

Abstract
Objective
Investigate the contribution of PIG-A mutations to clonal expansion in paroxysmal nocturnal hemoglobinuria (PNH).
Methods
Primary CD34+ hematopoietic progenitors from PNH patients were assayed for annexin V positivity by flow cytometry in a cell-mediated killing assay using autologous effectors from PNH patients or allogeneic effectors from healthy controls. To specifically assess the role of the PIG-A mutation in the development of clonal dominance and address confounders of secondary mutation and differential immune attack that can confound experiments using primary cells, we established an inducible PIG-A CD34+ myeloid cell line, TF-1. Apoptosis resistance was assessed after exposure to allogeneic effectors, NK92 cells (an IL-2 dependent cell line with the phenotype and function of activated NK cells), TNF-α, and γ-irradiation. Apoptosis was measured by annexin V staining and caspase 3/7 activity.
Results
In PNH patients, CD34+ hematopoietic progenitors lacking GPI-anchored proteins (GPI-AP-) were less susceptible than GPI-AP+ CD34+ precursors to autologous (8% versus 49%, p<0.05) and allogeneic (28% versus 58%, p<0.05) cell-mediated killing from the same patients. In the inducible PIG-A model, GPI-AP- TF-1 cells exhibited less apoptosis than induced, GPI-AP+ TF-1 cells in response to allogeneic cell-mediated killing, NK92-mediated killing, TNF-α, and γ-irradiation. GPI-AP- TF-1 cells maintained resistance to apoptosis when effectors were raised against GPI-AP- cells, arguing against a GPI-AP being the target of immune attack in PNH. NK92 mediated killing was partially inhibited with blockade by specific antibodies to the stress-inducible GPI-AP ULBP1 and ULBP2 that activate immune effectors. Clonal competition experiments demonstrate that the mutant clone expands over time under pro-apoptotic conditions with TNF-α.
Conclusion
PIG-A mutations contribute to the clonal expansion in PNH by conferring a survival advantage to hematopoietic progenitors under pro-apoptotic stresses.
Keywords: paroxysmal nocturnal hemoglobinuria, PIG-A, clonal expansion
Introduction
PNH is a clonal hematopoietic stem cell disorder manifested by complement-mediated intravascular hemolysis, thrombosis, smooth muscle dystonias, and bone marrow failure.[1] The disease results from the expansion of an abnormal clone that harbors a somatic mutation in the X-linked gene, PIG-A.[2-4] The PIG-A gene product is required for the initial step in the biosynthesis of glycosylphosphatidylinositol (GPI), a lipid moiety that attaches various proteins to the cell surface. The hallmark of PNH blood cells is a clonal population that are deficient in cell surface GPI anchored proteins (GPI-AP-).[5]
The mechanisms of clonal dominance in PNH are not entirely understood. One hypothesis to explain the close relationship between PNH and aplastic anemia, as well as the mechanisms whereby the PNH clone achieves dominance, involves a two-step model. This model first proposes that early hematopoietic stem cells randomly and spontaneously acquire PIG-A mutations. Indeed, PIG-A mutations can be found at low frequency in mature blood cells from most healthy controls; however, in contrast to PIG-A mutations in PNH, most of these mutations do not occur in hematopoietic stem cells.[6-8] Step two in this model proposes that the immunologic attack that targets hematopoietic stem cells, as in acquired aplastic anemia, spares PNH cells (i.e., immune escape), possibly because of the absence of one or more GPI-AP.[9, 10] There is evidence for such an autoimmune attack in the classical form of PNH that does not arise from aplastic anemia.[11, 12] Although the one feature common to all cases of PNH is a mutant PIG-A gene, it remains unclear to what degree the mutation contributes to the clonal expansion of GPI-AP- cells. It has been proposed that the expansion of PNH cells may involve a second somatic mutation in the PIG-A mutant stem cell[13]. Inoue et al. recently reported two PNH patients who possessed a chromosome 12 rearrangement that resulted in ectopic expression of the transcription factor HMGA2.[14] While the chromosome 12 rearrangement may have contributed to the clonal expansion in these two patients, most PNH patients have normal karyotypes.
Using hematopoietic progenitors from PNH patients and an inducible PIG-A myeloid cell line, we studied the mechanism of clonal dominance in PNH. We find that there is an ongoing immune attack against hematopoietic progenitors in both de novo PNH and that arising from aplastic anemia, and this attack spares GPI-AP- cells. Moreover, the PIG-A mutations appear to contribute to the resistance of GPI-AP- cells to the immune attack by conferring a global cellular resistance to pro-apoptotic stresses.
Methods
Patients
Peripheral blood and/or bone marrow samples from 3 normal controls and 6 patients with clinically evident PNH were collected after obtaining informed consent according to the requirements of the Institutional Review Board of the Johns Hopkins School of Medicine. The diagnosis of severe aplastic anemia was based on the criteria of Camitta et al.[15] The diagnosis of PNH was confirmed by flow cytometry as previously described.[16, 17]
TF-1 inducible cell line
TF-1 is a GM-CSF dependent, human CD34+ hematopoietic progenitor cell line (CRL-2003, ATCC, Manassas, VA) that expresses GPI-AP and possesses a wild-type PIG-A gene.[6] TF-1 cells were selected in growth medium containing 1nM proaerolysin[18] (Protox Biotech, Victoria, Canada). Proaerolysin binds the glycan core of the GPI anchor and lyses cells expressing GPI-AP. Thus, proaerolysin does not directly induce DNA damage; rather, it selects for spontaneous PIG gene family mutants that lead to failed expression of GPI anchored proteins. A proaerolysin resistant clone was expanded and shown to be GPI-AP deficient. TF-1 proaerolysin resistant cells had a 7 nucleotide deletion at position 291-297 (TTGTCAC) in exon 2 of PIG-A, which results in a frameshift mutation (data not shown). Inducible TF-1 cells were prepared as previously described.[20] Briefly, the PIG-A inducible TF-1 cells were transduced by the two lentiviral vectors comprising the tet-SUPER inducible transgene expression system. In this system, the first vector expresses a transcriptional suppressor (TS) domain fused to the tetracycline (tet) repressor DNA binding domain. The chimeric transcription suppressor (tTS) binds to its cognate DNA site, tetO, and inhibits transcription from nearby promoters. The second lentiviral vector expresses the human PIG-A cDNA controlled by the human EF1α promoter adjacent to the tetO DNA site. The binding of tTS to the tetO site suppresses the EF1α promoter-mediated transcription of the PIG-A transgene. Addition of tetracycline or its synthetic analogue doxycycline (Dox) will abolish the binding of the tTS, and thereafter releases its suppression of PIG-A transgene expression. Thus, we established an isogenic system for induced PIG-A expression: PIG-A expression is absent when Dox is absent; conversely, once Dox is added, PIG-A expression is turned on, and maximal GPI-AP expression is reached after 2 days. Cells were maintained in RPMI 1640 medium supplemented with 2 ng/mL GM-CSF (Biosource, Camarillo, CA), 2mM l-glutamine, penicillin/streptomycin, and 10% fetal calf serum under BL2 lab containment. Phenotyping of TF-1 cells was performed with the following reagents: fluorescent aerolysin (FLAER, Protox Biotech, Victoria, Canada; mouse monoclonal antibodies to human ULBP1 and ULBP2 (R&D Systems, Minneapolis, MN) with secondary goat anti-mouse IgG2a FITC conjugated (Invitrogen, Carlsbad, CA) according to manufacturers recommendations.
