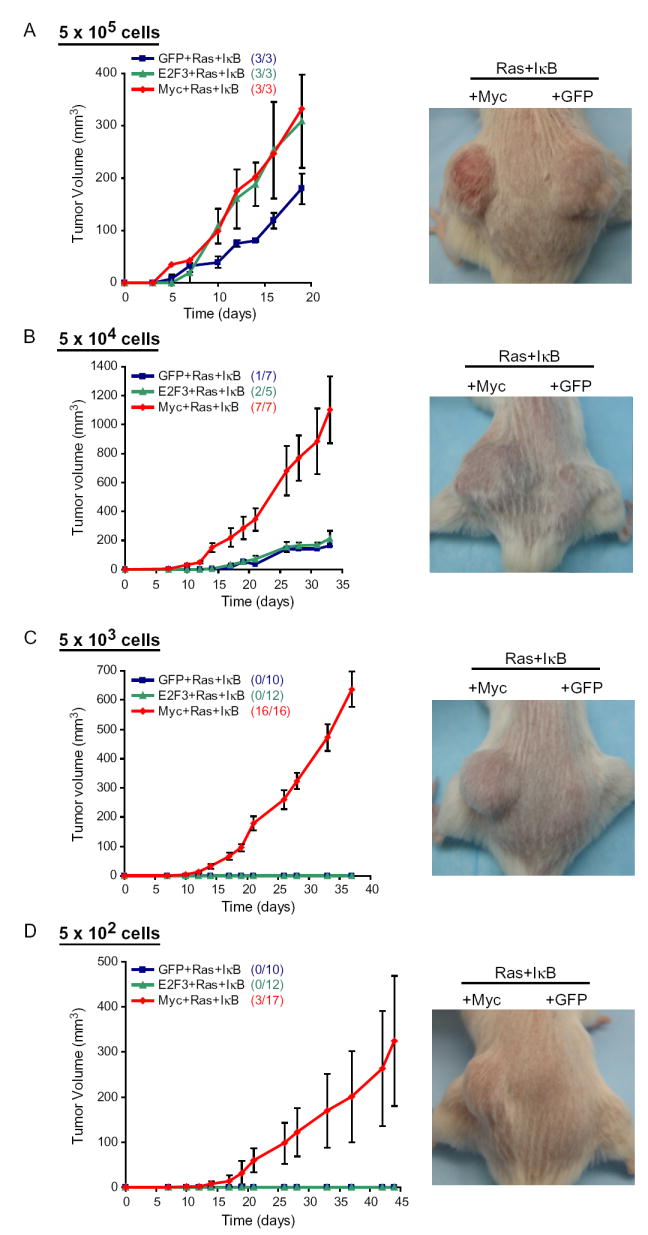
Left: Growth kinetics of tumors formed after subcutaneous injection of transduced primary human keratinocytes expressing the indicated proteins (mean ± SEM). Right: Representative pictures of subcutaneous tumors in scid mice: c-Myc+Ras+IκB tumors on left flank and GFP+Ras+IκB tumors on right flank. (A) 5×105 cells per subcutaneous injection. Picture is at day 18 post-injection. (B) 5×104 cells. The growth kinetics shown for GFP+Ras+IκB is of the one tumor that formed. The growth kinetics shown for E2F3+Ras+IκB is the mean ± SEM of the two tumors that formed. Picture is at day 20. (C) 5×103 cells. Picture is at day 28. (D) 5×102 cells. The growth kinetics shown for Myc+Ras+IκB is the mean ± SEM of the three tumors that formed. Picture is at day 45.
c-Myc-mediated tumor initation is cell-autonomous and not linked with genomic instability
c-Myc may increase the success of tumor initiation by increasing the fraction or efficiency of tumor-initiating cells (the seed); alternatively, c-Myc may enhance the cancer stem cell niche by altering the tumor microenvironment (the soil). To distinguish between these possibilities, we performed a cell-mixing experiment. As shown above in the limiting dilution analysis, 5 × 104 GFP-Ras-IκBα cells forms a tumor at a substantially slower rate and efficiency than 5 × 104 c-Myc-Ras-IκBα cells. We mixed 5 × 104 c-Myc-Ras-IκBα cells and 5 × 104 GFP-Ras-IκBα cells, and then injected these cells subcutaneously into scid mice (n=3). The resulting tumors appeared histologically identical to tumors injected with c-Myc-Ras-IκBα cells alone and were derived exclusively from c-Myc-transduced cells (Figure 6A). The lack of promotion of GFP-marked, non-c-Myc transduced cells suggests that the remarkable improvement in tumor initiation is a cell-autonomous effect of c-Myc, and thus is likely due to an effect on inducing tumor-initiating cells.
