

Abstract
Phosphatidylserine (PS)-dependent erythrocyte adhesion to endothelium and sub-endothelial matrix components is mediated in part via thrombospondin (TSP). While TSP exhibits multiple cell-binding domains, the PS-binding site on TSP is unknown. Since a cell-binding domain for anionic heparin is located at the amino-terminus, we hypothesized that PS-positive red cells (PS+ve-RBCs) bind to this domain. We demonstrate that both heparin and its low-molecular-weight derivative enoxaparin (0.5-50u/ml) inhibited PS+ve-RBC adhesion to immobilized TSP in a concentration-dependent manner (21-77% inhibition, P<0.05). Pre-incubation of immobilized TSP with an antibody against the heparin-binding domain blocked PS+ve-RBC adhesion to TSP. Antibodies that recognize the collagen- and the carboxy-terminal CD47-binding domain on TSP had no effect on this process. While pre-incubation of PS+ve-RBCs with TSP-peptides from the heparin-binding domain containing the specific heparin-binding motif KKTRG inhibited PS+ve-erythrocyte adhesion to matrix TSP (P<0.001), these peptides in the immobilized form supported PS-mediated erythrocyte adhesion. A TSP-peptide lacking the binding-motif neither inhibited nor supported PS+ve-RBC adhesion. Additional experiments show that soluble-TSP also interacted with PS+ve-RBCs via its heparin-binding domain. Our results demonstrate that PS-positive erythrocytes bind to both immobilized and soluble TSP via its heparin-binding domain and that both heparin and enoxaparin, at clinically relevant concentrations, block this interaction. Other studies have shown that heparin inhibited P-selectin- and soluble-TSP-mediated sickle erythrocyte adhesion to endothelial cells. Our results taken together with the previously documented findings provide a rational basis for clinical use of heparin or its low-molecular-weight derivatives as therapeutic agents in treating vaso-occlusive pain in patients with sickle cell disease.
INTRODUCTION
Phosphatidylserine (PS), an anionic phospholipid present exclusively in the inner leaflet of the plasma membrane of normal cells, is externalized following cell activation by both physiologic and pathologic stimuli.1,2 It has been well recognized that PS exposure on the cell surface serves as a signal for phagocytic recognition and removal of apoptotic cells.3 It can also function as an adhesion ligand mediating cell-cell interaction. PS-mediated erythrocyte adhesion to endothelial cells and/or sub-endothelial matrix components has been documented in patients with many hemolytic anemias including sickle cell disease (SCD),4 malaria,5 and uremia6 with documented positive correlation in SCD between the levels of percent PS-positivity and red cell-endothelial adhesion.4 Abnormal erythrocyte adhesion appears to play an important role in vascular complications seen not only in patients with SCD,7 but also in malaria5 and uremia.6 PS-dependent erythrocyte adhesion appears to be mediated in part via thrombospondin (TSP),8 a multifunctional and a matricellular glycoprotein.9-13 TSP is synthesized and released by a variety of mammalian cells including endothelial cells, and is incorporated into their matrix, becoming exposed following endothelial injury or cell retraction induced by agonists such as thrombin.14-17 As shown in Figure-1, while TSP can interact with a variety of cells via specific cell-binding domains on the molecule,9-13 the binding site for the anionic PS on the TSP molecule has not been identified to date. In this study, we demonstrate that PS-positive erythrocytes bind to both soluble and immobilized TSP via its heparin-binding domain.
Figure-1. Structure of thrombospondin subunit.
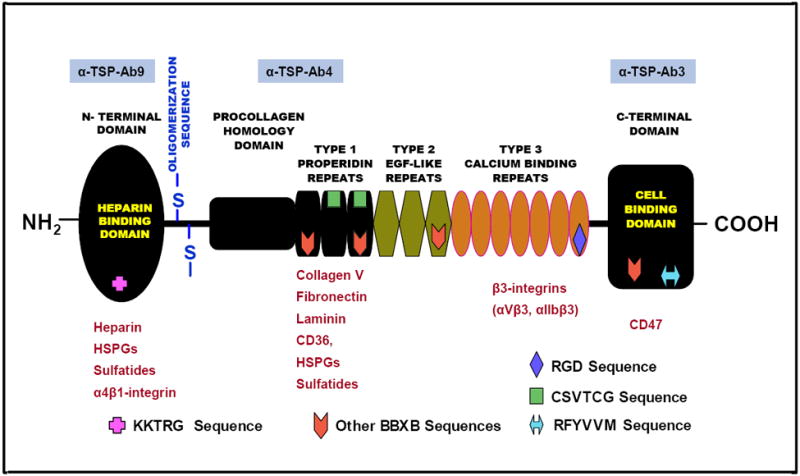
Schematic diagram modified from Gupta et al18 depicting the different structural domains and cell binding regions of the TSP subunit relevant to erythrocyte adhesion to endothelial cells and/or to the components of sub-endothelial matrix. Each subunit of the TSP molecule contains several structural domains including the N-terminal, the C-terminal and the pro-collagen homology domains, the oligomerization sequence, and three type 1 properidine repeats, three type 2 EGF-like repeats and seven type 3 calcium binding repeats. Erythrocyte-related cell surface receptors, proteins and adhesion markers that have been reported to interact with various regions of TSP molecule are shown in red. Anti-TSP antibodies used in this study are shown in blue boxes above their respective TSP interacting domains. HSPGs: heparan sulfate proteoglycans. In other BBXB sequences, B is a basic amino acid and X is any amino acid.