Induction of apoptosis in inducible TF-1 cells
Inducible TF-1 cells were split into 2 equal volumes during log phase growth and 10 ng/mL doxycycline was added to one flask for at least 2 days to induce GPI anchor expression. For TNF-∝ induced apoptosis, at least 106 cells in log phase growth were subjected to TNF-∝ (Promega, Madison, WI) for 2 hours at 37°C and then washed in cold PBS. For irradiation experiments, at least 106 cells from each induced and uninduced sample were washed × 1 in RPMI 1640 to remove serum prior to radiation and assayed 6 hours after irradiation exposure at 2 or 4.5 Gy using a cesium GammaCell irradiator (GammaCell, Ontario, Canada).
Caspase 3/7 activity was measured in cell lysates using a CaspAce colorimetric assay (Promega). All steps after incubation were performed at 4°C. Caspase activity was normalized to the total protein content of each lysate, as measured by the Bradford method. Time-course experiments were done to determine the optimal sampling time for caspase activity (2 hours for TNF-α and 6 hours for irradiation).
Flow cytometric cell-mediated lysis assay
Effector peripheral blood mononuclear cells (PBMC) isolated by Ficoll/Hypaque density gradient (sg=1.077) were co-cultured at 37°C, 5%CO2, and 100% humidity for 5 to 7 days with normal, irradiated allogeneic PBMC (12 Gy), or with original or PIG-A mutant TF-1 cells (40 Gy) as stimulators at a concentration of 1×106/ml (each cell type) in RPMI-1640 medium supplemented with 20% FBS, 50μM 2-mercaptoethanol, and 2mM l-glutamine. CD34+ bone marrow mononuclear cells were isolated by iron-colloid conjugated anti-CD34 antibody and double passage through a magnetically activated column (Miltenyi Biotech, Auburn, CA). CD34+ targets were then labeled with 4μM PKH-26 (Sigma, St. Louis, MO); induced or uninduced TF-1 cells were labeled with 10μM carboxyfluorescein diacetate succinimidyl ester (CFSE). Stimulated effectors were mixed with the PKH-26+ or CFSE+ targets at a 20:1 effector:target ratio, lightly pelleted (100 × g for 3 minutes) in round-bottom 12×75mm tubes, and co-cultured for 4 - 7 hours at 37°C. Primary CD34+ cells were then stained with fluorescent aerolysin (FLAER, Protox Biotech), propidium iodide (PI), and annexin V-biotin/streptavidin-PerCP (Pharmingen, San Diego, CA). TF-1 targets were stained with PI and annexin V-APC. Data was acquired on a FACScan or FACSCalibur instrument (Becton Dickinson, Mansfield, MA). Apoptosis was measured as the sum of all PI positive and annexin-only positive PKH-26+ or CFSE+ target cells. Results for TF-1 experiments represent apoptosis of the treated cells minus the background apoptosis untreated induced and uninduced controls.
NK92 cell-mediated lysis assay with the inducible TF-1 cell line
The NK cell line NK92 (ATCC) was incubated in alpha-MEM medium without ribonucleosides and deoxyribonucleosides, supplemented with 12.5% horse serum, 12.5% fetal bovine serum, 2mM L-glutamine, 0.2mM inositol, 0.02mM folic acid, and 0.1mM 2-mercaptoethanol. NK92 cells were incubated with CFSE-labeled induced or uninduced TF-1 cells at 37° and 5% CO2 after 3 minute centrifugation at 100g in 12×75mm round bottom tubes. Cells were stained after incubation with annexin V-APC and PI. CFSE+ gated events were analyzed for apoptosis (annexin and/or PI positive events) and reported as the percent apoptosis minus the background apoptosis in untreated, CFSE-labeled controls. Time-course and dosing experiments showed that a two hour incubation with an effector:target ratio of 1:1 yielded the most consistent apoptosis in the range of 30-70% by annexin V and PI staining. For NK92 inhibition experiments, target cells were incubated with mouse monoclonal antibodies to ULBP1 and ULBP2 (25 μg/mL each) for 15 minutes at room temperature prior to incubation with NK92 cells.
Clonal competition assay
Original and PIG-A mutant cells were maintained at similar cell concentrations, counted, and resuspended in RPMI 1640 media (serum-free) at 1:1 or 19:1 ratios of original to mutant cells. Cell mixtures were treated with nothing or TNF- α (1 or 10 ng/mL). Cell clone sizes were measured as above by flow cytometry with FLAER and PI exclusion at 1 and 2 days after exposure. Percent GPI negative populations were compared to the untreated control mixtures sampled at the same time points.
Statistics
Groups were compared using a two-tailed paired t test using Microsoft Excel software.
Results
Sensitivity of CD34+ cells from PNH patients to immune cell-mediated killing
In order to assess differences in apoptosis resistance in primary PNH cells, the sensitivity of GPI-AP- and GPI-AP+ CD34+ targets from PNH patients to both autologous effectors from PNH patients and allogeneic effectors from healthy controls was analyzed using a flow cytometry-based cell-mediated killing assay. Patient characteristics and the results of cell-mediated killing from the six PNH patients are listed in Table 1. The diagnosis of PNH was preceded by aplastic anemia in patients 1-3; all of these patients received immunosuppressive therapy (ATG/CSA) for their aplastic anemia before being diagnosed with PNH. In these patients, autologous effectors killed a mean of 55% CD34+GPI+ cells compared to only 5% of the CD34+GPI- cells. CD34+ cells from patients with classical PNH (Patients 4 - 6) were also studied. Similar to the patients whose PNH evolved from aplastic anemia, autologous effectors killed 42% CD34+GPI+ cells compared to only 11% of the CD34+GPI- cells. Overall (patients 1-6), the CD34+GPI- cells were more resistant than the CD34+GPI+ cells to autologous cell-mediated killing (8% versus 49%, p<0.05). Allogeneic effectors from normal controls kill based on MHC disparities rather than specific GPI-AP expression[24-26]. When allogeneic effectors were incubated with CD34+ cells from PNH patients, the CD34+GPI- cells again were more resistant than the CD34+GPI+ cells (28% versus 58%, p<0.05). Differences in apoptosis are unlikely due to FLAER staining because the channel forming portion of proaerolysin has been modified and previously shown not to induce apoptosis at this concentration[27], and control experiments show no differences in apoptosis between FLAER stained and unstained cells (supplemental Figure 1).
