

Abstract
Tumors in bone are associated with pain in humans. Data generated in a murine model of bone cancer pain suggest that a disturbance of local endocannabinoid signaling contributes to the pain. When tumors formed after injection of osteolytic fibrosarcoma cells into the calcaneus bone of mice, cutaneous mechanical hyperalgesia was associated with a decrease in the level of anandamide (AEA) in plantar paw skin ipsilateral to tumors. The decrease in AEA occurred in conjunction with increased degradation of AEA by fatty acid amide hydrolase (FAAH). Intraplantar injection of AEA reduced the hyperalgesia, and intraplantar injection of URB597, an inhibitor of FAAH, increased the local level of AEA and also reduced hyperalgesia. An increase in FAAH mRNA and enzyme activity in dorsal root ganglia (DRG) L3–L5 ipsilateral to the affected paw suggests DRG neurons contribute to the increased FAAH activity in skin in tumor-bearing mice. Importantly, the anti-hyperalgesic effects of AEA and URB597 were blocked by a CB1 receptor antagonist. Increased expression of CB1 receptors by DRG neurons ipsilateral to tumor-bearing limbs may contribute to the anti-hyperalgesic effect of elevated AEA levels. Furthermore, CB1 receptor protein-immunoreactivity as well as inhibitory effects of AEA and URB597 on the depolarization-evoked Ca2+ transient were increased in small DRG neurons cocultured with fibrosarcoma cells indicating that fibrosarcoma cells are sufficient to evoke phenotypic changes in AEA signaling in DRG neurons. Together, the data provide evidence that manipulation of peripheral endocannabinoid signaling is a promising strategy for the management of bone cancer pain.
Keywords: cannabinoid, CB1 receptor, dorsal root ganglion (Drg), hyperalgesia, mice, skin, culture
Introduction
Bone destruction from primary or metastatic tumors is associated with severe pain (Coward and Wilkie, 2000; Serafini, 2001). Tumor growth within the bone is accompanied by infiltration and compression of peripheral nerves as well as release of inflammatory and tumorigenic components, and these factors lead to development of ongoing and movement-evoked pain (McBride, 1997; Mercadante and Arcuri, 1998; Wacnik et al., 2001, 2005; Luger et al., 2005). Because of its complex nature and multifactor origin, bone cancer pain is difficult to manage and is relatively resistant to suppression by morphine (Luger et al., 2002; Srivastava and Walsh, 2003; Wacnik et al., 2003; Mancini et al., 2004; Yamamoto et al., 2008). Cannabinoids offer an alternative opportunity for management of cancer pain.
Constituents of the cannabis plant as well as synthetic cannabinoids are effective analgesics in the management of inflammatory, neuropathic (for review, see Cheng and Hitchcock, 2007) and cancer pain (Kehl et al., 2003; Hamamoto et al., 2007; Guerrero et al., 2008; Maida et al., 2008; Potenzieri et al., 2008). The effectiveness of these agents occurs through the activation of central and peripheral cannabinoid receptors (Pertwee, 2001; Sagar et al., 2005; Gutierrez et al., 2007). These compounds mimic the effects of endogenous cannabinoid ligands which have been shown to tonically modulate nociceptive processing (Strangman et al., 1998; Chapman, 1999; Agarwal et al., 2007).
N-arachidonoyl ethanolamine (anandamide, AEA), a well characterized endocannabinoid, is produced by neuronal cells on demand (Di Marzo et al., 1994) and acts as a partial agonist at cannabinoid (CB)-1 and -2 receptors (Hillard, 2000; Pertwee and Ross, 2002; Padgett et al., 2008). Inhibitory effects after activation of CB1 receptors on DRG neurons are linked to the suppression of Ca2+ and Na+ channels (Vásquez and Lewis, 1999; Ross et al., 2001; Khasabova et al., 2002, 2004; Kim et al., 2005). Although the intraplantar injection of AEA suppresses neuropathic (Guindon and Beaulieu, 2006) and inflammatory pain (Guindon et al., 2006), its rapid catabolism in vivo limits its therapeutic use. Thus, compounds that decrease the degradation of endogenous AEA are generating great interest. AEA is catabolized by the enzyme fatty acid amide hydrolase (FAAH, Cravatt et al., 2001). The FAAH inhibitor cyclohexylcarbamic acid 3-carbamoyl biphenyl-3-yl ester (URB597) reduces hyperalgesia in models of inflammatory and neuropathic pain, and its effectiveness is associated with increased levels of anandamide (Lichtman et al., 2004; Fegley et al., 2005; Hohmann et al., 2005; Holt et al., 2005; Jhaveri et al., 2006).
Here we describe biochemical, cellular and molecular changes in peripheral endocannabinoid signaling that contribute to mechanical hyperalgesia in a murine model of bone cancer pain. Parallel studies in vivo and in vitro resolved a contribution of the endocannabinoid system in primary afferent neurons to the development of cancer-related cutaneous mechanical hyperalgesia and provide a rationale for inhibition of AEA degradation in the management of bone cancer pain. The data support the strategy of applying therapeutic interventions at the site of pathophysiology in the periphery to minimize centrally mediated side effects.
Materials and Methods
Animals.
Adult, male C3H/HeN mice (25–30 g, National Cancer Institute) were used in the studies. This strain is syngeneic to the fibrosarcoma cells used in the experiments and allows the fibrosarcoma cells to grow tumors without rejection (Clohisy et al., 1995). All procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Implantation of fibrosarcoma cells.
NCTC clone 2472 fibrosarcoma cells (American Type Culture Collection) were maintained as described previously (Clohisy et al., 1995). Fibrosarcoma cells (2 × 105) were injected unilaterally into and around the calcaneus bone of mice in 10 μl of PBS, pH 7.3, under isoflurane anesthesia (Cain et al., 2001). Histological studies documented that this approach produces a tumor with bone osteolysis (Wacnik et al., 2001).
Assay of mechanical hyperalgesia in tumor-bearing mice.
Hyperalgesia was defined as an increase in the frequency of paw withdrawal in response to a von Frey monofilament with a bending force of 3.7 mN. This filament evoked a paw withdrawal frequency of ∼11% in naive mice. Access to the plantar surface of hindpaws was managed by placing animals individually on a raised mesh platform under a glass container. Mice were allowed to acclimate to the environment for at least 30 min before testing. The monofilament was applied 10× for 2 s each time to random locations on the plantar surface of each hindpaw, and the number of withdrawal responses evoked by the monofilament was counted and expressed as a percentage of total stimuli (withdrawal frequency). In tumor-bearing mice, withdrawal responses to this monofilament are reproducible and sufficiently large to allow detection of dose-dependent effects of analgesics (Wacnik et al., 2001; Hamamoto et al., 2007). Baseline responses were determined daily for each animal for 3 d before implantation of fibrosarcoma cells or vehicle. Mice with baseline withdrawal frequencies of 50% or more (< 5% of mice) were eliminated from further experiments. On day 10 after implantation of fibrosarcoma cells, mechanical hyperalgesia was confirmed before drug administration. Thirty minutes after collection of baseline withdrawal responses, drugs or vehicle were injected subcutaneously into the plantar surface [intraplantar (ipl)] of one hindpaw in a volume of 10 μl. Withdrawal responses evoked by the monofilament were measured every 30 min for at least 3 h after the injection. The individual determining mechanical responses after injection of drugs was blinded to the treatment.
Assay of mechanical sensitivity in naive mice.
Sensitivity of the hindpaws of naive mice to mechanical stimuli was monitored using a repetitive stimulation paradigm (Joseph et al., 2003). Six von Frey monofilaments with ascending bending forces of 3.7, 5.9, 9.8, 13.7, 19.6, and 34 mN were used. Baseline withdrawal responses to each monofilament were determined for each animal daily for 4 d before drug administration. The frequency of withdrawal to each mechanical stimulus was determined before and 120 min after intraplantar injection of drugs or vehicle. For this report, mechanical withdrawal threshold was defined as the force that produced a 50% frequency of withdrawal. The individual determining mechanical responses after injection of drugs was blinded to the treatment.
Measurement of endogenous AEA.
Lipids were extracted from tissue samples as described by Maione et al. (2007). Immediately after decapitation under a surgical level of anesthesia, samples of hindpaw plantar skin were removed from naive mice and ipsilateral to tumors in tumor-bearing mice, frozen in liquid nitrogen, and kept frozen at −80°C until the time of processing. On the first day of processing, tissues were weighed, minced, and extracted with five volumes of chloroform at 4°C overnight. Mixtures were then homogenized with an equal volume of methanol: Tris-HCl 50 mm (1:1) containing 10 pmol of deuterated d8-AEA (Cayman Chemical) as an internal standard. Homogenates were centrifuged at 3000 × g for 15 min (4°C); the aqueous phase plus debris were collected and extracted again with 1 volume of chloroform. The organic phases were pooled and evaporated with a gentle stream of nitrogen gas. Dried samples were stored at −80°C until analyzed. Targeted isotope-dilution HPLC/atmospheric pressure chemical ionization/mass spectrometry was conducted on each sample. A Phenomex Prodigy (ODS-3, 150 × 2 mm) column was used. Mobile phase A was 0.5% formic acid in water and phase B was 0.5% formic acid in acetonitrile. The flow rate was 0.1 ml/min with a gradient that began with 95% A:5% B for 1 min followed by a ramp to 100% B over 10 min and maintenance in buffer B for another 16 min. Analyses were performed by the Analytical Biochemistry Core facility of the University of Minnesota Cancer Center (see Carmella et al., 2007 for equipment specifications). The AEA level in unknown samples was estimated from the ratio of the area of the signals of d8-AEA and AEA standards (0.2–20 pmol) and was expressed as picomoles per gram tissue.