MATERIALS and METHODS
Materials
Purified thrombospondin-1 from human platelets (referred to as TSP in this manuscript), annexin-V-pure (product A9460) and unfractionated heparin (from porcine intestine) were purchased from Sigma Chemical (St Louis, MO). Enoxaparin, a low molecular weight heparin derivative (Aventis Pharmaceuticals, Sanofi-Aventis, Bridgewater, NJ) was obtained through Jefferson University Hospital Pharmacy. High molecular weight dextran sulfate or HDS (ICN Biochemicals, Cleveland, OH), chondroitin sulfate A or CSA (from bovine trachea), calcium ionophore A23187 (Calbiochem, La Jolla, CA) and fluorescein isothiocyanate (FITC)-labeled annexin-V (R & D Systems, Minneapolis, MN) were also obtained. Mouse monoclonal antibodies against human thrombospondin: TSP Ab-9 (isotype IgG1, clone MBC200.1), TSP Ab-4 (isotype IgG1, clone A6.1), and TSP Ab-3 (isotype IgG1, clone C6.7) were procured from Lab Vision Corporation (Fremont, CA). These anti-TSP antibodies have previously been demonstrated to specifically recognize the N-terminal heparin-, the collagen-, and the C-terminal CD47-binding domain on TSP, respectively,18-21 as depicted in Figure-1. Both TSP-Ab9 and TSP-Ab3 functionally block red cell, platelet and melanoma cell adhesion to TSP.18-21 Antibodies against human CD36 (clone FA6.152), CD49d (α-chain of the VLA4 or very late activation antigen-4, clone HP2.1), CD47 (integrin-associated protein or IAP, clone BRIC126), CD239 (basal cell adhesion molecule/Lutheran protein or BCAM/LU, clone BRIC221), isotype-matched negative control antibody (clone 679.1Mc7), and FITC- and Tri-Color (TC)-labeled goat anti-mouse IgG were obtained from Immunotech (Beckman Coulter, Miami, FL), Serotec (Oxford, UK), or Caltag Laboratories (Burlingame, CA). Three TSP peptides from the amino-terminal heparin-binding domain of TSP containing the amino acid sequences KKTRGTLLALERKDHS (the heparin-binding motif is italicized in bold, residues 80-95, TSP peptide-1), VDAVRTEKGFLLLASRQMKKTRGT (residues 61-85, peptide-2), and a TSP peptide without the binding motif TLLALERKDHS (residues 85-95, peptide-3) were synthesized through Sigma Genosys (Woodlands, TX). The TSP peptides -1 and -2 (positive peptides) support adhesion of a wide variety of cells including murine 3T3 fibroblasts, several human breast carcinoma cells, human Bowes melanoma cells, human osteoblastic cells and Chinese hamster ovary cells,22,23 and inhibit binding of Chinese hamster ovary cells to immobilized N-terminal domain of TSP.23 The negative TSP peptide (peptide-3) neither supported cell adhesion nor inhibited cell binding to immobilized N-terminal domain of TSP.22,23
Preparation of PS-positive Red Cells
For the majority of experiments, PS positive HbAA erythrocytes were generated artificially from fresh adult control blood. PS-positive red cells were prepared by treating control erythrocytes with A23187,24,25 which routinely yielded a red cell preparation with PS positivity in the range of 60 to 75%. The ionophore-activated erythrocytes were suspended to a final concentration of 2×108 cells/ml in adherence buffer (10mM HEPES in Hanks balanced salt solution [buffer-A], pH 7.4 containing 1.3mM CaCl2, 0.7mM MgCl2 and 0.5% BSA), unless otherwise indicated. Untreated washed control erythrocytes (2×108 cells/ml in adherence buffer) served as the PS-negative control. Ionophore-treated PS-positive red cells were diluted with untreated PS-negative erythrocytes from the same donor to obtain PS-positivity in the range of 0.5 to 17.5%. Cell surface PS and other adhesion markers on test erythrocytes were analyzed by flow cytometry.26,27 Select experiments also were performed using erythrocytes obtained from 12 SCD patients with HbSS genotype, without subjecting these cells to any in vitro treatment. Blood samples were collected from patients with steady state disease (ages 6 to 15 years) during their routine out-patient clinic visit. This study was reviewed and approved by the Institutional Review Committee for the protection of human subjects at Thomas Jefferson University and at St Christopher’s Hospital for Children, Drexel University. In accordance with the Declaration of Helsinki, blood samples were obtained following informed consent. For minors, patient assent where appropriate was obtained in addition to parental permission.