Table I. Autologous and allogeneic cell-mediated killing of wild type and PNH CD34+ hematopoietic progenitors.
| Patient | Age / Sex | Diagnosis | Disease Duration, years | % GPI− CD34+ Target Cells | % Apoptosis | |||
|---|---|---|---|---|---|---|---|---|
| Autologous Effectors vs. CD34+GPI− |
Autologous Effectors vs. CD34+GPI+ |
Allogeneic Effectors vs. CD34+GPI− |
Allogeneic Effectors vs. CD34+GPI+ |
|||||
| 1 | 31 / F | AA / PNH | 1 | 81 | 3 | 34 | ND | ND |
| 2 | 13 / F | AA / PNH | 1 | 65 | 8 | 50 | 18 | 52 |
| 3 | 52 / M | AA / PNH | 20 | 65 | 3 | 83 | 53 | 93 |
| 4 | 19 / F | PNH | 1 | 75 | 5 | 21 | 6 | 42 |
| 5 | 34 / M | PNH | 5 | 70 | 18 | 52 | 24 | 67 |
| 6 | 61 / F | PNH | 0.2 | 65 | 10 | 54 | 27 | 53 |
| Mean | 35 | 4.8 | 70 | 8± 2 | 49± 8 | 28± 8 | 58± 10 | |
ND= not done
Effects of PIG-A mutations on immune cell-mediated killing of TF-1 cells
The relative resistance of PNH progenitors to allogeneic killing suggests that a GPI-anchored epitope is not the target of immune attack, since such killing is mediated by major histocompatibility differences. However, it is possible that the increased in vitro T cell killing[25] of normal progenitors from PNH patients is the result of injury the cells had suffered during the in vivo autoimmune attack.[28] To control for genetic heterogeneity and mitigate differences in background apoptosis, a PIG-A mutant myeloid cell line was established by proaerolysin selection of the CD34+, GM-CSF dependent TF-1 cell line. The PIG-A mutant TF-1 cells were then transduced with PIG-A cDNA using a lentiviral vector under the control of a doxycycline inducible promoter to generate inducible PIG-A TF-1 cells (Figure 1A). The inducible TF-1 cell line becomes >90% GPI-AP positive after PIG-A induction with doxycycline (Figure 1B). PNH and wild type cells in this system are isogenic and are not subject to ongoing immune selection.
Figure 1. PIG-A inducible TF-1 cell line.
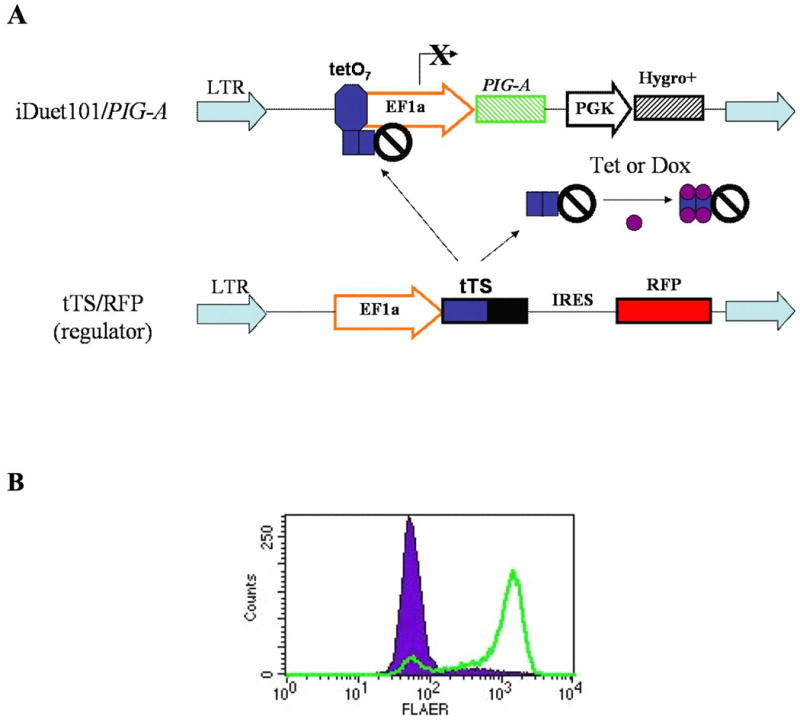
(A) The tet-SUPER PIG-A inducible lentiviral system. A tetracycline transcriptional suppressor (tTS), located on a regulator vector, blocks EF1a driven expression of PIG-A, until tetracycline or its analogue, doxycycline, is added. (B) Inducible TF1 cells are <5% GPI anchor positive, as identified by fluorescent aerolysin (FLAER, solid). After 2 days induction with 10 ng/mL doxycycline, cells become >90% GPI anchor positive (outline).
The effects of both NK92 cells and primed effector PBMCs on inducible PIG-A TF-1 cells were studied. NK92 is an IL-2 dependent NK cell line with the phenotype and function of activated NK cells; like NK cells, NK92 cells kill targets in an MHC independent manner by either granule exocytosis or binding TNF family receptors.[29] Compared to the induced, GPI-AP+ TF-1 cells, the uninduced, GPI-AP- cells were relatively resistant to NK92 mediated killing (44% vs. 58%, p<0.05, Figure 2), in agreement with prior results[30]. The induced and uninduced cells exhibited similar amounts of background apoptosis, demonstrating that doxycycline was not responsible for the differences in apoptosis (Figure 2A).
Figure 2. GPI- TF-1 cells are more resistant to NK92-mediated killing.