Measurement of FAAH enzyme activity.
FAAH activity was assayed as described (Hillard et al., 1995; Pratt et al., 1998; Holt et al., 2005). Samples of paw skin and L3–L5 DRGs were removed from naive mice and tumor-bearing mice, frozen in liquid nitrogen, and stored at −80°C until the time of processing. On the day of the assay, tissues were weighed and homogenized in 5 ml of TME buffer (50 mm Tris-HCl, 3 mm MgCl2, 1 mm EDTA, pH 7.4). Samples were sonicated and Bradford protein assays were performed. Aliquots of skin samples (25 μg of protein per assay) were incubated in assay buffer (TME buffer with 1 mg/ml fatty acid-free bovine serum albumin) for 20 min at 37°C with [3H]AEA (100,000 dpm/total assay volume of 500 μl, American Radiolabeled Chemicals). DRGs from the same side of 2 mice were pooled for assay of enzyme activity. Under these conditions [3H]AEA hydrolysis was within the linear range with respect to time and protein concentration (data not shown). The enzyme reaction was stopped by the addition of 2 ml of chloroform:methanol (1:2). After standing at room temperature for 30 min, the phases were separated with the addition of 0.67 ml of chloroform and 0.6 ml of water and centrifugation at 280 × g for 10 min. The amount of [3H] in 1 ml of each of the aqueous and organic phases was determined using liquid scintillation spectrometry. FAAH activity was expressed as percentage hydrolysis of [3H]AEA taking into account that the radioactivity in the aqueous phase was [3H]ethanolamine, whereas [3H]AEA remained in the organic phase (Pratt et al., 1998).
Isolation of adult murine dorsal root ganglion neurons.
Primary cultures of dissociated dorsal root ganglia (DRG) were prepared from DRGs dissected from all levels of the spinal cord of adult C3H male mice, unless otherwise specified in results, as described previously (Scott, 1977; Khasabova et al., 2007). After enzymatic and mechanical dissociation, the final cell suspension was plated at a density of 10,000 cells/25 mm2 on laminin-coated glass coverslips (Fisher Scientific) and maintained in 5 ml of Ham's F12/DMEM medium supplemented with l-glutamine (2 mm), glucose (40 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), and DNase I (0.15 mg/ml, Sigma). Cells were placed in a humidified atmosphere of 5% CO2 at 37°C for 40–48 h before use unless otherwise noted.
Fibrosarcoma cell–DRG neuron coculture preparation.
Three days before preparation of cocultures, fibrosarcoma cells were harvested, rinsed with PBS, pH 7.4, and plated at a density of 50,000 cells/150 μl/25 mm2 on glass coverslips. Cells were maintained in Ham's F12/DMEM medium supplemented with l-glutamine (2 mm), glucose (40 mm), penicillin (100 U/ml), and streptomycin (100 μg/ml) in a humidified atmosphere of 5% CO2 at 37°C. On the day of DRG isolation, a coverslip with freshly isolated neurons was combined in one Petri dish with a coverslip of preplated fibrosarcoma cells. Cells were maintained in 3 ml of fresh Ham's F12/DMEM with 2 ml of cancer cell-conditioned medium at 37°C in a humidified atmosphere of 5% CO2. Neurons were maintained in vitro for 40–48 h before use. No physical contact occurred between neurons and fibrosarcoma cells during this time. Neurons maintained under these conditions develop some phenotypic changes that parallel those of L3–L5 DRG neurons ipsilateral to the tumor in tumor-bearing mice (Khasabova et al., 2007).
Measurement of free intracellular calcium concentration ([Ca2+]i).
The maximum and minimum diameters of each neuron were estimated using a grid mounted in the eyepiece of the microscope, and the average was used to calculate somal area. Measures of [Ca2+]i were made in soma of single neurons using a dual-emission microfluorimeter (Photoscan, Photon Technology International) to monitor fluorescence of indo-1 (3 μm, Molecular Probes) as previously described in our laboratory (Stucky et al., 1996). Counts from the photomultiplier tubes were summed over 0.5 s and recorded on a computer. Values for [Ca2+]i were calculated using Fluorescence System Hardware and Felix Software (Photon Technology International) from the equation [Ca2+]i =K Dβ(R − R min)/(R max − R), where R = 405 nm/485 nm fluorescence emission ratio corrected for background fluorescence. The dissociation constant (K D) for indo-1 was 250 nm (Grynkiewicz et al., 1985) and β was the ratio of fluorescence at 485 nm in the absence and presence of a saturating concentration of Ca2+. Other values, empirically determined in adult murine DRG neurons, were as follows: R min = 0.275; R max = 4.73; β = 4.85.
The basic protocol used to study the effect of cannabinoids on responses of DRG neurons was three applications of KCl (50 mm, 10 s) separated by superfusion for 5 min with HEPES buffer containing (in mm) 25 HEPES, 135 NaCl, 2.5 CaCl2, 3.5 KCl, 1 MgCl2, and 3.3 glucose, pH 7.4 and 335–340 mOsm. Drugs were included in the superfusate after the first application of KCl and were diluted in the HEPES buffer to the final concentrations reported in the results. The amplitude of each KCl-evoked change in Ca2+ transient was calculated as the difference between baseline (average of values for 2 min before KCl) and the peak. To minimize variability within treatment groups, data are reported as relative response, which was defined as the amplitude of the third KCl-evoked Ca2+ transient (in the presence of drug) divided by the amplitude of the first response (in the absence of drug). Relative responses for KCl alone (i.e., control group) as well as an agonist or enzyme inhibitor plus KCl (i.e., drug alone) were determined on each day of an experiment when testing interaction with a third drug (i.e., receptor antagonist) to control for variation among preparations of neurons. To be scored as inhibited by a cannabinoid agonist or vehicle, the relative response of a neuron to KCl in the presence of cannabinoid had to be <0.75. This value is equivalent to the mean of the relative response of the vehicle group minus 1 SD and minimizes occurrence of an effect attributable to chance.
Immunocytochemistry.
Immunocytochemistry was used to determine CB1 receptor-, CB2 receptor-, and NeuN-immunoreactivity (ir) associated with neurons that were maintained in vitro for 40 h. After a wash in HEPES buffer, cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and then rinsed with PBS. Cultures were incubated for 1 h in PBS containing 0.1% sodium azide, 0.3% Triton X-100, 5% normal donkey serum (Jackson ImmunoResearch Laboratories) and then incubated with rabbit anti-CB1 receptor (1:500; Sigma) or rabbit anti-CB2 receptor (1:500; Cayman Chemical). To confirm the association of CB1 receptor-ir with neurons, double labeling was performed by combining the rabbit anti-CB1 receptor antibody with mouse anti-NeuN antibody (1:100; Chemicon International) that binds a neuron-specific nuclear protein. Specimens were incubated with primary antibodies overnight at room temperature. After rinses with PBS, samples were incubated for 1 h in Cy3-labeled donkey anti-rabbit IgG (1:400; Jackson ImmunoResearch Laboratories) combined with AMCA-labeled donkey anti-mouse IgG (1:100; Jackson ImmunoResearch Laboratories). Finally, coverslips were rinsed in PBS and bound fluorophores were protected by mounting to slides in a medium of glycerol/PBS (5/1, v/v), containing 0.1% p-phenylenediamide. CB1 and CB2 receptor antibodies were characterized by Tsou et al. (1998) and Munro et al. (1993) respectively. No immunofluorescence was seen in the absence of primary antibodies.
Images of CB1R-immunofluorescence were collected on a CARV nonlaser Confocal Microscope System in successive frames of 1 μm optical sections (Z-series) using a 20× or 40× oil plan Apochromat objective. MetaMorph software (Universal Imaging) was used for image analysis. Neurons were identified on the basis of NeuN-ir. The cross-sectional area of the soma of a neuron was determined by tracing the plasma membrane. Intensity of fluorescence at the membrane was calculated as the difference in the average intensity for the total area of the soma minus the average value for the cytoplasmic plus nuclear compartments. A neuron was scored “immunopositive” if its average intensity was >2 SDs greater than the mean intensity of neurons labeled with the secondary antibody alone. This strategy was consistent with scores determined independently by a naive observer who judged immunofluorescence in neurons labeled by the primary antibodies.
Western blot analysis of CB1 receptor protein.
Samples of L3–L5 DRGs and tibial nerves (∼0.5 cm) from naive and tumor-bearing mice were sonicated in single-detergent lysis buffer [50 mm Tris-HCl, pH 8.0 with 1% Triton X-100, 150 mm NaCl, 0.02% Na azide, 100 μg/ml PMSF, and 1 μg/ml protease inhibitor mixture (Sigma)], and the particulate fraction of the supernatant was obtained after serial centrifugation at 800 × g for 10 min and 14,000 × g for 25 min. Samples of tibial nerves were pooled from 3 mice. Western blot analysis was performed on 30 μg of protein which were loaded onto a 10% SDS-PAGE gel, subjected to electrophoresis and then transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories). Nonspecific binding to membranes was blocked by incubation in Tris-buffered saline with 3% defatted dry milk for 1 h at room temperature. The membranes were probed with a rabbit anti-CB1 receptor antibody (1:1000, Sigma) overnight at 4°C. Detection was performed using a peroxidase conjugate of goat anti-rabbit IgG (1:20,000; Amersham Biosciences). Immunoreactivity was visualized using the enhanced chemifluorescence detection reagent (Pierce). Loading controls were performed with an anti-β-tubulin antibody (1:20,000, Sigma).
Quantification of mRNA by real-time PCR.