Flow Adhesion Assay
For flow adhesion, matrices were prepared by coating pre-washed glass slides (25mm × 75mm) with desired concentrations of TSP (25 to 200 ng protein per cm2 in bicarbonate buffer, 0.5M, pH 9.6), air-dried, rinsed with the adherence buffer, and used. Erythrocyte adherence to immobilized TSP was evaluated using a parallel plate flow chamber as described by Hillery and coworkers.28 A shear rate of 1 dyne/cm2, a force equivalent to that in the post-capillary venule was employed, and adherence buffer was used in all steps of the assay. The matrix was preconditioned with the wash buffer under flow for 5-minutes, erythrocytes were then allowed to flow over the matrix for 3-minutes followed by a wash-off of non-adherent cells for 10-minutes. To enumerate adherent erythrocytes, each matrix was divided vertically into five equal segments, the central area of each segment photographed, and the images analyzed using Image-Pro Software (Media Cybernetics, Silver Spring, MD), with each data point representing the mean of these five evaluations. Results are presented as the number of adherent cells/mm2. To evaluate the effects on adhesion of heparin, enoxaparin, other glycosaminoglycans or GAGs including CSA and HDS, and TSP-peptides, erythrocytes were pre-incubated for 30-minutes at 37°C in the presence or absence of the desired agent prior to adhesion. To evaluate the effects of antibodies and sulfatides, immobilized TSP matrix was pre-incubated with the desired test reagent at room temperature for 30-minutes by flooding the matrix with the desired antibody at 10 μg/ml or sulfatide at 200μg/ml. Matrices were washed and adhesive potential assessed. To verify the involvement of PS in adhesion, erythrocytes were pre-incubated at 37°C in the presence of the indicated concentrations of annexin-V for 30-minutes to cover surface PS and then tested for their adhesive potential to immobilized TSP. Red cell surface PS was evaluated by flow cytometry using annexin-V-FITC.
Binding Studies with Soluble Thrombospondin
Erythrocytes (1×108 cells/ml containing 12% PS-positive verses PS-negative cells) were pre-incubated at 37°C in the presence of the indicated concentrations of soluble TSP for 60-minutes, and then evaluated for erythrocyte-bound TSP using indirect immunostaining, and employing the monoclonal antibodies TSP-Ab9 and TSP-Ab4. In brief, erythrocytes that had been pre-incubated with soluble TSP were washed twice with buffer-A containing 1% bovine serum albumin, 1.3mM CaCl2 and 0.7mM MgCl2. Washed red cells (1×106 cells) were incubated with 2μg primary antibody (TSP-Ab9, TSP-Ab4, or isotype-matched negative control) for 30-minutes at room temperature. After two washes, the antibody-labeled erythrocytes were incubated with 1μg TC-labeled goat anti-mouse IgG for 30-minutes at room temperature, washed, and analyzed by flow cytometry.
Statistical Analysis
Statistical evaluation was performed using Sigmastat Statistical Package (Systat, Richmond, CA). All results are presented as mean±SD. Comparison between controls and paired treatment groups (effects of GAGs and TSP peptides on red cell adhesion to TSP, erythrocyte adhesion to TSP peptides, and adhesion marker levels on control verses ionophore-activated erythrocytes) was performed using the paired student t-test or the Mann-Whitney rank sum test on the medians, if the data showed non-normal distribution. Unpaired student t-test or the Mann-Whitney rank sum test was employed to compare the differences in levels of adhesion markers between high PS and low PS groups. Multiple group comparison (dose response effects with heparin and enoxaparin, and effects of TSP antibodies and divalent cations on PS-TSP adhesion) was performed using either one-way ANOVA (for data with normal distribution) or the Kruskal-Wallis test (for data with non-normal distribution). If the P-value for this overall comparison was significant at P<0.05, then group-wise comparison was performed using the Dunnett’s or the Dunn’s test.