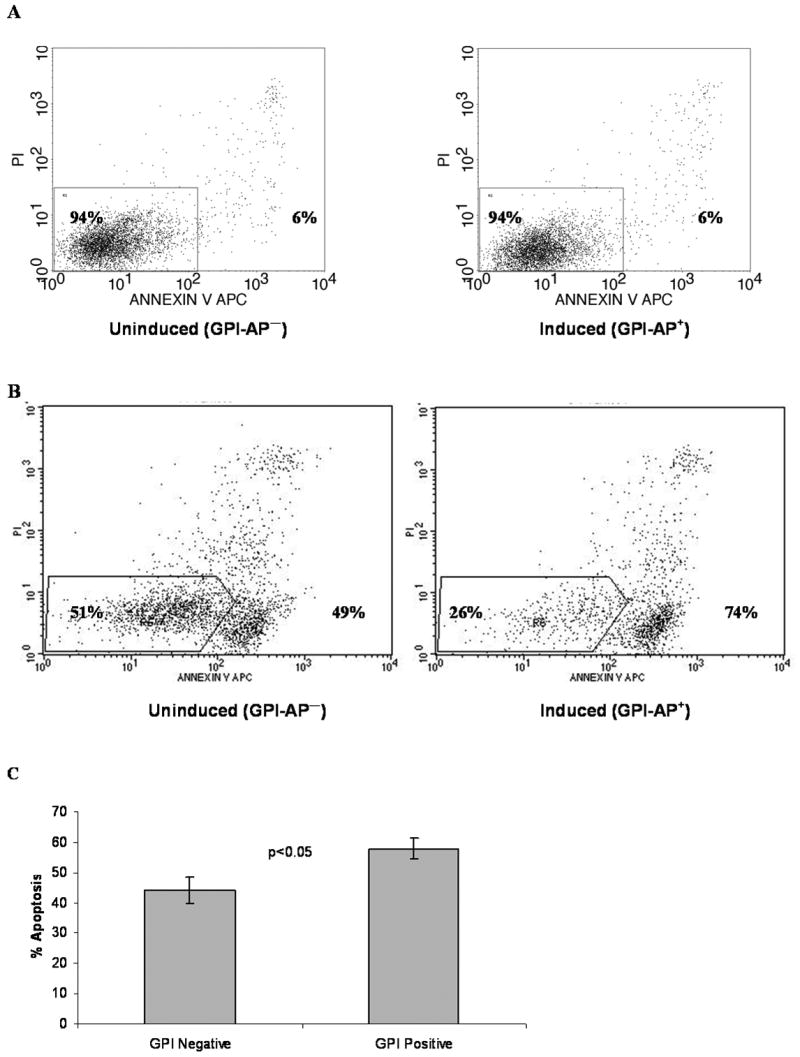
(A) An example of annexin V-APC and PI staining of CFSE-labeled, control uninduced (GPI-, left) and induced (GPI+, right) TF1 cells, showing similar background apoptosis. Numbers on the left inside the gate represent % viable cells, and numbers on the right represent % apoptotic cells. (B) An example of NK92 mediated cell lysis. NK92 cells were mixed 1:1 with CFSE labeled uninduced (GPI-) or induced (GPI+) TF-1 cells for 2 hours. Apoptosis is measured as percent annexin+ and/or PI+ of CFSE+ events. (C) Results of 5 experiments +/- SEM. Results represent percent apoptosis minus background apoptosis of untreated, CFSE-labeled controls.
Allogeneic PBMCs from normal controls were primed against either GPI-AP replete or deficient TF-1 cells and used to test resistance to allogeneic cell-mediated killing. Using GPI-AP- TF-1 cells to stimulate effectors yielded a mean 18% apoptosis in uninduced TF-1 targets and 28% apoptosis in induced TF-1 targets. Using GPI-AP replete TF-1 cells to stimulate effectors yielded a mean 16% apoptosis in uninduced TF-1 targets and 28% apoptosis in induced TF-1 targets. Induced (GPI-AP+) TF-1 targets consistently underwent more apoptosis than uninduced (GPI-AP-) targets, regardless of whether wild-type or PNH TF-1 cells were used to stimulate effectors (Figure 3). Since the allogeneic cell-mediated lysis results were independent of whether GPI-AP- or GPI-AP+ cells were used to stimulate the effectors, the difference in apoptosis does not appear to be a function of the immunogenicity of specific GPI-AP epitopes.
Figure 3. GPI- TF-1 cells are more resistant to allogeneic cell-mediated killing.
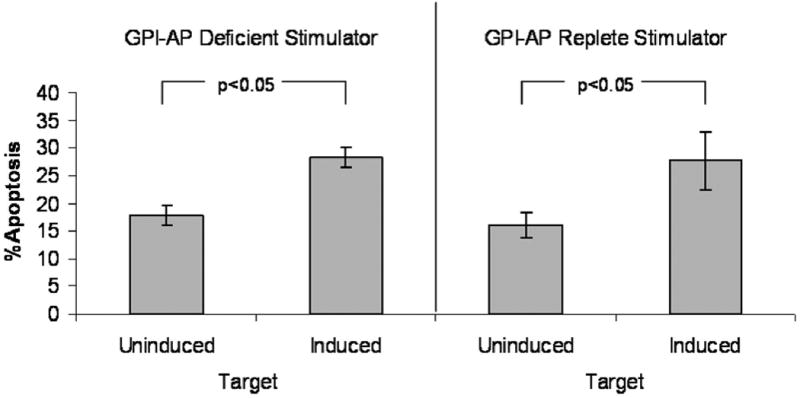
Allogeneic effectors (peripheral blood mononuclear cells) were raised against either original (WT) or PIG-A mutant TF-1 cells as stimulators. Effectors were then incubated with either uninduced (GPI deficient) or induced (GPI replete) CFSE-labeled TF-1 cells. Bars represent apoptosis (annexin+ and/or PI+) caused by cell-mediated killing minus background apoptosis of CFSE-labeled, untreated controls. GPI+ TF-1 cells had more apoptosis regardless of whether GPI deficient or replete cells were used to stimulate effectors. Results represent the mean ± SEM 5 experiments.
Effects of PIG-A mutations on TNF-α and radiation mediated apoptosis in TF-1 cells
Resistance to cell-mediated apoptosis in PNH could result from the absence of a GPI-AP required for immune recognition or killing, or alternatively from an intrinsic resistance to apoptosis caused by PIG-A mutations. To investigate the effects of PIG-A mutations on resistance to alternate mechanisms of apoptosis induction, caspase 3/7 activity was analyzed in the inducible TF-1 cells treated with either TNF-α or γ-irradiation (Figure 4). Induced cells (GPI-AP+) showed dose-dependent, increased caspase activity as compared to the uninduced (GPI-AP-) cells to both irradiation (Figure 4A) and TNF-α (Figure 4B).