Figure 6. c-Myc-mediated Tumor Initation Is Cell-Autonomous and Not Linked with Genomic Instability.
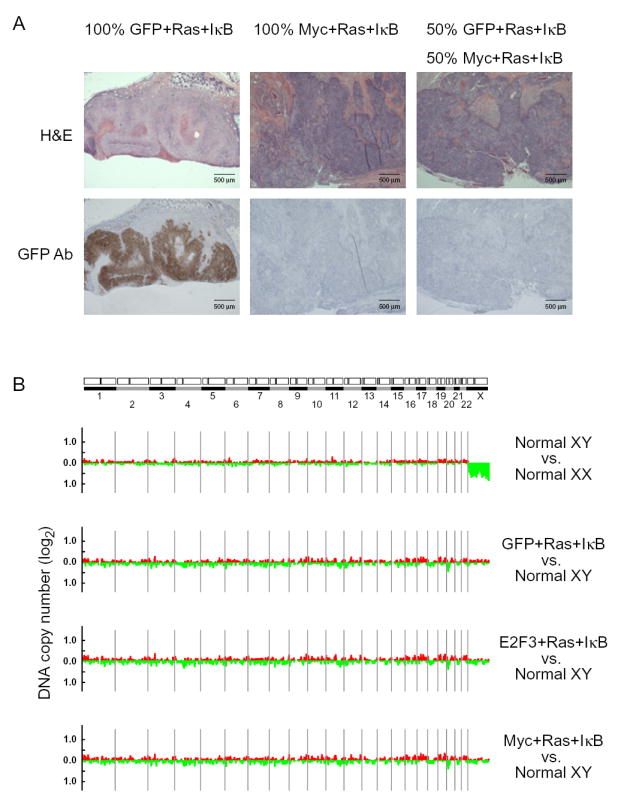
(A) Subcutaneous scid mouse tumors derived from injection of 5×104 transduced primary human keratinocytes expressing GFP, Ras, and IκB (100% GFP+Ras+IκB), 5×104 cells expressing c-Myc, Ras, and IκB (100% Myc+Ras+IκB), or 2.5×104 cells expressing GFP, Ras, and IκB plus 2.5×104 cells expressing c-Myc, Ras, and IκB (50% GFP+Ras+IκB, 50% Myc+Ras+IκB). Histology and immunohistochemistry with antibody against GFP. (B) Graphical displays of genome-wide DNA copy number alteration of subcutaneous tumors using array-based comparative genomic hybridization. Ratios are plotted on a log2 scale according to chromosome position.
In addition to its role in gene regulation, c-Myc expression can also induce genomic instability in certain settings (Felsher and Bishop, 1999). A possible alternative explanation of these results is the accumulation of de novo mutations in c-Myc-transduced cells, leading to the acquisition of tumor-initiation activity. To evaluate this possibility, we performed array-based comparative genomic hybridization of wild-type cells and tumors with all three genotypes and found no evidence of significant genomic amplifications or deletions in any of the tumors (Figure 6B). Thus, the increase in tumor-initiating cells in the c-Myc-Ras-IκBα tumors is not due to induction of genomic instability. While these results do not exclude the possibility of additional functions of c-Myc that may contribute to tumor initiation, they support the idea that c-Myc-mediated activation of tumor initiation likely occurs in a direct manner.