RESULTS
Binding of PS-positive Erythrocyte to Immobilized Thrombospondin
Initial binding experiments (n=4) were performed in adherence buffer using erythrocyte preparations containing 0.5 to 17.5% PS-positive cells and immobilized TSP matrices containing 25 to 200 ng protein per cm2. Binding of PS-positive erythrocytes to matrix TSP increased almost linearly up to 12% (Figure-2A). Similarly, red cell binding increased linearly between 25 and 100 ng matrix TSP with a marginal increase noted between 100 and 200 ng protein (Figure-2B). In all further studies to be described we have employed a matrix concentration of 50 ng/cm2 and a red cell PS-positivity of 12%, since this experimental condition produced ~25-fold increase in erythrocyte binding compared to the control matrix without TSP permitting us to evaluate both inhibitory and stimulatory responses within the same experiment without changing experimental parameters between treatments. Requirement of divalent cations including Ca2+ and Mg2+ in adhesion was next evaluated. While neither Ca2+ nor Mg2+ alone significantly modulated red cell interaction with TSP, presence of both cations in the binding buffer increased PS-mediated erythrocyte adhesion by 12-fold (p<0.01). All binding assays described in this study were, therefore, performed in medium containing Ca2+ (1.3mM) and Mg2+ (0.7mM). In the presence of both Ca2+ and Mg2+, 644 ± 123 red cells adhered per mm2 when PS-positive cells were used in adhesion, in contrast to 20 ± 8 cells with PS-negative control erythrocytes. PS specificity in adhesion was tested using the calcium-dependent PS-specific binding protein, annexin-V.29 As shown in Figure-3A, pre-treatment of red cell PS with annexin-V blocked PS-mediated cell binding to TSP matrix and reduced surface PS in parallel, confirming PS-TSP adhesion specificity.
Figure 2. Adhesion of PS-positive Erythrocytes to Immobilized Thrombospondin.
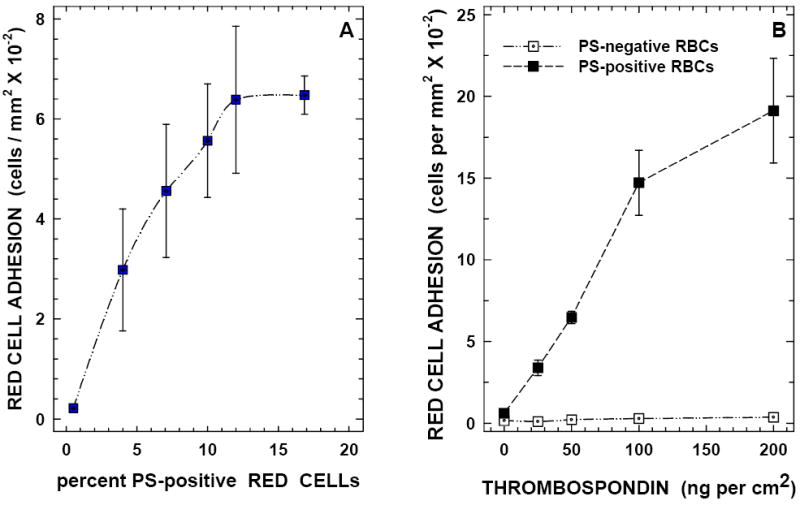
Panel A: PS-positive erythrocytes bind to immobilized TSP matrix in a concentration-dependent manner. Adhesion of erythrocytes (2×108 cells per ml containing 0.5 to 17% PS-positive red cells) was evaluated using a TSP matrix (containing 50 ng of protein per square cm) as described in methods.
Panel B: Immobilized TSP supports erythrocyte binding in a concentration-dependent Manner. Red cells (2×108 cells per ml containing 12% PS positivity) were evaluated in adhesion assays employing matrices containing 25 to 200 ng of protein per square cm.
Figure-3. Modulation of PS-mediated erythrocyte binding to immobilized TSP by annexin V, heparin, enoxaparin, HDS, CSA, and sulfatide.