Figure 4. TF-1 cells lacking PIG-A expression are resistant to apoptosis.
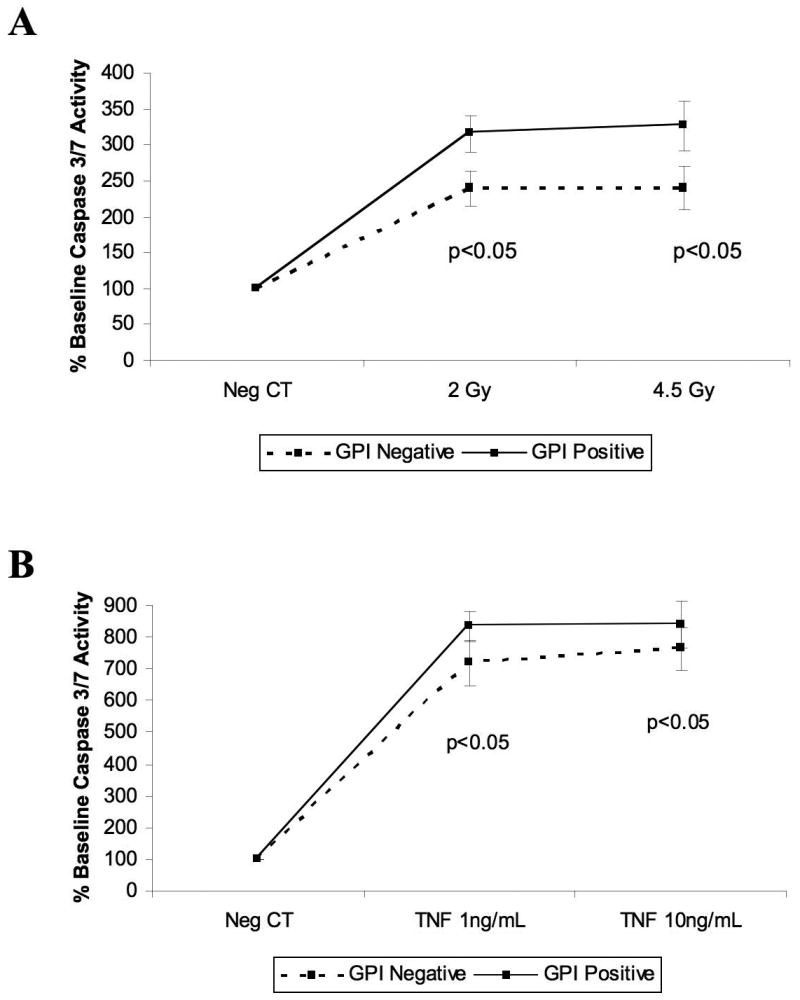
(A) Relative induction of caspase 3/7 activity 6h after radiation exposure. (B) Relative induction of caspase 3/7 activity 2h after TNF-α exposure. Time-course experiments were done to determine the optimal sampling time for caspase activity. Results represent the mean ±SEM of 6 experiments.
ULBP1 and ULBP2 in TF-1 apoptosis resistance
ULBP1 and ULBP2 are stress-inducible GPI-AP that activate immune effectors. Hanaoka et al. demonstrated that PIG-A mutant K562 cells are resistant to NK mediated killing because of the lack of expression of these proteins[31]. Similar to K562 cells, PIG-A induced TF-1 cells express ULBP1 and ULBP2, whereas uninduced TF-1 cells do not (Figure 5a). In response to gamma irradiation and TNF-α no increase in expression is noted (Figure 5b,c). When induced TF-1 cells have ULBP1 and ULBP2 blocked with antibody and are incubated with NK92 cells, a 3.4% decrease in apoptosis is observed (p< 0.05) when assayed with annexin V and PI positivity as above. This decrease is less than the 14% decrease observed with total absence of GPI-AP.
Figure 5. ULBP 1 and 2 expression on PIGA inducible TF-1 cells.
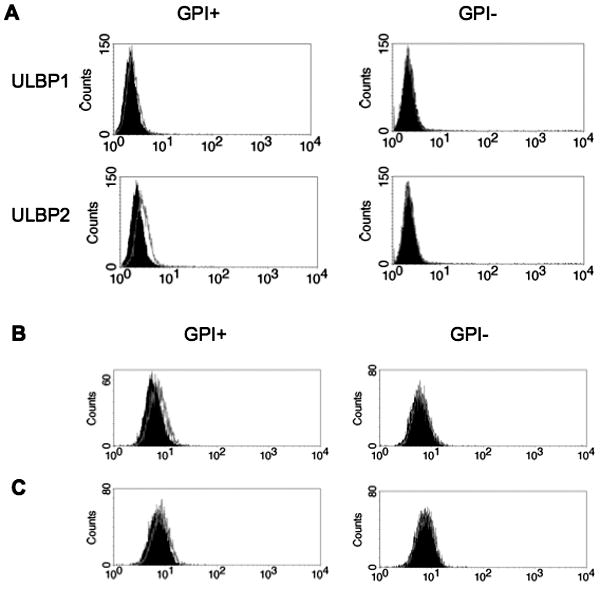
(A) PIG-A inducible TF-1 cells were treated with (GPI+) or without (GPI-) doxycycline for 2 days. Expression of ULBP1 and ULBP2 was determined by comparison to isotype control staining (solid) by flow cytometry. (B) ULBP2 expression is not increased after 16 hour exposure to TNF-α (outline) or (C) 4.5 Gy radiation (outline). Isotype controls are displayed as solid histograms.
Apoptosis resistance and clonal selection
PIG-A inducible TF-1 cells showed differences in apoptosis under a variety of stressors, but it is important to demonstrate that apoptosis resistance translates into clonal expansion, as is often the case in the natural history of PNH. Since PIG-A inducible TF-1 cells need constant induction with doxycycline to maintain PIG-A expression, it is not feasible to mix induced and uninduced TF-1 cells in clonal competition experiments. We therefore mixed original and PIG-A mutant TF-1 cells and exposed them to TNF-α as a surrogate for autoimmune attack. After exposure to TNF-α, mutant TF-1 cells outgrew wild-type cells in a time and dose-dependent relationship at a 19:1 original to mutant TF-1 ratio (Figure 6). Similar results were observed using a 1:1 original to mutant ratio (data not shown).
Figure 6. Clonal competition of wild type and PIG-A mutant TF-1 cells.