DISCUSSION
Module map of stem cells as an engine of biological discovery
By assembling and relating the gene expression signatures of ESC and diverse tissue stem cells, we have created a module map where diverse sources of data can be integrated and higher order relationships among the transcriptional programs of stem cells populations may be revealed. Where gene-level analyses failed to observe commonalities in gene expression between stem cell signatures identified by different labs (Fortunel et al., 2003), the module map was able to systematically organize ESCs and various adult tissue stem cells profiled by at least two or more laboratories with their expected biological neighbors. Moreover, the map confirms that there is no single “stemness” program shared across all stem cells. Instead, select tissue stem cells, notably fetal liver HSCs, retinal stem cells, and a subset of NSCs, share a core transcriptional program with ESCs. The finding that NSCs and retinal stem cells are the only normal adult stem cells examined to share the ESC module is intriguing and may relate to the fact that neuralization is the default, presumably ground state, ectodermal fate during development (Weinstein and Hemmati-Brivanlou, 1999). In contrast, the majority of adult tissue stem cells examined, including neural crest, hair bulge, bone marrow HSCs, and mammary stem cells share a distinct transcriptional program. The map also defined the gene composition of the two stem cell modules, which opens future investigation into the unique molecular networks of these two distinct stem cell classes.
In addition to classifying different types of stem cells, the map of gene modules also facilitated the connection of stem cell transcriptional programs to human cancers and mechanistic investigations of their control. We found that the ESC-like module was induced in many aggressive human epithelial cancers; the ESC module can be induced by c-Myc, creating highly tumorigenic cells that can initiate cancer with as few as 500 cells. The map can be used to establish improved standards for classifying and defining stem cells, by using the expression signature modules as “fingerprints” rather than reliance on just a few molecular markers. Further studies will be needed to determine the specificity of the ESC-like signature as a marker for cancer stem cells. In addition, this map can be used to ask additional questions about the role of stem cell expression programs in other disease states as well as normal development.
Experimental induction of epithelial tumor initiating cells by reactivation of an ESC transcriptional program
Because purified fractions of putative cancer stem cells can initiate tumor growth with a few hundred cells while hundreds of thousands of other cells from the bulk tumor cannot, cancer stem cells are likely key cells-of-origin for cancer establishment and metastasis. We show that the reactivation of an ESC-like transcriptional program in primary human keratinocytes, mediated by c-Myc, is able to create cells that can initiate epithelial tumor growth with comparable efficiency as bona fide cancer stem cells, at least as defined by current standards. The ESC-like transcriptional program we defined is clinically relevant because it is frequently activated in human epithelial cancers and predicts cancer metastasis and death. Further, the ESC-like gene module is enriched in tumor initiating cells purified from authentic human cancers. Our findings suggest that inappropriate activation of the ESC-like transcriptional program in adult epithelial cells can induce epithelial tumor initiating cells. Because previous studies in leukemia stem cells have demonstrated reactivation of HSC transcriptional programs (Krivtsov et al., 2006), reactivation of tissue-specific, adult stem cell programs may have been predicted to underlie cancer stem cells from different epithelial cancers. However, ectopic activation of Oct4, an ESC-specific transcription factor, in adult epithelia induces dysplasia (Hochedlinger et al., 2005), suggesting that off-lineage activation of an ESC-like transcriptional program can promote cancer. Our results add to the latter idea and suggest that ESC-like module activation is prevalent in common human epithelial cancers. A relationship between increased tumor initiating cell numbers and metastatic potential is intriguing, but its mechanistic basis will require further investigation.
The ability to experimentally create cells with many properties of epithelial cancer stem cells has several potentially important implications. Currently, purification of cancer stem cells from human tumors is limited by patient heterogeneity and availability of fresh and sizable tumor samples. Moreover, current markers enrich for but do not allow complete purification of human cancer stem cells. Our findings suggest an alternative route to obtain epithelial cancer stem cells, offering many new possibilities to improve cancer diagnosis and treatment. For instance, the gene expression profiles of genetically induced tumor-initiating cells can be used to identify cell surface or secreted proteins enriched in this population, which may be used to guide the identification, further purification, and blood-based detection of incipient cancer. Uniform sources of experimentally created tumor initiating cells may also be used for drug screening to identify compounds that specifically target cancer stem cells. Further, a key goal in cancer research is to identify the mechanism by which cancer stem cells arise and self-renew. Experimentally created and genetically defined tumor initiating cells offers the possibility of dissecting the temporal and functional dynamics of these processes.