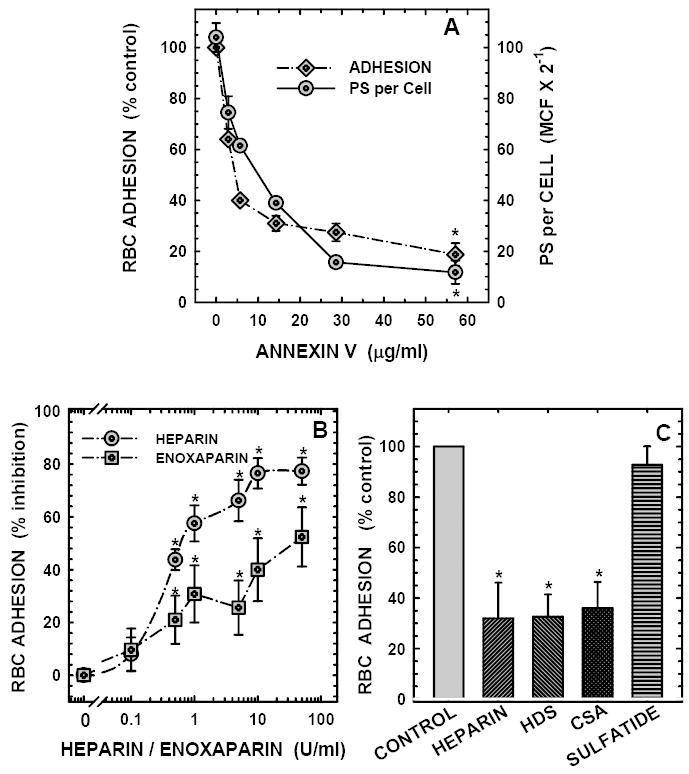
Panel A: Annexin V reduces PS binding sites on the red cell surface (measured as mean cell fluorescence or MCF) and inhibits erythrocyte adhesion to immobilized TSP. Erythrocytes (1×108 cells/ml containing 12% PS-positive cells) were pre-incubated in the presence or absence of the indicated concentrations of annexin-V, and then assessed for their adhesive potential (diamonds) and residual surface PS (circles) as described. Values presented are the means ± SD from 3 (annexin V between 3 and 28 μg/ml) to 5 (annexin V at 57 μg/ml) different experiments. The number of PS-positive erythrocytes adhered in the absence of annexin V was 639 ± 147 cells/mm2. *Changes noted with annexin V at 57 μg/ml were statistically significant at P<0.05.
Panel B: Heparin and enoxaparin inhibit PS-mediated erythrocyte binding to immobilized thrombospondin in a concentration-dependent manner. Red cells (2×108 cells/ml containing 12% PS-positive cells) were pre-treated with the indicated concentrations (0.1 to 50 U/ml) of heparin (circles) or Lovenox (squares), and then assessed for their adhesive potential using a TSP matrix containing 50 ng protein per cm2 as described in methods. Values presented are the means ± SD from 5 (Lovenox), or 9 (heparin) different experiments. *Inhibitory effects noted with heparin (between 0.5 and 50 U/ml) and Lovenox (between 0.5 and 50 U/ml) were statistically significant at P<0.05. The number of PS-positive erythrocytes adhered in the absence of modulators were 632 ± 241 cells/mm2.
Panel C:Effects of glycosaminoglycans (GAGs), and sulfatide on PS-mediated erythrocyte binding to immobilized thrombospondin. Effects of high molecular weight dextran sulfate, MW 500,000 (HDS, 200 μg/ml), chondroitin sulfate A (CSA, 200 μg/ml), and bovine brain sulfatide (200 μg/ml) on PS-mediated RBC-TSP binding were evaluated using erythrocytes (2×108 cells/ml) containing 12% PS and TSP matrix at 50 ng/cm2. Values presented are the means ± SD from 6 different experiments. Heparin (200 μg/ml, equivalent to 50 units/ml) tested in parallel with other GAGs is shown for paired comparison with HDS and CSA. *Red cell adhesion in the presence of heparin, HDS and CSA were different from the respective paired controls as assessed by the paired student’s t-test at P<0.01.