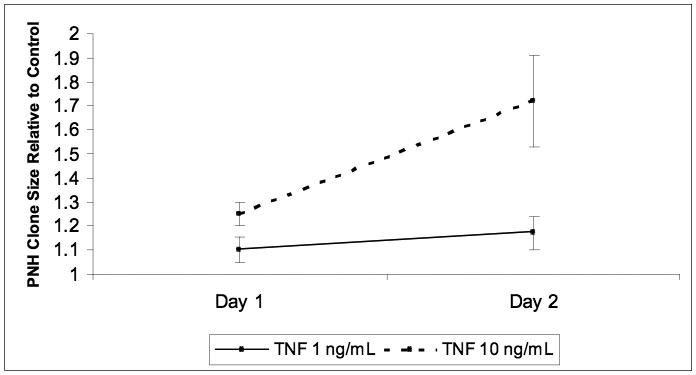
Original and PIG-A mutant TF-1 cells were mixed 19:1, respectively, and exposed daily to nothing, 1 ng/mL or 10 ng/mL TNF-α. The percentage of GPI− cells in treated and untreated controls were measured on days 1 and 2. Data are reported as the ratio of GPI− cells in treated groups to percent GPI− cells in untreated control cell mixtures. Both treatment groups showed statistically significant expansion over controls (p<0.05).
Discussion
The mechanisms underlying clonal expansion of PNH in vivo remain unclear. There are three leading hypotheses to explain such clonal expansion: 1) PNH cells evade immune attack possibly because a cell surface GPI-AP is the target of the immune attack[10, 11], 2) the PIG-A mutation itself confers an intrinsic resistance to apoptosis[32], and 3) a second anti-apoptotic mutation occurs in the PNH clone[14]. There are data that support each of these potential mechanisms for clonal expansion, as well data that conflict. For example, it would seem unlikely that a specific GPI-AP is the target of an autoimmune attack: GPI-AP are synthesized and degraded intracellularly in the setting of a PIG-A mutation[33]; therefore, protein epitopes should be displayed extracellularly by histocompatibility antigens and be immunogenic even if the intact protein is not on the cell surface.
Whether PNH cells are inherently less susceptible to apoptosis also remains controversial. No survival advantage has been found under a variety of conditions in the mouse model of PNH [34]. Moreover, several studies using human lymphoid cell lines found that the PIG-A mutant lymphocytes had equivalent apoptosis to wild type cells [35-38]. In contrast, reports consistently have found that primary GPI-AP- myeloid cells from PNH patients were relatively resistant to apoptosis [12, 32, 35, 39-41], even when the GPI-AP- lymphocytes were not [35]. However, two of these studies concluded that, since the degree of resistance was not proportional to PNH clone size, the resistance could be independent of the PIG-A mutation[35, 39]. Because primary GPI-AP- CD34+ progenitors exhibited similar proliferative and survival rates to CD34+ progenitors from healthy controls, other studies concluded that the survival advantage of the primary GPI-AP- cells resulted from diminished survival of GPI-AP+ progenitors in a “hostile” PNH environment[12, 40, 41]. However, it is possible that primary GPI-AP- CD34+ progenitors may also be damaged by the same hostile environment present in the PNH patients, and an intrinsic survival advantage in PNH cells may be obscured when PNH progenitors are compared to normal control progenitors which had not been exposed to such an environment. Thus, we designed experiments to address the potential methodological issues associated with studying lymphocytes, which are rarely affected in PNH, as well with comparing normal and PNH progenitors that had experienced different in vivo environments.
PNH CD34+ progenitors displayed less apoptosis than their wild type counterparts when incubated with both autologous and allogeneic effectors (Table 1). This finding suggested to us that the resistance to apoptosis of the PNH cells was not the result of the absence of a particular GPI-AP. Allogeneic effectors kill based on HLA incompatibility recognition, not GPI-AP expression. Thus, we would expect allogeneic effectors to kill GPI-AP- and GPI-AP+ to the same degree. However, the GPI-AP- cells have less apoptosis, suggesting that the improved survival is not due to a specific GPI-AP.
Experiments with primary cells cannot control for additional mutations that could contribute to a survival advantage or an in vivo immune attack that targets both the GPI-AP- as well as the GPI-AP+ cells in PNH patients. To specifically test the role of mutant PIG-A in the apoptosis resistance of myeloid cells, we developed an inducible PIG-A myeloid cell line, TF-1. PNH and wild type TF-1 cells in this system are isogenic as there are no other genetic differences between cells in which PIG-A is induced or suppressed. Additionally, the background levels of apoptosis are similar in both the PIG-A induced and uninduced cells, and the similar apoptosis controls for confounding effects of an autoimmune attack targeting the PNH cells. Thus, any differences in susceptibility to apoptosis could only be attributed to effects of the PIG-A gene. We found that PNH TF-1 cells consistently displayed less apoptosis than their isogenic, wild type counterparts when subjected to radiation, TNF-α, NK92, and allogeneic T cell-mediated apoptosis. This resistance to apoptosis translated into clonal expansion of PIG-A mutant TF-1 cells under immune selection by TNF-α.
Resistance to apoptosis was observed regardless of whether PIG-A mutant or original TF-1 cells were used to stimulate the allogeneic effectors, providing further evidence that the resistance to apoptosis of the PNH cells is not due to the absence of a particular GPI-AP. It is possible that stimulating effectors with the original TF-1 cell line could generate effector clones that target a specific GPI-AP. By using mutant TF-1 cells, which do not express GPI-AP, to stimulate effectors, we exclude the possibility that effectors are primed against a GPI-AP. The observed differences in apoptosis attributable to a PIG-A mutation were small, but consistent across experiments and statistically significant. This is not surprising, given that clonal expansion in PNH often occurs over many years.
Collectively, these data support an immune escape model in which an autoimmune attack provides a selective pressure on all hematopoietic progenitors, and under this stress the PNH clone expands because of an intrinsic survival advantage. These results further indicate that a second hit is not required to confer apoptotic resistance to PNH cells, although a second genetic alteration could certainly further augment survival and proliferation in a subset of PNH, as has been described[14].
A potential mechanism by which PIG-A mutations may produce resistance to apoptosis is through the disruption of lipid rafts by GPI anchor depletion. Lipid rafts are cell surface microdomains consisting primarily of sphingolipid, cholesterol, and GPI-AP.[42] It has been observed that cell signaling for Fas and TNF receptors is localized to lipid rafts,[43, 44] and that disruption of lipid rafts by depletion of sphingolipid or cholesterol can lead to apoptosis resistance[45-47]. Disruption of GPI anchors may destabilize lipid rafts in the same way, as has been described with the mutation of the PIG-A homologue in yeast[48]. Dispersion of lipid rafts may also explain the reported decreased Fas receptor expression on PNH cells[12].