c-Myc as a switch of stem cell transcriptional programs
c-Myc represents one of likely several oncogenes that play a role in normal stem cell biology and may also promote the formation of cancer stem cells. Our finding that c-Myc activates the ESC-like program but down-regulates the adult tissue stem program is consistent with previous studies showing that sustained c-Myc promotes self-renewal and blocks differentiation in ESCs and cancer, but conversely promotes differentiation with depletion of stem cells in hair bulge and bone marrow (Arnold and Watt, 2001; Arvanitis and Felsher, 2006; Cartwright et al., 2005; Waikel et al., 2001; Wilson et al., 2004). The recent finding of c-Myc as one of the four key genes sufficient to reprogram mouse or human fibroblasts into embryonic stem cells highlights the importance of c-Myc as an enforcer of ESC fate. This unique function may relate to c-Myc’s ability to globally reprogram chromatin (Knoepfler et al., 2006; Wu et al., 2007). The concept of cancer stem cells has recently been challenged by the observation of frequent (>1/10) tumor initiating cells in murine lymphoma (Kelly et al., 2007); interestingly, the strongest effect came from the Eμ-Myc model. Our data provide a potential explanation for these findings and reinforce the unique role of c-Myc as an oncogene.
Our results suggest that the ESC-like program is not simply a proliferation signature. The ESC-like program, derived from a gene module map without gene sets associated with proliferation, was activated in diverse human cancers and predictive of mortality and metastasis. Moreover, enforced cell cycle entry by E2F3 overexpression was able to activate a proliferation signature but unable to induce the ESC-like signature or enhance tumor initiation, indicating that c-Myc’s role is more than simply driving cell proliferation. However, we were not able to demonstrate that the ESC-like signature provides independent prognostic value over a proliferation signature; nor is it clear whether the ESC-like signature is expressed in rare cancer stem cells or the bulk tumor in human cancer samples. Thus, additional studies will be needed to address these important issues.
EXPERIMENTAL PROCEDURES
Gene Module Map
Gene module map was implemented with the program Genomica (Segal et al., 2004). We downloaded data available on GEO database or as attachments to manuscripts on journal websites as well as those given directly from authors upon request (Table S1). We normalized the expression of each gene in every data set separately. Genomica and the full gene module maps can be downloaded and interactively queried at http://genie.weizmann.ac.il.
Data Analysis
All of the analyses of the ESC-like gene module were performed both including and excluding the proliferation genes within the module and showed consistent results regardless of inclusion or exclusion of those genes. In all of the figures except for Figure 3, the ESC-like gene module including the proliferation genes is shown. For clarity, in Figure 3, the results of the analyses are shown using the ESC-like module excluding the proliferation genes because E2F3 induces the proliferation genes (Figure S4) but not the remainder of the ESC-like module.
Cell Culture and Gene Transfer
Coding regions of human c-Myc, E2F3, Ha-Ras G12V, and IκBαM, as well as GFP were subcloned into the LZRS retroviral vector. Virus was produced as described previously (Robbins et al., 2001). Primary human keratinocytes isolated from normal human skin were serially transduced at intervals of 12-24 hours. We verified that gene transfer efficiency was higher than 99% by immunostaining.
Animal Studies
8-week-old scid mice were injected subcutaneously with cells resuspended in 100 μl PBS and 50 μl Matrigel. Tumor growth was followed for 10 weeks. For serial propagation assays, subcutaneous tumors were excised, minced, and digested into a single cell suspension, prior to subcutaneous injection into scid mice. All animal studies were conducted in compliance with appropriate Stanford institutional review boards.
Microarrays
Total RNA from the subcutaneous tumors was purified, amplified and then labeled by direct incorporation of Cy5. Universal human RNA reference from pooled cell lines (Stratagene) was amplified and labeled by direct incorporation of Cy3. These fluorescent cDNA probes were hybridized to human cDNA arrays containing over 41,000 elements, representing over 27,000 unique genes. Array data is publicly available at GEO GSE10423.
Tissue Analysis
For histological analysis, a portion of each subcutaneous tumor was fixed in 10% formalin (Sigma-Aldrich) and embedded in paraffin. Immunohistochemistry was performed on the formalin-fixed paraffin sections with antibody specific to GFP (Abcam). Another portion of each subcutaneous tumor was snap-frozen in OCT for immunofluorescence. Nuclei were stained with Hoeschst dye 33342. Primary antibodies used were specific to keratin 5 (Covance), keratin 8 (Abcam), keratin 10 (Neomarkers), keratin 14 (Covance), and transglutaminase 1 (Biomedical Tech).