Effects of Heparin, Enoxaparin, HDS, CSA and Sulfatide on PS-mediated Erythrocyte Binding to Immobilized Thrombospondin
To test the hypothesis that PS-positive erythrocytes interacted with the N-terminal domain of TSP which supports binding of anionic polysaccharides including heparin, we evaluated the effects of heparin (0.1 to 50 units/ml) on PS-positive red cell adhesion to immobilized TSP. We demonstrate that heparin blocked erythrocyte binding to immobilized TSP in a concentration-dependent manner with significant inhibition of 44 to 77% noted at 0.5 to 50 units/ml heparin (Figure-3B). We next tested whether other anionic polysaccharides including HDS and CSA modulated PS-positive erythrocyte binding to TSP. Results presented in Figure-3C demonstrate that both HDS and CSA inhibited PS-mediated cell binding to TSP with the magnitude of the inhibitory effects comparable to that of heparin (~70% inhibition). These findings demonstrate that a charge-based interaction plays a role in erythrocyte binding to TSP. Enoxaparin, a low molecular weight heparin derivative, and an anticoagulant and antithrombotic therapeutic agent, also inhibited PS-mediated red cell adhesion to immobilized TSP in a concentration-dependent manner with significant inhibitory effects of 21 to 52% noted at concentrations between 0.5 and 50 units/ml (Figure-3B). The inhibitory effects noted with enoxaparin were lower when compared to that seen with un-fractionated heparin. In contrast to GAGs, bovine brain sulfatide had no effect on PS-mediated erythrocyte adhesion to immobilized TSP (Figure-3C).
Effects of anti-Thrombospondin Antibodies on PS-mediated Erythrocyte Binding to Immobilized Thrombospondin
To test whether the heparin-binding domain on TSP was involved in PS-mediated red cell binding to TSP, we evaluated the effects of three specific monoclonal anti-TSP antibodies: TSP-Ab3, TSP-Ab4 and TSP-Ab9, which recognize the C-terminal CD47-, the collagen-, and the N-terminal heparin-binding domain on TSP, respectively (Figure-1). As depicted in Figure-4A, pre-treatment of TSP matrix with TSP-Ab9 (antibody recognizing the heparin-binding domain) showed a significant reduction in erythrocyte binding compared to either the vehicle control or the IgG control (approximately 70 to 80% inhibition, P<0.05). TSP-Ab3, and TSP-Ab4, which recognize the CD47- and the collagen-binding domain on TSP, had no effect. To test whether PS-positive erythrocytes from a pathologic milieu interact with immobilized TSP in a manner similar to artificially generated PS-positive cells, in binding experiments, erythrocytes from SCD patients with HbSS disease containing various levels of PS positivity ranging from 0.6% to 8.1% were evaluated. These erythrocytes were divided into two experimental groups including low versus high PS-positive sickle erythrocytes (1.1±0.5% versus 6.6±1.7% PS positivity, P<0.001, Table-1) as we have done in our previous studies.4,25 As depicted in Figure-4B, while the anti-TSP antibody Ab9 had no significant effect on adhesion of low percent PS positive HbSS erythrocytes to immobilized TSP, a significant inhibition in adhesion to TSP of HbSS erythrocytes containing high percent PS positive cells was noted (38 ± 8% inhibition, P<0.05).
Figure-4. Effects of anti-TSP antibodies on PS-mediated erythrocyte binding to immobilized TSP.
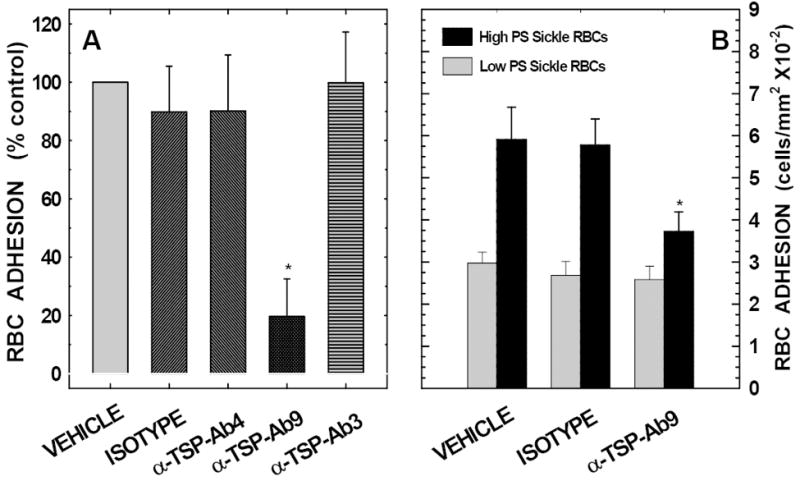
Panel A:Anti-TSP antibody that recognizes TSP’s heparin-binding domain (α-TSP-Ab9) inhibits PS-mediated erythrocyte binding to immobilized thrombospondin. Antibody-treated TSP matrices were tested for their adhesive potential using erythrocytes (2×108 cells/ml) containing 12% PS-positive cells. Values presented are the means ± SD from 7 different experiments. *Inhibition noted with TSP-Ab9 was statistically significant at P<0.05 when compared to either TSP-Ab3, TSP-Ab4, or the isotype-matched negative immunoglobulin control. Both anti-TSP-Ab4 and the negative immunoglobulin control demonstrated minimal inhibitory effects on red cell binding which were not statistically significant. The antibodies, TSP-Ab3 and TSP-Ab4 recognize the C-terminal CD47- and the collagen- binding domain on TSP, respectively.