Dorsal root ganglia L3–L5 and skin samples were isolated from mice, placed in RNAlater (Qiagen) and stored at 4°C. Total RNA was isolated from DRG samples using RNeasy Lipid Tissue Mini Kits (Qiagen) and plantar paw skin using RNeasy Fibrous Tissue Mini Kits (Qiagen). RNA was reverse transcribed into cDNA using QuantiTect RT-PCR kits (Qiagen) as per the manufacturer's instructions. Real-time PCR studies were performed with DyNAmo HS SYBR Green Master Mix (Finnzymes) using the DNA engine Opticon 2 (MJ Research) through 45 PCR cycles (94°C for 10 s, 57°C for 20 s, 72°C for 30 s). Each cDNA sample was run in triplicate for the murine CB1 receptor, FAAH, and the reference gene (S15). Primer pair sequences were as follows: CB1 receptor (GenBank Accession number NM_007726) forward primer 5′-GTT CTG ATC CTG GTG GTG TTG-3′ and reverse primer 5′-GTT CAG CAG GCA GAG CAT AC-3′; FAAH (NM_010173) forward primer 5′-GCT GTG CTC TTT ACC TAC CTG-3′ and reverse primer 5′-GAA GCA TTC CTT GAG GCT CAC-3′; S15 (BC094409) forward primer 5′-CCG AAG TGG AGC AGA AGA AG-3′ and reverse primer 5′-CTC CAC CTG GTT GAA GGT C-3′. All primers were synthesized by Operon Biotechnologies. Specificity of each amplicon was confirmed by melting curve analysis, evidence of a single band after gel electrophoresis, authenticity of the DNA sequence of the band isolated from the gel, and resolution by BLAST analysis that the sequence of the amplicon was unique to each target. The ratio of fold change in expression of the mRNA of interest for each sample was calculated by normalization of cycle threshold (Ct) values to S15 using the equation derived by Pfaffl (2001) to correct for potential differences in PCR primer efficiencies between the target and reference genes. This approach was validated by the lack of an effect of treatments on the expression of S15 and confirmation that data for tissue samples were within the linear range used to determine the efficiency of the primers. The efficiency of the primer set was derived from the slope of its linear graph using the following equation: efficiency = 10 (−1/slope). Samples were collected from normal and tumor-bearing mice four times. To control for variability in preparations and assays, data for CB1 receptor and FAAH mRNA (corrected to the reference gene) were normalized to the mean of the control group for each date of sample collection.
Drugs.
In behavioral studies, arachidonoylethanolamide (anandamide, AEA) was dissolved in Tocrisolve100. Stock solutions of the CB1 receptor antagonist AM281 (10 mg/ml) and the CB2 receptor antagonist AM630 (10 mg/ml) were prepared in DMSO. AEA, AM281, AM630, and Tocrisolve were purchased from Tocris. A stock solution of the FAAH inhibitor, cyclohexylcarbamic acid 3′-carbamoyl-biphenyl-3-yl ester (8 mg/ml, URB597, Cayman Chemical) was prepared in ethanol. All drugs were diluted in saline to achieve the final dose. For in vitro experiments, stock solutions of cannabinoid receptor ligands AEA (14 mm), arachidonyl-2′-chloroethylamide (ACEA, 14 mm, Tocris) and URB597 (10 mm) were prepared in ethanol. The CB1 receptor antagonist SR141716A (10 mm, RTI International) was dissolved in 1-methyl-2-pyrrolidinone. Pertussis toxin was purchased from Calbiochem Novabiochem. All chemicals were diluted in the HEPES buffer to the final concentrations indicated for superfusion. Superfusion with any vehicle by itself at the highest concentration used with a drug did not alter basal or KCl-evoked increases in [Ca2+]i.
Statistical analyses.
Data are presented as proportions or the mean ± SEM for each group. Differences among mean withdrawal frequencies to mechanical stimuli were analyzed by two-way ANOVA or two-way repeated-measures ANOVA with Bonferroni multiple-comparisons test. Nonlinear regression analysis was used to determine the mechanical force that would produce a withdrawal response in a mouse with a frequency of 50%. Fisher's exact test was used to determine differences in proportions of neurons exhibiting a trait. In measures of Ca2+ transients, statistical differences among groups were identified using one-way ANOVA or two-way ANOVA followed by the Tukey multiple-comparisons test. In some instances, data were transformed to log10 (Kolmogorov–Smirnov test) before conducting an analysis to meet the normality requirement. Student's t test was used to determine differences between two groups. A value of p < 0.05 was considered significant.
Results
Cutaneous mechanical hyperalgesia in a model of cancer pain was associated with a decreased level of AEA in plantar paw skin ipsilateral to the tumor
Cutaneous mechanical hyperalgesia develops within a few days after implantation of mouse NCTC clone 2472 fibrosarcoma cells unilaterally into and around the calcaneus bone (Cain et al., 2001; Wacnik et al., 2001). In the present experiments, withdrawal to a force of 3.7 mN applied to the plantar surface of the hindpaw increased from a pretumor (PT) frequency of 11 ± 2% to ∼80% in the paw ipsilateral to the tumor on day 10 after implantation of fibrosarcoma cells (Fig. 1 A), whereas no change occurred in the contralateral paw (9.8 ± 1.6%, n = 31). It was noteworthy that the level of AEA in plantar skin from the hindpaw ipsilateral to the tumor in tumor-bearing mice was only 22% of the level in naive animals (8.8 ± 1.12 pmol/g tissue in tumor-bearing mice compared with 40.1 ± 7.98 pmol/g in naive mice, n = 8–9; p < 0.005, Student's t test).
Figure 1.
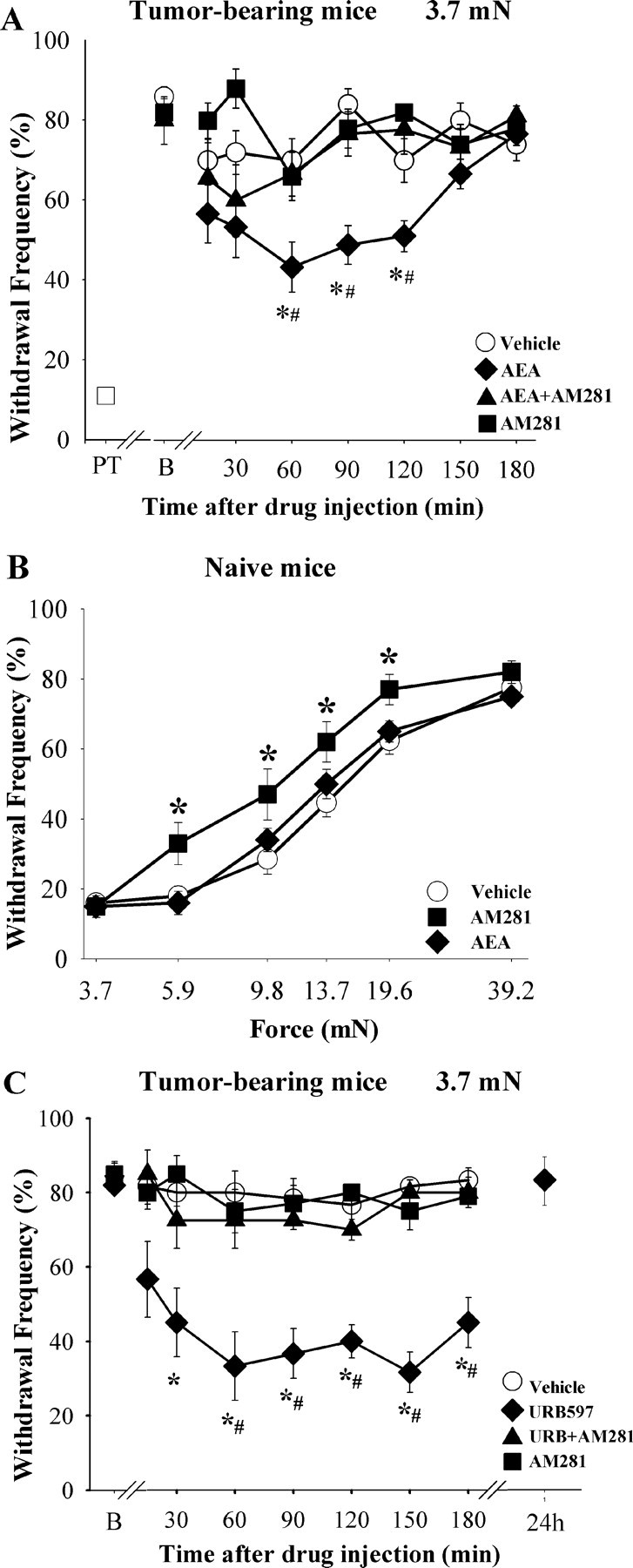
AEA activation of CB1 receptors in plantar skin decreases sensitivity to mechanical stimuli in tumor-bearing and naive mice. A, Injection of AEA (1 μg, ipl) ipsilateral to the tumor attenuated the mechanical hyperalgesia in tumor-bearing mice. On day 10 after injection of fibrosarcoma cells unilaterally into and around the calcaneus bone (B, baseline), mice exhibited increased withdrawal to a mechanical stimulus (monofilament with a bending force of 3.7 mN) compared with responses before implantation of tumor cells (PT, pretumor). The effect of AEA was blocked by coinjection of the CB1 receptor antagonist AM281 (10 μg). B, The dose of AEA that suppressed hyperalgesia in tumor-bearing mice did not change mechanical sensitivity in naive mice. However, the dose of AM281 that blocked the anti-hyperalgesic effect of AEA in tumor-bearing mice increased mechanical sensitivity in naive mice. The x-axis is presented on a log scale. C, Injection of the URB597 (9 μg, ipl) into the hindpaw ipsilateral to the tumor produced a long-lasting reduction in the mechanical hyperalgesia. The effect of URB597 was blocked by coinjection of the CB1 receptor antagonist AM281 (10 μg, ipl). *Significantly different from vehicle within each drug group; #significantly different from AEA+AM281 or URB597+AM281 (p < 0.05, two-way ANOVA with Bonferroni test, n = 6–9 mice/treatment group).