Comparative Genomic Hybridization
Genomic DNA from the subcutaneous tumors was isolated, digested, and then labeled by direct incorporation with Cy5. DNA isolated from the blood of a normal donor was used as reference, which was labeled with Cy3. Labeled tumor and reference DNAs were competitively hybridized to human cDNA microarrays as previously described (Pollack et al., 1999).
Supplementary Material
Acknowledgments
We thank P.A. Khavari, D.W. Felsher, J.L. Rinn, and A.S. Adler for comments on the manuscript. Supported by Dermatology Foundation Research Career Development Award (D.J.W.), American Cancer Society RSG 07-084-01 (H.Y.C.), NIH R01-CA118750 (H.Y.C.) and R01-CA119176 (E.S.). E.S. is the incumbent of the Soretta and Henry Shapiro career development chair. H.Y.C. is a Kenneth G. and Elaine A. Langone Scholar of the Damon Runyon Cancer Research Foundation.
References
- Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–430. doi: 10.1038/ng1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold I, Watt FM. c-Myc activation in transgenic mouse epidermis results in mobilization of stem cells and differentiation of their progeny. Curr Biol. 2001;11:558–568. doi: 10.1016/s0960-9822(01)00154-3. [DOI] [PubMed] [Google Scholar]
- Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313–317. doi: 10.1016/j.semcancer.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–13795. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild A, Febbo PG. Application of a priori established gene sets to discover biologically important differential expression in microarray data. Proc Natl Acad Sci U S A. 2005;102:15278–15279. doi: 10.1073/pnas.0507477102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y, et al. NF-kappaB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature. 2003;421:639–643. doi: 10.1038/nature01283. [DOI] [PubMed] [Google Scholar]
- Fazekas de St G. The evaluation of limiting dilution assays. J Immunol Methods. 1982;49:R11–23. doi: 10.1016/0022-1759(82)90269-1. [DOI] [PubMed] [Google Scholar]
- Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunel NO, Otu HH, Ng HH, Chen J, Mu X, Chevassut T, Li X, Joseph M, Bailey C, Hatzfeld JA, et al. Comment on “ ‘Stemness’: transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science. 2003;302:393. doi: 10.1126/science.1086384. author reply 393. [DOI] [PubMed] [Google Scholar]
- Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development. 2003;130:2793–2808. doi: 10.1242/dev.00462. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR. A stem cell molecular signature. Science. 2002;298:601–604. doi: 10.1126/science.1073823. [DOI] [PubMed] [Google Scholar]
- Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317:337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc influences global chromatin structure. Embo J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Lawlor ER, Soucek L, Brown-Swigart L, Shchors K, Bialucha CU, Evan GI. Reversible kinetic analysis of Myc targets in vivo provides novel insights into Myc-mediated tumorigenesis. Cancer Res. 2006;66:4591–4601. doi: 10.1158/0008-5472.CAN-05-3826. [DOI] [PubMed] [Google Scholar]
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- Robbins PB, Lin Q, Goodnough JB, Tian H, Chen X, Khavari PA. In vivo restoration of laminin 5 beta 3 expression and function in junctional epidermolysis bullosa. Proc. Natl Acad Sci U S A. 2001;98:5193–5198. doi: 10.1073/pnas.091484998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Sinha S, Adler AS, Field Y, Chang HY, Segal E. Systematic functional characterization of cis-regulatory motifs in human core promoters. Genome Res. 2008 doi: 10.1101/gr.6828808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack JM. Metaplasia and transdifferentiation: from pure biology to the clinic. Nat Rev Mol Cell Biol. 2007;8:369–378. doi: 10.1038/nrm2146. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:1–12. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet. 2001;28:165–168. doi: 10.1038/88889. [DOI] [PubMed] [Google Scholar]
- Weinstein DC, Hemmati-Brivanlou A. Neural induction. Annu Rev Cell Dev Biol. 1999;15:411–433. doi: 10.1146/annurev.cellbio.15.1.411. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.