Panel B:Effects of anti-TSP antibody Ab9 on binding of sickle erythrocytes to immobilized thrombospondin: TSP matrices were pre-incubated in the presence or absence of anti-TSP-Ab9 (an antibody that recognizes the heparin binding domain on TSP), or an equivalent amount of an isotype-matched negative immunoglobulin control for 30 minutes prior to their use in flow adhesion. Pre-treated matrices were then tested for their binding potential using red cells from patients with sickle cell disease, HbSS genotype (2×108 cells per ml). The gray and black bars represent adhesion of HbSS erythrocytes with low (1.1 ± 0.5% positivity) and high (6.6 ± 1.7% positivity) PS positivity, respectively. Results presented are the means ± SD from 5 (low PS) or 7 (high PS) experiments. *P<0.05 compared to the respective vehicle or the isotype control.
Table-1. Expression of adhesion molecules on HbAA control erythrocytes and HbSS red cells from patients with sickle cell disease.
Control HbAA erythrocytes (n=6) were made PS-positive by treating with A23187. PS-positivity was adjusted to 12% by diluting ionophore-treated red cells with untreated erythrocytes. No significant differences in the relevant red cell adhesion markers were noted in control untreated verses ionophore-treated cells exclusive of the changes in PS-positivity induced by ionophore treatment.
HbSS patients were deliberately cohorted into low PS (n=5) verses high PS (n=7) groups for our experimental purpose as we have done previously4,25. When the “low” PS group was compared to the “high” PS group, significant differences only in PS-positivity were noted (P<0.001). All other adhesion markers were comparable.
Cell surface PS and other erythrocyte adhesion markers were evaluated using two-color flow cytometry and employing glycophorin A as an erythrocyte marker. Values presented are the means ± SD from 5 to 7 experiments.
| Adhesion Marker | HbAA Erythrocytes | HbSS Erythrocytes | ||
|---|---|---|---|---|
| Untreated | A23187-treated Erythrocytes | Low PS group | High PS group | |
| CD36+ve | Not present | Not present | 0.6 ± 0.4% | 2.7 ± 2.1% |
| VLA4+ve | Not present | Not present | 0.11 ± 0.12% | 0.13 ± 0.20% |
| PS+ve | <0.3% | 12% | 1.1 ± 0.5% | 6.6 ± 1.7% |
| IAP (or CD47) +ve | 98.7 ± 1.7% | 99.6 ± 0.4% | 99.2 ± 0.5% | 98.3 ± 1.6% |
| BCAM/LU (or CD239) +ve | 19.7 ± 10.8% | 17.3 ± 12.9% | 35.2 ± 12.5% | 20.8 ± 13.9% |
Effects of Thrombospondin Peptides on PS-mediated Erythrocyte Binding to Immobilized Thrombospondin
To confirm that PS-positive red cells bind to heparin-binding domain on TSP, in binding assays we evaluated the effects of three previously characterized TSP peptides: two peptides containing the specific heparin binding motif KKTRG (positive peptides, peptides -1 and -2), and a third peptide lacking the binding motif (negative peptide, peptide-3).22,23 In preliminary experiments TSP peptide-1 blocked PS-mediated erythrocyte adhesion to immobilized TSP in a concentration-dependent manner with 30, 53, and 54% inhibition noted at 25, 50, and 100 μM, respectively. Our subsequent experiments were therefore performed using TSP peptides at 50 μM, since maximal inhibitory effects on adhesion were noted at this peptide concentration. In paired experiments (n=7), pretreatment of PS-positive red cells with TSP peptides -1 and -2 inhibited cell binding to immobilized TSP by 53% and 45%, respectively (P<0.001, Figure-5A). The negative peptide had no effect on this process (P>0.75). Inhibition of erythrocyte adhesion to immobilized TSP by TSP peptides indicated that these peptides interact directly with PS-positive red cells and suggested that the TSP peptides might support cell adhesion. We next evaluated whether red cells bind to immobilized TSP peptides and demonstrate that while no erythrocytes adhered to immobilized negative peptide (peptide-3), both positive TSP peptides (peptides-1 and -2) supported PS-mediated red cell binding (Figure-5B). These results taken together demonstrate that the heparin-binding domain on TSP supports PS-mediated erythrocyte binding to TSP.