Several lines of evidence indicated that the low level of AEA in the plantar paw skin contributed to the mechanical hyperalgesia. First, a single injection of AEA (1 μg, ipl) into the tumor-bearing paw attenuated mechanical hyperalgesia when compared with the vehicle-treated group or baseline (Fig. 1 A). No effect was observed with lower doses (10 and 100 ng/paw, data not shown). The anti-hyperalgesic effect of AEA was delayed in onset and persisted for 1 h. There was no evidence of anti-hyperalgesia when AEA (1 μg) was injected in the contralateral paw (n = 4, p = 0.160, two-way repeated-measures ANOVA with Bonferroni test). These data suggest that the paw tissue in tumor-bearing mice retained the capacity to respond to AEA and confirmed that the anti-hyperalgesic effect of AEA was not mediated systemically.
Injection of the selective CB1 receptor antagonist AM281 (10 μg, ipl) with AEA (1 μg) blocked the anti-hyperalgesic effect of AEA (Fig. 1 A). The antagonistic effect of AM281 was not mediated systemically because injection of AM281 into the contralateral paw did not alter the effect of AEA on cutaneous mechanical hyperalgesia when AEA was injected ipsilateral to the tumor. The CB2 receptor antagonist AM630 (4 or 25 μg, ipl) did not alter the anti-hyperalgesic effect of AEA (p = 0.182, two-way ANOVA with Bonferroni test). These doses of AM630 were effective in vivo in other studies (Guindon et al., 2006; Potenzieri et al., 2008). The intraplantar injection of neither AM281 nor AM630 alone had an effect on the withdrawal response to the 3.7 mN stimulus in tumor-bearing mice (n = 5–6; p = 0.945). Thus, CB1 receptors in the paw tissue of tumor-bearing mice have the capacity to mediate anti-hyperalgesia.
Consistent with an earlier study (Sokal et al., 2003), injection of AEA (1 μg, ipl) into a hindpaw of naive mice did not affect mechanical sensitivity (Fig. 1 B). However, the dose of the CB1 receptor antagonist AM281 that blocked the anti-hyperalgesic effect of AEA in tumor-bearing mice increased sensitivity to a mechanical stimulus in naive mice. In the present experiment, injection of AM281 (10 μg, ipl) increased the frequency of withdrawal to forces ranging from 5.9–19.6 mN (n = 5 mice/group; p = 0.05, two-way ANOVA with Bonferroni test). Thus, the mechanical threshold after administration of AM281 was lower than that of the vehicle-treated group (8.97 ± 0.55 mN for AM281 versus 11.77 ± 0.53 mN for vehicle; p < 0.005, one-way ANOVA). The increase in mechanical sensitivity after local injection of a CB1 receptor antagonist suggests the existence of tonic activity of CB1 receptors in naive mice. This effect was not present in tumor-bearing mice.
FAAH activity increased in plantar skin in tumor-bearing mice
The low level of AEA in paw skin of tumor-bearing mice suggested a shift in the balance of activity of synthetic and catabolic enzymes that regulate the level of AEA in tissues. Because one enzyme, FAAH, is largely responsible for the degradation of AEA, we tested whether an increase in FAAH activity in the paw skin contributed to the reduced level of AEA in skin ipsilateral to the tumor in tumor-bearing mice. Indeed, hydrolysis of AEA was ∼3× greater in hindpaw plantar skin ipsilateral to tumors from tumor-bearing mice relative to samples from naive mice (Table 1). Similarly, there was a fourfold increase of FAAH activity in L3–L5 DRGs ipsilateral to tumors in tumor-bearing mice compared with the same DRGs from naive mice, suggesting that sensory neurons contributed to the increased activity of FAAH in the paw skin. This possibility was supported by evidence that the level of FAAH mRNA was twofold greater in L3–L5 DRGs ipsilateral to tumors in tumor-bearing mice compared with the same DRGs from naive mice, whereas there was no difference in the amount of FAAH mRNA in samples of plantar paw skin isolated from the two groups (Table 1).
Table 1.
FAAH enzyme activity and mRNA in skin and DRGs
| Tissue | FAAH activity |
FAAH mRNA |
||
|---|---|---|---|---|
| Naive (%) | Tumor-bearing (% of naive) | Naive (%) | Tumor-bearing (% of naive) | |
| Skin | 100 ± 14 (11) | 301 ± 36* (17) | 100 ± 30 (4) | 118 ± 52 (6) |
| L3–L5 DRGs | 100 ± 14 (5) | 399 ± 95* (6) | 100 ± 11 (11) | 206 ± 29* (11) |
Enzyme activity and mRNA were measured in separate samples taken from plantar skin of the hindpaw and DRGs. Samples from tumor-bearing animals were taken ipsilateral to tumors. FAAH enzyme activity was expressed as percent hydrolysis of [3H]AEA per microgram of protein for skin and per pooled DRGs and then normalized to the mean for naive mice. Values for FAAH mRNA were corrected to the amount of mRNA for the reference gene within each sample and then were normalized to the mean for naive mice for each isolation of total RNA.
*Significantly different from respective naive group at p < 0.01 (Student's t test). n values are given in parentheses.
Although mean values for markers of FAAH expression were elevated in the contralateral paw in tumor-bearing mice, none were statistically different from values for naive mice. The level of FAAH mRNA in the L3–L5 DRGs contralateral to tumors (210 ± 61% of DRGs from naive mice, n = 11) was comparable to that in DRGs ipsilateral to tumors (Table 1), but it was not different from the level in the naive group (p = 0.10, Student's t test). A clearer difference occurred in the level of enzyme activity. FAAH activity in the hindpaw skin contralateral to tumors (160 ± 20% of paw skin from naive mice, n = 10) was not greater than activity in skin from naive mice (p = 0.398) and the level was significantly lower than FAAH activity in skin ipsilateral to tumors (p < 0.005, ANOVA with Tukey's multiple-comparisons test).
To test whether the increased FAAH activity was important in maintaining mechanical hyperalgesia in this model of cancer pain, we determined whether inhibition of FAAH would attenuate the hyperalgesia. Injection of the FAAH inhibitor URB597 (9 μg, ipl) into the hindpaw ipsilateral to the tumor produced a long-lasting reduction in mechanical hyperalgesia (Fig. 1 C) that was accompanied by an increase in the level of endogenous AEA at 1 h after drug injection (122.27 ± 38.01 pmol/g tissue with URB597 compared with 8.8 ± 1.12 pmol/g tissue with vehicle in tumor-bearing mice, n = 5–9; p = 0.002, Student's t test). Subsequent studies determined that this dose of URB597 was equivalent to the ED60 of the drug in reducing mechanical hyperalgesia after intraplantar injection in this model. Recovery to baseline frequency of withdrawal to the suprathreshold stimulus was confirmed 24 h after injection of drug. Importantly, the anti-hyperalgesic effect of URB597 was mediated by activation of CB1 receptors because coadministration of the CB1 antagonist AM281 (10 μg, ipl) with URB597 blocked the inhibitory effect of URB597 (Fig. 1 C). Coinjection of the CB2 receptor antagonist AM630 with URB597 did not alter the anti-hyperalgesic effect of URB597 (p = 0.1892, two-way ANOVA with Bonferroni test). Furthermore, intraplantar injection of URB597 into the paw contralateral to the tumor did not alter mechanical hyperalgesia in the paw ipsilateral to the tumor (n = 4, p = 0.278, two-way repeated-measures ANOVA with Bonferroni test). Together these data support the conclusion that increased degradation of AEA contributed to the maintenance of mechanical hyperalgesia in a murine model of bone cancer pain.
Expression of CB1 receptors increased in DRG neurons in tumor-bearing mice
A factor that may contribute to the anti-hyperalgesic effect of AEA in tumor-bearing mice is the increased expression of CB1 receptors by somatosensory neurons. Using quantitative real time RT-PCR, a twofold increase in CB1 receptor mRNA was measured in L3–L5 DRGs ipsilateral to tumors in tumor-bearing mice compared with the same DRGs isolated from naive mice (Fig. 2 A). The level of CB1 mRNA in L3–L5 DRGS ipsilateral to the tumors was greater than that in the same DRGs from the contralateral side. The increase in mRNA was associated with a comparable increase in expression of CB1 receptor protein in L3–L5 DRGs as well as the tibial nerve (a cutaneous nerve that innervates the plantar surface of the hind foot) ipsilateral to tumors in tumor-bearing mice (Fig. 2 B,C). Thus, an increase in the expression of CB1 receptors by somatosensory neurons in tumor-bearing mice may contribute to the analgesic efficacy of the local injection of URB597. Studies of individual DRG neurons were required to determine whether a change in functional receptors was associated with small diameter DRG neurons.
Figure 2.

Expression of CB1 receptors increased in DRG neurons ipsilateral to tumors in tumor-bearing mice. A, Quantitative real time RT-PCR was used to measure CB1 receptor mRNA in DRGs L3–L5 isolated from naive and tumor-bearing mice. Abundance of CB1 receptor mRNA was quantified with respect to S15 as an internal control and was then normalized to the average value for the control group for each preparation and assay. B, Western blot analysis was used to determine the expression of CB1 receptor protein in L3–L5 DRGs as well as the tibial nerve. The amount of CB1 receptor protein was expressed as a percentage of β-tubulin-immunoreactivity that was used as a loading control for each sample. C, Representative examples of CB1 receptor protein detected by Western blot. *Significantly different from naive mice at p < 0.05 (Student's t test). #Significantly different from contralateral side at p < 0.05 (paired Student's t test). Values inside the bars represent the sample size.