The absence of ULBP1 and ULBP2 on GPI-AP- TF-1 cells explains at least some of this resistance to apoptosis, as well. In the PIG-A inducible TF-1 model system, blocking ULBP1 and ULBP2 did not completely overcome the change in apoptosis; nor was a significant change in ULBP1 or ULBP2 expression observed in apoptosis resistance in response to radiation and TNF-α. Nevertheless, it is feasible that the absence of ULBPs may play a larger role in apoptosis resistance in vivo.
Supplementary Material
Peripheral blood mononuclear cells were stained with nothing or 1 nM fluorescent aerolysin (FLAER) and incubated at 37 degrees, 5% CO2 in RPMI 1640 + 10% fetal calf serum. Apoptosis was measured by annexin V staining among CD45+ cells at 24 hours. Trypan blue exclusion demonstrated 94.2% viability of control cells and 95.0% viability of FLAER stained cells at 24 hours.
Acknowledgments
We wish to thank the patients who volunteered to contribute to this study and the thoughtful comments of the reviewers. Supported in part by National Institutes of Health Grants CA70970 and CA15396.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004;126:133–138. doi: 10.1111/j.1365-2141.2004.04992.x. [DOI] [PubMed] [Google Scholar]
- 2.Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 3.Miyata T, Takeda J, Iida Y, et al. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259:1318–1320. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 4.Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. Embo J. 1994;13:110–117. doi: 10.1002/j.1460-2075.1994.tb06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosse WF, Ware RE. The molecular basis of paroxysmal nocturnal hemoglobinuria. Blood. 1995;86:3277–3286. [PubMed] [Google Scholar]
- 6.Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA. PIG-A mutations in normal hematopoiesis. Blood. 2005;105:3848–3854. doi: 10.1182/blood-2004-04-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci U S A. 1999;96:5209–5214. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ware RE, Pickens CV, DeCastro CM, Howard TA. Circulating PIG-A mutant T lymphocytes in healthy adults and patients with bone marrow failure syndromes. Exp Hematol. 2001;29:1403–1409. doi: 10.1016/s0301-472x(01)00746-9. [DOI] [PubMed] [Google Scholar]
- 9.Zeng W, Miyazato A, Chen G, Kajigaya S, Young NS, Maciejewski JP. Interferon-gamma-induced gene expression in CD34 cells: identification of pathologic cytokine-specific signature profiles. Blood. 2006;107:167–175. doi: 10.1182/blood-2005-05-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young NS, Maciejewski J. The pathophysiology of acquired aplastic anemia. N Engl J Med. 1997;336:1365–1372. doi: 10.1056/NEJM199705083361906. [DOI] [PubMed] [Google Scholar]
- 11.Karadimitris A, Manavalan JS, Thaler HT, et al. Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2000;96:2613–2620. [PubMed] [Google Scholar]
- 12.Chen R, Nagarajan S, Prince GM, et al. Impaired growth and elevated fas receptor expression in PIGA(+) stem cells in primary paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2000;106:689–696. doi: 10.1172/JCI8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker CJ. The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2007;35:523–533. doi: 10.1016/j.exphem.2007.01.046. [DOI] [PubMed] [Google Scholar]
- 14.Inoue N, Izui-Sarumaru T, Murakami Y, et al. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2006;108:4232–4236. doi: 10.1182/blood-2006-05-025148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camitta BM, Thomas ED, Nathan DG, et al. A prospective study of androgens and bone marrow transplantation for treatment of severe aplastic anemia. Blood. 1979;53:504–514. [PubMed] [Google Scholar]
- 16.Mukhina GL, Buckley JT, Barber JP, Jones RJ, Brodsky RA. Multilineage glycosylphosphatidylinositol anchor-deficient haematopoiesis in untreated aplastic anaemia. Br J Haematol. 2001;115:476–482. doi: 10.1046/j.1365-2141.2001.03127.x. [DOI] [PubMed] [Google Scholar]
- 17.Brodsky RA, Mukhina GL, Li S, et al. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol. 2000;114:459–466. doi: 10.1093/ajcp/114.3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brodsky RA, Mukhina GL, Nelson KL, Lawrence TS, Jones RJ, Buckley JT. Resistance of paroxysmal nocturnal hemoglobinuria cells to the glycosylphosphatidylinositol-binding toxin aerolysin. Blood. 1999;93:1749–1756. [PubMed] [Google Scholar]
- 19.Beier F, Balabanov S, Buckley T, et al. Accelerated telomere shortening in glycosylphosphatidylinositol (GPI)-negative compared with GPI-positive granulocytes from patients with paroxysmal nocturnal hemoglobinuria (PNH) detected by proaerolysin flow-FISH. Blood. 2005;106:531–533. doi: 10.1182/blood-2004-10-3996. [DOI] [PubMed] [Google Scholar]
- 20.Zhou BY, Ye Z, Chen G, Gao ZP, Zhang YA, Cheng L. Inducible and Reversible Transgene Expression in Human Stem Cells after Efficient and Stable Gene Transfer. Stem Cells. 2006 doi: 10.1634/stemcells.2006-0128. [DOI] [PubMed] [Google Scholar]
- 21.Zeng W, Nakao S, Takamatsu H, et al. Characterization of T-cell repertoire of the bone marrow in immune-mediated aplastic anemia: evidence for the involvement of antigen-driven T-cell response in cyclosporine-dependent aplastic anemia. Blood. 1999;93:3008–3016. [PubMed] [Google Scholar]
- 22.Melenhorst JJ, Fibbe WE, Struyk L, van der Elsen PJ, Willemze R, Landegent JE. Analysis of T-cell clonality in bone marrow of patients with acquired aplastic anaemia. Br J Haematol. 1997;96:85–91. doi: 10.1046/j.1365-2141.1997.d01-1989.x. [DOI] [PubMed] [Google Scholar]
- 23.Zeng W, Maciejewski JP, Chen G, Young NS. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest. 2001;108:765–773. doi: 10.1172/JCI12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hess AD, Donnenberg AD, Tutschka PJ, Santos GW. Effect of cyclosporin A on human lymphocyte responses in vitro. V. Analysis of responding T lymphocyte subpopulations in primary MLR with monoclonal antibodies. J Immunol. 1983;130:717–721. [PubMed] [Google Scholar]