Response to cannabinoids increased in small DRG neurons isolated from tumor-bearing mice
Because cannabinoids inhibit voltage-dependent Ca2+ channels in DRG neurons (Ross et al., 2001; Khasabova et al., 2004), a decrease in the Ca2+ transient evoked by KCl (50 mm) was used as a bioassay for activation of CB1 receptors by the selective CB1 receptor agonist ACEA. DRG neurons with somal areas <500 μm2 were selected for study because murine DRG neurons of this size give rise to C-fibers with a variety of nociceptor properties (Hiura and Sakamoto, 1987; Urban and Dray, 1993; Pearce and Duchen, 1994; Dirajlal et al., 2003). This population is referred to as “small” in this report. Each neuron was superfused with KCl (50 mm, 10 s) three times at intervals of 5 min, and drugs diluted in the HEPES buffer were included in the superfusate after the first application of KCl (Fig. 3 A). In the vehicle control condition, the amplitude of the depolarization-evoked transient did not change between the first and third applications of KCl in neurons either from naive or tumor-bearing mice (p = 0.152; one-way ANOVA with repeated measures). Consistent with data reported for rat (Khasabova et al., 2004), ACEA (1 μm, 10 min) had no effect on the KCl-evoked Ca2+ transient in small DRG neurons from naive mice, but it did inhibit the evoked transient in larger DRG neurons by 40% (p < 0.05, one-way ANOVA with Tukey's multiple-comparisons test, data not shown). Furthermore, 1 μm was the minimally effective concentration of ACEA in large DRG neurons (concentrations of 10 and 100 nm were also tested). Importantly, a change occurred in DRG neurons isolated from L3–L5 DRGs of tumor-bearing mice: ACEA (1 μm) attenuated the KCl-evoked Ca2+ transient in small DRG neurons by 60% (Fig. 3 A,B). This effect was CB1 receptor-mediated because the CB1 receptor antagonist SR141716A (1 μm) blocked the inhibitory effect of ACEA. The inhibitory effect of ACEA on the mean amplitude of the depolarization evoked Ca2+ transient in small DRG neurons isolated from tumor-bearing mice was paralleled by an increase in the proportion of neurons that was inhibited by ACEA (7/8 of neurons from tumor-bearing mice compared with 2/8 neurons from naive mice, p < 0.05, Fisher's exact test; see methods for the criterion for an inhibitory effect). ACEA had no independent effect on basal [Ca2+]i at any concentration tested nor in any neurons sampled from naive or tumor-bearing mice. These data support the conclusion that the increased expression of CB1 receptor mRNA and protein in DRG neurons from tumor-bearing mice contributed to a functional increase in coupling of CB1 receptors to inhibition of voltage-dependent Ca2+ channels in small DRG neurons.
Figure 3.

Small DRG neurons (< 500 μm2) isolated from DRGs L3–L5 of tumor-bearing mice and maintained in vitro in control medium for 20–28 h exhibited a change in sensitivity to the CB1 agonist ACEA. A, The trace represents cannabinoid agonist inhibition of the Ca2+ transient evoked by brief superfusion with KCl (50 mm, 10 s, arrows). ACEA (1 μm) was included in the superfusate after the first test with KCl. B, ACEA attenuated the Ca2+ transient evoked by KCl (50 mm) in neurons isolated from L3–L5 DRGs of tumor-bearing mice, but not in those from naive mice. Involvement of CB1 receptors in the response to ACEA was confirmed by blocking the inhibitory effect by coapplication of the CB1 receptor antagonist SR141716A (SR, 1 μm). Relative response was defined as the amplitude of the response of a neuron to KCl in the presence of ACEA divided by the amplitude of the response in the absence of ACEA. *Significantly different at p < 0.001; #significantly different at p < 0.01 (one-way ANOVA with Tukey's multiple-comparisons test).
Cancer cells were sufficient to evoke changes in CB1 receptor function in small DRG neurons
We developed an in vitro model to test for direct effects of cancer cells on DRG neurons (Khasabova et al., 2007). In this model, fibrosarcoma cells and isolated DRG neurons are maintained in one culture dish on separate coverslips for 48 h such that the cancer cells condition the medium for the DRG neurons. Biochemical and functional changes that contribute to excitability occur in small DRG neurons in this model and parallel changes that occur in small DRG neurons isolated from tumor-bearing mice. In the present experiments, the CB1 agonist ACEA (1 μm) inhibited the amplitude of the depolarization-evoked Ca2+ transient in small DRG neurons in the coculture model, and the effect was blocked by coperfusion of the CB1 receptor antagonist SR141716A (Fig. 4 A). Similarly, coculture of DRG neurons with fibrosarcoma cells increased the proportion of neurons inhibited by ACEA (Table 2). It is noteworthy that in both analyses the inhibitory effect of ACEA was significantly different from both the vehicle control in the coculture condition as well as ACEA-treated neurons maintained in the control condition.
Figure 4.

Coculture of murine DRG neurons with fibrosarcoma cells for 40–48 h altered the effect of the cannabinoid agonist ACEA on the KCl-evoked Ca2+ transient in small DRG neurons. A, Superfusion with ACEA (1 μm) had no effect on the depolarization-evoked Ca2+ transient in small-diameter neurons in the control cultures. After maintenance in medium conditioned by fibrosarcoma cells, the Ca2+ transient was attenuated by ACEA (1 μm), and the inhibitory effect was blocked by SR141716A (1 μm). Drugs were included in the superfusate after the first test with KCl (50 mm). Relative response was defined as the amplitude of the response of a neuron to KCl in the presence of drug divided by the amplitude of the response in the absence of drug. *Significantly different at p < 0.05 (one-way ANOVA with Tukey's multiple comparison test). B, Composite of confocal images (1 μm optical thickness) illustrating CB1 receptor-immunofluorescence associated with the plasma membrane of the neuronal soma and axons. Scale bar, 10 μm. C, Coculture of DRG neurons with fibrosarcoma cells increased the proportion of small DRG neurons exhibiting CB1 receptor-ir on the plasma membrane of the soma. *Significantly greater from control at p < 0.001 (χ2 test, n = 94–122).
Table 2.
The number of small DRG neurons inhibited by cannabinoid agonists increased when they were cocultured with fibrosarcoma cells
| Culture condition | Percentage of neurons inhibited |
||
|---|---|---|---|
| Vehicle | ACEA | AEA | |
| Control | 10 (31) | 24 (17) | 61 (23)* |
| Coculture | 12 (33) | 57 (35)* , ** | 94 (16)* , ** |
ACEA and AEA were used at concentration of 1 μm and 10 nm, respectively. In order to be scored as inhibited by a cannabinoid agonist or vehicle, the relative response of a neuron to KCl in the presence of cannabinoid had to be <0.75. Relative response was defined as the amplitude of the response to 50 mm KCl in the presence of drug divided by the amplitude of the response in the absence of drug. The value of 0.75 was selected because it is equivalent to the mean of the relative response of the vehicle group minus 1 SD in order to minimize occurrence of an effect due to chance.
*Significantly different from vehicle within a culture condition at p < 0.05 (Fisher's exact text).
**Significantly different from the control culture within a drug treatment at p < 0.05 (Fisher's exact test). n values are given in parentheses.
Immunocytochemical studies confirmed that the CB1 receptor mediated inhibition of the Ca2+-evoked transient in small DRG neurons maintained in vitro with fibrosarcoma cells was associated with an increase in CB1 receptors on the plasma membrane. Although 27% (n = 122) of small DRG neurons maintained in vitro in the control condition for 40 h exhibited intracellular CB1 receptor-ir, less than one-half of those (i.e., 11% of all small neurons) expressed CB1 receptor-ir on the plasma membrane. However, maintenance of dissociated DRG neurons with fibrosarcoma cells for 40–48 h in vitro increased the occurrence of CB1 receptor-ir on the plasma membrane of small neurons nearly threefold such that CB1 receptor-ir was observed primarily on the plasma membrane of neuronal cell bodies and processes (Fig. 4 B,C). It is noteworthy that CB1 receptor-ir was absent among non-neuronal cells in both culture preparations. Together, these data demonstrate that the coculture condition promoted the expression in small DRG neurons of functional CB1 receptors that coupled to inhibition of voltage-dependent Ca2+ channels.
Of the known cannabinoid receptors, the change in inhibition of the evoked Ca2+ transient in small DRG neurons was specific to CB1 receptors. The CB2 receptor agonist JWH015 (1 μm) had no effect on the depolarization-evoked Ca2+ transient after maintenance of dissociated DRG neurons in either control or fibrosarcoma cell-conditioned medium (p = 0.845, two-way ANOVA with Turkey test; n = 8–9 neurons/group). This concentration of JWH015 effectively activates CB2 receptors in vitro in other models (Chin et al., 1999; Mulè et al., 2007). Morphological data were consistent with the absence of a pharmacological effect of the CB2 agonist. No specific neuronal CB2 receptor-ir was noted in either the control or coculture preparation.
Coculture of DRG neurons with fibrosarcoma cells altered anandamide signaling in small DRG neurons
AEA (1 nm to 1 μm) did not have an independent effect on basal [Ca2+]i at any concentration tested in adult murine DRG neurons maintained in vitro. For example, after superfusion of small neurons with 1 μm AEA for 2 min, at no point was the mean [Ca2+]i (81.1 ± 8.0 nm; n = 10) different from control (80.0 ± 7.8 nm; n = 10; p=0.922, Student's t test). However, a low concentration of AEA (10 nm) decreased the depolarization-evoked Ca2+ transient by 28% (Fig. 5 A), but only one concentration among those tested was effective.
Figure 5.

Maintenance of murine DRG neurons with fibrosarcoma cells for 40–48 h altered AEA signaling in small neurons. A, The concentration-response curve for AEA-mediated inhibition of the depolarization-evoked Ca2+ transient in small DRG neurons cultured alone. Only one concentration of AEA (10 nm) among those tested inhibited the KCl evoked Ca2+ transient. *Significantly different from the vehicle treated group (Cont) at p < 0.01 (one-way ANOVA with Tukey's multiple comparison test; n = 10–30 neurons/concentration). B, The inhibitory effect of AEA in neurons cultured alone was not blocked by either the CB1 receptor antagonist SR141716A (SR) or pretreatment with pertussis toxin (PTX). SR141716A (1 μm) was included in the superfusate with AEA (10 nm) after the first test with KCl (50 mm). Neurons were pretreated with PTX (500 ng/ml) overnight. *Significantly different at p < 0.05 (one-way ANOVA with Tukey's multiple comparison test). C, When neurons were cocultured with fibrosarcoma cells, the KCl-evoked Ca2+ transient in small DRG neurons was also attenuated by AEA (10 nm), but the inhibitory effect was blocked by SR141716A (1 μm). *Significantly different at p < 0.01 (two-way ANOVA with Tukey's multiple comparison test). D, Superfusion with URB597 (30 nm) did not alter the KCl-evoked Ca2+ transient in small DRG neurons cultured alone. However, after maintenance of neurons with fibrosarcoma cells, URB597 (30 nm) attenuated the KCl-evoked Ca2+ transient, and the inhibitory effect was blocked by SR141716A (SR, 1 μm). *Significantly different at p < 0.01 (two-way ANOVA with Tukey's multiple comparison test). Relative response was defined as the amplitude of the response of a neuron to KCl in the presence of drug, divided by the amplitude of the response in the absence of drug.
Several strategies were used to characterize the inhibitory effect of AEA on neurons maintained in control condition (Fig. 5 B). The CB1 receptor antagonist SR141716A (1 μm) did not block the effect of AEA (10 nm). These results are also consistent with the low proportion of small DRG neurons (4/17) inhibited by ACEA (1 μm) and the low occurrence of membrane-associated CB1 receptor-ir among small DRG neurons (Fig. 4 C). It is unlikely that the inhibitory effect of AEA was mediated by CB2 receptors, or any receptor that couples to an inhibitory G-protein, because pretreatment of cultures with pertussis toxin (PTX, 500 ng/ml, overnight) did not block the effect of AEA (Fig. 5 B). As a positive control, pretreatment with PTX blocked the ACEA-mediated inhibition of the depolarization-evoked Ca2+ transient in larger neurons under the same conditions (data not shown). None of the drugs altered basal [Ca2+]i in DRG neurons. Together, these data confirmed that the inhibitory effect of AEA on the KCl-evoked Ca2+ transient in small DRG neurons in the control condition was independent of inhibitory G-protein coupled receptors.
Coculture of DRG neurons with fibrosarcoma cells dramatically altered their response to AEA. There was a significant increase in the proportion of small DRG neurons that were inhibited by AEA when the neurons were cocultured with fibrosarcoma cells (Table 2). Similarly, the inhibitory effect of AEA on the amplitude of the Ca2+ transient increased from 28% in small DRG neurons cultured alone to 55% in the coculture condition (Fig. 5 B,C). It is noteworthy that the effect of AEA was now blocked by SR141716A. The enhanced inhibitory effect of AEA on voltage-dependent Ca2+ channels in small DRG neurons cocultured with cancer cells is consistent with the increased occurrence of CB1 receptor-ir on the plasma membrane of neuronal cell bodies and processes (Fig. 4 C). In addition to a change in AEA signaling, there was a change in response to inhibition of FAAH with URB597. In the control condition, URB597 (30 nm) had no effect on the basal concentration of intracellular Ca2+ or on the KCl-evoked Ca2+ transient in small DRG neurons. However, when DRG neurons were cocultured with fibrosarcoma cells, the same concentration of URB597 was sufficient to inhibit the depolarization evoked Ca2+ transient in small DRG neurons. Consistent with data generated in vivo, the inhibitory effect of URB597 in vitro was blocked by SR141716A (Fig. 5 D) indicating the inhibitory effect was mediated by CB1 receptors. Therefore, inhibition of neuronal FAAH was capable of attenuating the amplitude of the KCl-evoked Ca2+ transient in small DRG neurons cocultured with fibrosarcoma cells.
Discussion
Mechanical hypersensitivity in murine models of bone cancer pain is associated with phenotypic changes in primary afferent neurons (Honore et al., 2000; Khasabova et al., 2007; Niiyama et al., 2007) that contribute to increased excitability of nociceptors that innervate the tumor-bearing limb (Cain et al., 2001). In this report we demonstrate that a decrease in peripheral AEA, resulting from increased degradation by FAAH, also contributes to the maintenance of mechanical hyperalgesia in bone cancer pain. The decrease in AEA occurred in conjunction with increased expression of FAAH and CB1 receptors by DRG neurons innervating the affected paw, such that local injection of the FAAH inhibitor URB597 increased the level of AEA in the skin and markedly reduced the mechanical hyperalgesia. Functional assays of small DRG neurons cocultured with fibrosarcoma cells paralleled effects observed in vivo demonstrating that the fibrosarcoma cells are sufficient to evoke the phenotypic and functional changes in endocannabinoid signaling in DRG neurons.
AEA has been measured in rodent paw skin by several laboratories (Felder et al., 1996; Calignano et al., 1998; Maione et al., 2007), and our data fall within the range of earlier reports. Generation of AEA by cells in skin relevant to mechanotransduction include keratinocytes (Maccarrone et al., 2003) and terminals of DRG neurons (Ahluwalia et al., 2003; van der Stelt et al., 2005). The decrease in AEA content contrasts with increased AEA in skin ipsilateral to persistent peripheral inflammation (Agarwal et al., 2007) and nerve injury (Jhaveri et al., 2006). Thus, a low level of AEA in the peripheral field of DRG neurons may be unique to the pathophysiology of cancer pain.
It is noteworthy that DRG neurons have the capacity to bind AEA in addition to the ability to synthesize this endocannabinoid, thereby supporting an autocrine role for AEA signaling in sensory neurons. CB1 receptors have been detected in primary afferent neurons, with variable localization of CB1 mRNA and protein among subtypes of sensory neurons (Hohmann and Herkenham, 1999; Ahluwalia et al., 2000, 2002; Salio et al., 2002; Bridges et al., 2003; Price et al., 2003; Agarwal et al., 2007). Basal activation of CB1 receptors on nociceptors has a fundamental role in determining the sensitivity of the neurons to thermal and mechanical stimuli. This assertion is based on evidence that selective knockdown of the CB1 receptor in nociceptors lowers thresholds for sensory transduction (Agarwal et al., 2007). Similarly, intraplantar injection of the CB1 receptor antagonist AM281 lowered the mechanical threshold in naive mice in the present experiments. The effect of endocannabinoids is also apparent in peripheral inflammatory pain when nocifensive behaviors are exaggerated after the intraplantar injection of a CB1 receptor antagonist (Calignano et al., 1998) or selective deletion of the CB1 receptor in nociceptors (Agarwal et al., 2007).
A significant finding was the association of a low level of AEA with increased FAAH activity in plantar skin ipsilateral to tumors. An inverse relationship between tissue levels of AEA and FAAH activity occurs in other pathophysiological conditions of the nervous system (Maccarrone et al., 2004; Nucci et al., 2007). It is likely that the decreased level of AEA in the paw skin contributes to the maintenance of mechanical hyperalgesia in this model of cancer pain because local inhibition of FAAH reduced the hyperalgesia. Intraplantar injection of URB597 is also anti-hyperalgesic in a model of neuropathic pain (Jhaveri et al., 2006). The peripheral CB1 receptor mediated anti-hyperalgesia in response to AEA and inhibition of FAAH is consistent with the effect of CB1 receptor agonists in murine models of cancer pain (Guerrero et al., 2008; Potenzieri et al., 2008).
Although the anti-hyperalgesic effects of AEA and inhibition of FAAH by URB597 were completely blocked by the CB1 receptor antagonist, elevated levels of fatty acid amides after administration of these compounds may temper their anti-hyperalgesic effects. High concentrations of AEA (Huang et al., 2002) and N-acyltaurines (Saghatelian et al., 2006), also substrates for FAAH, activate TRPV1 receptors which excite DRG neurons. Furthermore, fatty acid amides may be catabolized by cyclooxygenase 2 when FAAH is inhibited (Vandevoorde and Lambert, 2007), and some endocannabinoid derivatives generated by cyclooxygenase 2 promote hyperalgesia (Hu et al., 2008).
FAAH enzyme is widely expressed in the nervous system (Egertová et al., 2003) and increased expression of FAAH in DRG neurons contributes to the increased FAAH activity in plantar paw skin of tumor-bearing mice. This conclusion is based on evidence that FAAH mRNA did not increase in skin but doubled in DRGs ipsilateral to tumors in tumor-bearing mice. Furthermore, an increase in FAAH activity in the DRG accompanied the increase in mRNA. Finally, expression of FAAH by DRG neurons is functionally significant in that inhibition of FAAH in DRG neurons cocultured with fibrosarcoma cells resulted in CB1 receptor-mediated inhibition of DRG function. It is not likely that endocannabinoids generated by cells on the coverslip other than the one undergoing recording would contribute to the inhibition of the Ca2+ transient in the presence of URB597 because recordings were measured during continuous superfusion. Therefore, downregulation of FAAH in DRG neurons may be an effective clinical strategy to restore endogenous AEA levels in tissues affected by tumors thereby producing analgesia.