- 25.Hess AD, Tutschka PJ. Effect of cyclosporin A on human lymphocyte responses in vitro. I. CsA allows for the expression of alloantigen-activated suppressor cells while preferentially inhibiting the induction of cytolytic effector lymphocytes in MLR. J Immunol. 1980;124:2601–2608. [PubMed] [Google Scholar]
- 26.Petersdorf EW, Malkki M. Genetics of risk factors for graft-versus-host disease. Semin Hematol. 2006;43:11–23. doi: 10.1053/j.seminhematol.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 27.Nelson KL, Brodsky RA, Buckley JT. Channels formed by subnanomolar concentrations of the toxin aerolysin trigger apoptosis of T lymphomas. Cell Microbiol. 1999;1:69–74. doi: 10.1046/j.1462-5822.1999.00009.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen G, Zeng W, Maciejewski JP, Kcyvanfar K, Billings EM, Young NS. Differential gene expression in hematopoietic progenitors from paroxysmal nocturnal hemoglobinuria patients reveals an apoptosis/immune response in ‘normal’ phenotype cells. Leukemia. 2005;19:862–868. doi: 10.1038/sj.leu.2403678. [DOI] [PubMed] [Google Scholar]
- 29.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- 30.Nagakura S, Ishihara S, Dunn DE, et al. Decreased susceptibility of leukemic cells with PIG-A mutation to natural killer cells in vitro. Blood. 2002;100:1031–1037. doi: 10.1182/blood.v100.3.1031. [DOI] [PubMed] [Google Scholar]
- 31.Hanaoka N, Kawaguchi T, Horikawa K, Nagakura S, Mitsuya H, Nakakuma H. Immunoselection by natural killer cells of PIGA mutant cells missing stress-inducible ULBP. Blood. 2006;107:1184–1191. doi: 10.1182/blood-2005-03-1337. [DOI] [PubMed] [Google Scholar]
- 32.Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to apoptosis caused by PIG-A gene mutations in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci U S A. 1997;94:8756–8760. doi: 10.1073/pnas.94.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodukula K, Amthauer R, Cines D, et al. Biosynthesis of phosphatidylinositol-glycan (PI-G)-anchored membrane proteins in cell-free systems: PI-G is an obligatory cosubstrate for COOH-terminal processing of nascent proteins. Proc Natl Acad Sci U S A. 1992;89:4982–4985. doi: 10.1073/pnas.89.11.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulkarni S, Bessler M. The effect of GPI-anchor deficiency on apoptosis in mice carrying a Piga gene mutation in hematopoietic cells. J Leukoc Biol. 2002;72:1228–1233. [PubMed] [Google Scholar]
- 35.Ware RE, Nishimura J, Moody MA, Smith C, Rosse WF, Howard TA. The PIG-A mutation and absence of glycosylphosphatidylinositol-linked proteins do not confer resistance to apoptosis in paroxysmal nocturnal hemoglobinuria. Blood. 1998;92:2541–2550. [PubMed] [Google Scholar]
- 36.Karadimitris A, Notaro R, Koehne G, Roberts IA, Luzzatto L. PNH cells are as sensitive to T-cell-mediated lysis as their normal counterparts: implications for the pathogenesis of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2000;111:1158–1163. doi: 10.1046/j.1365-2141.2000.02494.x. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi Y, McCoy JP, Jr, Carvallo C, et al. In vitro and in vivo evidence of PNH cell sensitivity to immune attack after nonmyeloablative allogeneic hematopoietic cell transplantation. Blood. 2004;103:1383–1390. doi: 10.1182/blood-2003-04-1281. [DOI] [PubMed] [Google Scholar]
- 38.Bastisch I, Tiede A, Deckert M, Ziolek A, Schmidt RE, Schubert J. Glycosylphosphatidylinositol (GPI)-deficient Jurkat T cells as a model to study functions of GPI-anchored proteins. Clin Exp Immunol. 2000;122:49–54. doi: 10.1046/j.1365-2249.2000.01350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horikawa K, Nakakuma H, Kawaguchi T, et al. Apoptosis resistance of blood cells from patients with paroxysmal nocturnal hemoglobinuria, aplastic anemia, and myelodysplastic syndrome. Blood. 1997;90:2716–2722. [PubMed] [Google Scholar]
- 40.Chen G, Kirby M, Zeng W, Young NS, Maciejewski JP. Superior growth of glycophosphatidy linositol-anchored protein-deficient progenitor cells in vitro is due to the higher apoptotic rate of progenitors with normal phenotype in vivo. Exp Hematol. 2002;30:774–782. doi: 10.1016/s0301-472x(02)00811-1. [DOI] [PubMed] [Google Scholar]
- 41.Ismail MM, Tooze JA, Flynn JA, et al. Differential apoptosis and Fas expression on GPI-negative and GPI-positive stem cells: a mechanism for the evolution of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2003;123:545–551. doi: 10.1046/j.1365-2141.2003.04643.x. [DOI] [PubMed] [Google Scholar]
- 42.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 43.Hueber AO, Bernard AM, Herincs Z, Couzinet A, He HT. An essential role for membrane rafts in the initiation of Fas/CD95-triggered cell death in mouse thymocytes. EMBO Rep. 2002;3:190–196. doi: 10.1093/embo-reports/kvf022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Lacour S, Hammann A, Grazide S, et al. Cisplatin-induced CD95 redistribution into membrane lipid rafts of HT29 human colon cancer cells. Cancer Res. 2004;64:3593–3598. doi: 10.1158/0008-5472.CAN-03-2787. [DOI] [PubMed] [Google Scholar]
- 46.Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood. 2007;109:711–719. doi: 10.1182/blood-2006-04-016824. [DOI] [PubMed] [Google Scholar]
- 47.Nachbur U, Kassahn D, Yousefi S, Legler DF, Brunner T. Posttranscriptional regulation of Fas (CD95) ligand killing activity by lipid rafts. Blood. 2006;107:2790–2796. doi: 10.1182/blood-2005-07-2744. [DOI] [PubMed] [Google Scholar]
- 48.Schonbachler M, Horvath A, Fassler J, Riezman H. The yeast spt14 gene is homologous to the human PIG-A gene and is required for GPI anchor synthesis. Embo J. 1995;14:1637–1645. doi: 10.1002/j.1460-2075.1995.tb07152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peripheral blood mononuclear cells were stained with nothing or 1 nM fluorescent aerolysin (FLAER) and incubated at 37 degrees, 5% CO2 in RPMI 1640 + 10% fetal calf serum. Apoptosis was measured by annexin V staining among CD45+ cells at 24 hours. Trypan blue exclusion demonstrated 94.2% viability of control cells and 95.0% viability of FLAER stained cells at 24 hours.