In light of the role of CB1 receptors in setting the threshold for activation of nociceptors, it is noteworthy that expression of CB1 receptors increased in DRG neurons ipsilateral to tumors in tumor-bearing mice. Although DRGs are a mixture of neuronal and supporting cells, the increase in CB1 receptor mRNA most likely occurred in neurons because CB1 receptor-ir was not associated with non-neuronal cells from dissociated DRGs in vitro and a significant increase in protein immunoreactivity was observed in a related nerve. These data parallel a recent report of increased CB1 receptor protein in a model of skin cancer (Guerrero et al., 2008). It is possible that the increased expression of CB1 receptors contributed to increased receptor function observed in small DRG neurons from tumor-bearing mice. The fact that increased receptor function was evident after maintaining neurons from these mice in vitro in the absence of fibrosarcoma cells for 16–20 h indicates the change in function was the result of a change in gene expression and not a transient event of receptor modulation. Moreover, it is likely that factors released from the fibrosarcoma cells were sufficient to evoke the change in receptor function because coculture of dissociated DRG cells from naive mice with fibrosarcoma cells mimicked the change observed in neurons isolated from tumor-bearing mice. The increased expression of CB1 receptors by DRG neurons ipsilateral to tumors provides an important opportunity for management of cancer pain in humans. The significance of this finding is even greater in light of the clinical experience of refractoriness of cancer pain to opioids (Portenoy, 1999; Mancini et al., 2004) and a recent finding that expression of μ-opioid receptors is decreased in a population of nociceptive DRG neurons in a model of bone cancer pain (Yamamoto et al., 2008).
It is also possible that the increase in CB1 receptor function was secondary to a decrease in basal activation by endogenous AEA. We speculate that the absence of a CB1-mediated inhibition of the depolarization-evoked Ca2+ transient in small DRG neurons from naive mice maintained in vitro (present results and Evans et al., 2004) is attributable to low degradation of AEA in these neurons such that CB1 receptors are maximally activated. This interpretation is supported by evidence that CB1 receptor immunoreactivity was observed more frequently in the cytoplasmic compartment compared with the plasma membrane of small neurons maintained in vitro, and evidence that the CB1 receptor undergoes internalization after activation (Hsieh et al., 1999; Coutts et al., 2001; Leterrier et al., 2006). However, increased expression of FAAH enzyme in DRG neurons in the cancer condition would decrease the ambient level of AEA. Although we were not able to recover sufficient material to measure FAAH activity in the DRG cultures, given other parallels in phenotypic changes between DRG neurons ipsilateral to tumors and neurons maintained in the coculture model (TRPV1 receptors, α2δ1 subunit, CB1 receptors) this assumption is reasonable. Furthermore, superfusion with a higher concentration of URB597 (100 nm) was required to observe an inhibitory effect of exogenous AEA in large DRG neurons from naive mice (Khasabova et al., unpublished observations), which is consistent with high degradation of AEA in this population and our ability to detect CB1 receptor-mediated inhibition in larger DRG neurons.
Conclusion
The observations that mechanical hyperalgesia and a decreased level of AEA in the paw were associated with increased expression of FAAH provided the basis for demonstrating that the hyperalgesia was attenuated when the enzyme was inhibited pharmacologically. These data provide a rationale for the local, peripheral use of FAAH inhibitors in the management of cancer pain. Moreover, localization of an increase in FAAH expression to DRG neurons provides a rationale for extended analgesia in cancer pain by delivery of a FAAH siRNA to selectively suppress FAAH activity in DRG neurons. In addition, increased expression of CB1 receptors on DRG neurons ipsilateral to tumors in tumor-bearing mice provides a basis for local injection of CB1 receptor agonists to suppress tumor-evoked pain. These strategies may provide important alternatives in the management of cancer pain in light of the low effectiveness of opioids as well as side effects associated with systemic cannabinoid administration.
Footnotes
This work was supported by grants from the Academic Health Center, the Graduate School (I.A.K.), and the National Institutes of Health [CA091007 and DA11471 (D.A.S.)]. A.E.L. is a 3 M Science and Technology Fellow and predoctoral trainee on an institutional training grant from the National Institute of Drug Abuse (DA007234). We are grateful to P. Villalta and the University of Minnesota Cancer Center for expertise and resources in the measurement of anandamide and to J. Hodges for guidance in statistical analyses of the data.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Bevan S, Capogna M, Nagy I. Cannabinoid 1 receptors are expressed by nerve growth factor- and glial cell-derived neurotrophic factor-responsive primary sensory neurones. Neuroscience. 2002;110:747–753. doi: 10.1016/s0306-4522(01)00601-7. [DOI] [PubMed] [Google Scholar]
- Ahluwalia J, Yaqoob M, Urban L, Bevan S, Nagy I. Activation of capsaicin-sensitive primary sensory neurones induces anandamide production and release. J Neurochem. 2003;84:585–591. doi: 10.1046/j.1471-4159.2003.01550.x. [DOI] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertová M, Elphick MR, Winter J, Michael GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Cain DM, Wacnik PW, Turner M, Wendelschafer-Crabb G, Kennedy WR, Wilcox GL, Simone DA. Functional interactions between tumor and peripheral nerve: changes in excitability and morphology of primary afferent fibers in a murine model of cancer pain. J Neurosci. 2001;21:9367–9376. doi: 10.1523/JNEUROSCI.21-23-09367.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Carmella SG, Chen M, Zhang Y, Zhang S, Hatsukami DK, Hecht SS. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: effects of cigarette smoking. Chem Res Toxicol. 2007;20:986–990. doi: 10.1021/tx700075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman V. The cannabinoid CB1 receptor antagonist, SR141716A, selectively facilitates nociceptive responses of dorsal horn neurones in the rat. Br J Pharmacol. 1999;127:1765–1767. doi: 10.1038/sj.bjp.0702758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Hitchcock SA. Targeting cannabinoid agonists for inflammatory and neuropathic pain. Expert Opin Investig Drugs. 2007;16:951–965. doi: 10.1517/13543784.16.7.951. [DOI] [PubMed] [Google Scholar]
- Chin CN, Murphy JW, Huffman JW, Kendall DA. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J Pharmacol Exp Ther. 1999;291:837–844. [PubMed] [Google Scholar]
- Clohisy DR, Ogilvie CM, Ramnaraine ML. Tumor osteolysis in osteopetrotic mice. J Orthop Res. 1995;13:892–897. doi: 10.1002/jor.1100130613. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coward DD, Wilkie DJ. Metastatic bone pain. Meanings associated with self-report and self-management decision making. Cancer Nurs. 2000;23:101–108. doi: 10.1097/00002820-200004000-00005. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Dirajlal S, Pauers LE, Stucky CL. Differential response properties of IB(4)-positive and -negative unmyelinated sensory neurons to protons and capsaicin. J Neurophysiol. 2003;89:513–524. doi: 10.1152/jn.00371.2002. [DOI] [PubMed] [Google Scholar]
- Egertová M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- Evans RM, Scott RH, Ross RA. Multiple actions of anandamide on neonatal rat cultured sensory neurones. Br J Pharmacol. 2004;141:1223–1233. doi: 10.1038/sj.bjp.0705723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Felder CC, Nielsen A, Briley EM, Palkovits M, Priller J, Axelrod J, Nguyen DN, Richardson JM, Riggin RM, Koppel GA, Paul SM, Becker GW. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393:231–235. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guerrero AV, Quang P, Dekker N, Jordan RC, Schmidt BL. Peripheral cannabinoids attenuate carcinoma-induced nociception in mice. Neurosci Lett. 2008;433:77–81. doi: 10.1016/j.neulet.2007.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Beaulieu P. Antihyperalgesic effects of local injections of anandamide, ibuprofen, rofecoxib and their combinations in a model of neuropathic pain. Neuropharmacology. 2006;50:814–823. doi: 10.1016/j.neuropharm.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Guindon J, De Léan A, Beaulieu P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain. 2006;121:85–93. doi: 10.1016/j.pain.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, Hohmann AG. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. Br J Pharmacol. 2007;150:153–163. doi: 10.1038/sj.bjp.0706984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto DT, Giridharagopalan S, Simone DA. Acute and chronic administration of the cannabinoid receptor agonist CP 55,940 attenuates tumor-evoked hyperalgesia. Eur J Pharmacol. 2007;558:73–87. doi: 10.1016/j.ejphar.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim Biophys Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- Hiura A, Sakamoto Y. Quantitative estimation of the effects of capsaicin on the mouse primary sensory neurons. Neurosci Lett. 1987;76:101–106. doi: 10.1016/0304-3940(87)90200-x. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience. 2000;98:585–598. doi: 10.1016/s0306-4522(00)00110-x. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Hu SS, Bradshaw HB, Chen JS, Tan B, Walker JM. Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFkappaB activity. Br J Pharmacol. 2008;153:1538–1549. doi: 10.1038/bjp.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Parada CA, Levine JD. Hyperalgesic priming in the rat demonstrates marked sexual dimorphism. Pain. 2003;105:143–150. doi: 10.1016/s0304-3959(03)00175-1. [DOI] [PubMed] [Google Scholar]
- Kehl LJ, Hamamoto DT, Wacnik PW, Croft DL, Norsted BD, Wilcox GL, Simone DA. A cannabinoid agonist differentially attenuates deep tissue hyperalgesia in animal models of cancer and inflammatory muscle pain. Pain. 2003;103:175–186. doi: 10.1016/s0304-3959(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Simone DA, Seybold VS. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–625. doi: 10.1016/s0306-4522(02)00449-9. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Stucky CL, Harding-Rose C, Eikmeier L, Beitz AJ, Coicou LG, Hanson AE, Simone DA, Seybold VS. Chemical interactions between fibrosarcoma cancer cells and sensory neurons contribute to cancer pain. J Neurosci. 2007;27:10289–10298. doi: 10.1523/JNEUROSCI.2851-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HI, Kim TH, Shin YK, Lee CS, Park M, Song JH. Anandamide suppression of Na+ currents in rat dorsal root ganglion neurons. Brain Res. 2005;1062:39–47. doi: 10.1016/j.brainres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Lainé J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Luger NM, Sabino MA, Schwei MJ, Mach DB, Pomonis JD, Keyser CP, Rathbun M, Clohisy DR, Honore P, Yaksh TL, Mantyh PW. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs inflammatory pain. Pain. 2002;99:397–406. doi: 10.1016/S0304-3959(02)00102-1. [DOI] [PubMed] [Google Scholar]
- Luger NM, Mach DB, Sevcik MA, Mantyh PW. Bone cancer pain: from model to mechanism to therapy. J Pain Symptom Manage. 2005;29:S32–S46. doi: 10.1016/j.jpainsymman.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Di Rienzo M, Battista N, Gasperi V, Guerrieri P, Rossi A, Finazzi-Agrò A. The endocannabinoid system in human keratinocytes. Evidence that anandamide inhibits epidermal differentiation through CB1 receptor-dependent inhibition of protein kinase C, activation protein-1, and transglutaminase. J Biol Chem. 2003;278:33896–33903. doi: 10.1074/jbc.M303994200. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Piccirilli S, Battista N, Del Duca C, Nappi G, Corasaniti MT, Finazzi-Agrò A, Bagetta G. Enhanced anandamide degradation is associated with neuronal apoptosis induced by the HIV-1 coat glycoprotein gp120 in the rat neocortex. J Neurochem. 2004;89:1293–1300. doi: 10.1111/j.1471-4159.2004.02430.x. [DOI] [PubMed] [Google Scholar]
- Maida V, Ennis M, Irani S, Corbo M, Dolzhykov M. Adjunctive nabilone in cancer pain and symptom management: a prospective observational study using propensity scoring. J Support Oncol. 2008;6:119–124. [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, Rossi FS, Woodward DF, Di Marzo V. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini I, Dumon JC, Body JJ. Efficacy and safety of ibandronate in the treatment of opioid-resistant bone pain associated with metastatic bone disease: a pilot study. J Clin Oncol. 2004;22:3587–3592. doi: 10.1200/JCO.2004.07.054. [DOI] [PubMed] [Google Scholar]
- McBride G. Treating bone metastases: sparing the patient needless pain. J Natl Cancer Inst. 1997;89:418–419. doi: 10.1093/jnci/89.6.418. [DOI] [PubMed] [Google Scholar]
- Mercadante S, Arcuri E. Breakthrough pain in cancer patients: pathophysiology and treatment. Cancer Treat Rev. 1998;24:425–432. doi: 10.1016/s0305-7372(98)90005-6. [DOI] [PubMed] [Google Scholar]
- Mulè F, Amato A, Baldassano S, Serio R. Involvement of CB1 and CB2 receptors in the modulation of cholinergic neurotransmission in mouse gastric preparations. Pharmacol Res. 2007;56:185–192. doi: 10.1016/j.phrs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Niiyama Y, Kawamata T, Yamamoto J, Omote K, Namiki A. Bone cancer increases transient receptor potential vanilloid subfamily 1 expression within distinct subpopulations of dorsal root ganglion neurons. Neuroscience. 2007;148:560–572. doi: 10.1016/j.neuroscience.2007.05.049. [DOI] [PubMed] [Google Scholar]
- Nucci C, Gasperi V, Tartaglione R, Cerulli A, Terrinoni A, Bari M, De Simone C, Agrò AF, Morrone LA, Corasaniti MT, Bagetta G, Maccarrone M. Involvement of the endocannabinoid system in retinal damage after high intraocular pressure-induced ischemia in rats. Invest Ophthalmol Vis Sci. 2007;48:2997–3004. doi: 10.1167/iovs.06-1355. [DOI] [PubMed] [Google Scholar]
- Padgett LW, Howlett AC, Shim JY. Binding mode prediction of conformationally restricted anandamide analogs within the CB1 receptor. J Mol Signal. 2008;3:5. doi: 10.1186/1750-2187-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RJ, Duchen MR. Differential expression of membrane currents in dissociated mouse primary sensory neurons. Neuroscience. 1994;63:1041–1056. doi: 10.1016/0306-4522(94)90571-1. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids. 2002;66:101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portenoy RK. Managing cancer pain poorly responsive to systemic opioid therapy. Oncology (Williston Park) 1999;13:25–29. [PubMed] [Google Scholar]
- Potenzieri C, Harding-Rose C, Simone DA. The cannabinoid receptor agonist, WIN 55, 212–2, attenuates tumor-evoked hyperalgesia through peripheral mechanisms. Brain Res. 2008;1215:69–75. doi: 10.1016/j.brainres.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt PF, Hillard CJ, Edgemond WS, Campbell WB. N-arachidonylethanolamide relaxation of bovine coronary artery is not mediated by CB1 cannabinoid receptor. Am J Physiol. 1998;274:H375–H381. doi: 10.1152/ajpheart.1998.274.1.H375. [DOI] [PubMed] [Google Scholar]
- Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Coutts AA, McFarlane SM, Anavi-Goffer S, Irving AJ, Pertwee RG, MacEwan DJ, Scott RH. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: implications for antinociception. Neuropharmacology. 2001;40:221–232. doi: 10.1016/s0028-3908(00)00135-0. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Kelly S, Millns PJ, O'Shaughnessey CT, Kendall DA, Chapman V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci. 2005;22:371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. A FAAH-regulated class of N-acyl taurines that activates TRP ion channels. Biochemistry. 2006;45:9007–9015. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- Salio C, Fischer J, Franzoni MF, Conrath M. Pre- and postsynaptic localizations of the CB1 cannabinoid receptor in the dorsal horn of the rat spinal cord. Neuroscience. 2002;110:755–764. doi: 10.1016/s0306-4522(01)00584-x. [DOI] [PubMed] [Google Scholar]
- Scott BS. Adult mouse dorsal root ganglia neurons in cell culture. J Neurobiol. 1977;8:417–427. doi: 10.1002/neu.480080503. [DOI] [PubMed] [Google Scholar]
- Serafini AN. Therapy of metastatic bone pain. J Nucl Med. 2001;42:895–906. [PubMed] [Google Scholar]
- Sokal DM, Elmes SJ, Kendall DA, Chapman V. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurones via activation of CB2 receptors in anaesthetised rats. Neuropharmacology. 2003;45:404–411. doi: 10.1016/s0028-3908(03)00195-3. [DOI] [PubMed] [Google Scholar]
- Srivastava M, Walsh D. Diazepam as an adjuvant analgesic to morphine for pain due to skeletal muscle spasm. Support Care Cancer. 2003;11:66–69. doi: 10.1007/s00520-002-0386-8. [DOI] [PubMed] [Google Scholar]
- Strangman NM, Patrick SL, Hohmann AG, Tsou K, Walker JM. Evidence for a role of endogenous cannabinoids in the modulation of acute and tonic pain sensitivity. Brain Res. 1998;813:323–328. doi: 10.1016/s0006-8993(98)01031-2. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Thayer SA, Seybold VS. Prostaglandin E2 increases the proportion of neonatal rat dorsal root ganglion neurons that respond to bradykinin. Neuroscience. 1996;74:1111–1123. doi: 10.1016/0306-4522(96)00264-3. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Urban L, Dray A. Actions of capsaicin on mouse dorsal root ganglion cells in vitro. Neurosci Lett. 1993;157:187–190. doi: 10.1016/0304-3940(93)90733-2. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B, McNaughton P, Geppetti P, Di Marzo V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 2005;24:3026–3037. doi: 10.1038/sj.emboj.7600784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde S, Lambert DM. The multiple pathways of endocannabinoid metabolism: a zoom out. Chem Biodivers. 2007;4:1858–1881. doi: 10.1002/cbdv.200790156. [DOI] [PubMed] [Google Scholar]
- Vásquez C, Lewis DL. The CB1 cannabinoid receptor can sequester G-proteins, making them unavailable to couple to other receptors. J Neurosci. 1999;19:9271–9280. doi: 10.1523/JNEUROSCI.19-21-09271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacnik PW, Eikmeier LJ, Ruggles TR, Ramnaraine ML, Walcheck BK, Beitz AJ, Wilcox GL. Functional interactions between tumor and peripheral nerve: morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J Neurosci. 2001;21:9355–9366. doi: 10.1523/JNEUROSCI.21-23-09355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacnik PW, Kehl LJ, Trempe TM, Ramnaraine ML, Beitz AJ, Wilcox GL. Tumor implantation in mouse humerus evokes movement-related hyperalgesia exceeding that evoked by intramuscular carrageenan. Pain. 2003;101:175–186. doi: 10.1016/s0304-3959(02)00312-3. [DOI] [PubMed] [Google Scholar]
- Wacnik PW, Eikmeier LJ, Simone DA, Wilcox GL, Beitz AJ. Nociceptive characteristics of tumor necrosis factor-alpha in naive and tumor-bearing mice. Neuroscience. 2005;132:479–491. doi: 10.1016/j.neuroscience.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;151:843–853. doi: 10.1016/j.neuroscience.2007.11.025. [DOI] [PubMed] [Google Scholar]