Figure-5. Effects of TSP peptides on PS-mediated erythrocyte binding to immobilized TSP and adhesion of PS-positive red cells to immobilized peptides.
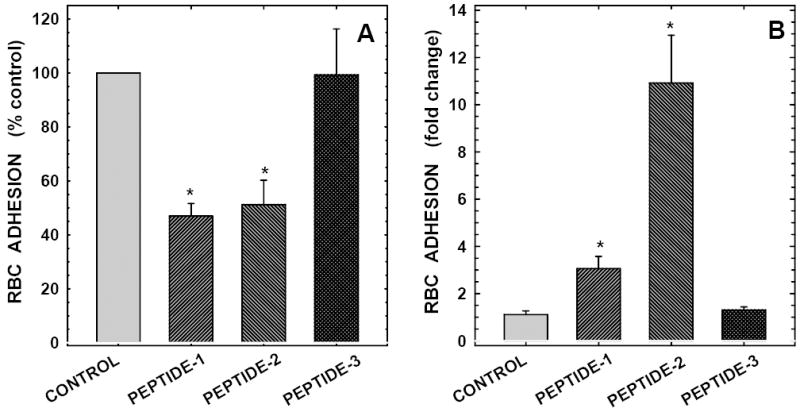
Panel A:Peptides from TSP’s heparin binding domain containing the heparin binding motif KKTRG inhibit PS-mediated erythrocyte binding to immobilized thrombospondin. Peptide (50μM)-treated erythrocytes (2×108 cells/ml containing 12% PS-positive cells) were tested for their adhesive potential to immobilized TSP. Both peptides -1 and -2 contained the heparin binding motif of TSP molecule (positive peptides) with the following amino acid sequences, peptide-1: KKTRGTLLALERKDHS (residues 80-95 of TSP subunit, the binding motif is shown in bold), peptide-2: VDAVRTEKGFLLLASRQMKKTRGT (residues 61-85 of TSP molecule). Peptide-3, TLLALERKDHS (residues 85-95 of TSP molecule, a negative peptide) lacked the heparin binding motif. Values presented are the means ± SD from 7 different experiments. *Erythrocyte adhesion in the presence of positive TSP peptides was significantly different from the respective controls as assessed by the paired student’s t-test at P<0.001.
Panel B:Immobilized peptides from TSP’s heparin binding domain containing the binding motif KKTRG support PS-mediated erythrocyte binding. Thrombospondin peptides were immobilized on glass slides using 0.5M bicarbonate buffer, pH 9.6 at 10 μg/cm2. Adhesion of erythrocytes (2×108 cells/ml containing 12% PS-positive cells) to immobilized TSP peptides was evaluated as described in methods. Values presented are the means ± SD from 3 different experiments. Erythrocyte binding to positive TSP peptides (peptides -1 and -2) was significantly different from the respective vehicle controls as assessed by the paired student’s t-test at P<0.02. There was no difference in red cell binding between the vehicle control and the negative peptide.
Soluble Thrombospondin Binds to PS-positive Erythrocytes
Since protein conformation of immobilized TSP may be different from that of soluble TSP, we investigated whether PS-positive erythrocytes bind to the same cell binding domain(s) on both soluble and immobilized TSP. Erythrocytes were incubated in the presence of increasing concentrations of soluble TSP (1 to 10 μg/ml), washed and then assessed for TSP positivity using the previously tested anti-TSP antibodies. As depicted in Figure-6A, while a minimal number of TSP-positive red cells were detected at all TSP concentrations tested using TSP-Ab9, an antibody that recognizes the heparin-binding domain on TSP (hatched bars), a concentration-dependent increase in TSP-positive red cells was observed with TSP-Ab4, an antibody that recognizes the collagen-binding domain (cross-hatched bars). No TSP-positive erythrocytes were detected when PS-negative red cells were incubated with soluble TSP (solid bars). Histogram profiles of TSP-positive red cells from a representative experiment are shown in Figure-6B demonstrating that TSP positivity on the red cell could be detected only with TSP-Ab4 (B2). TSP-positive red cells were almost undetectable when TSP-Ab9 was used as the detecting agent (B1). Neither TSP-Ab9 nor TSP-Ab4 detected any TSP-positivity on red cells that were not pre-treated with soluble TSP (B3 and B4).
Figure-6. Soluble thrombospondin binds to PS-positive erythrocytes and transforms PS-positive red cells to TSP-positive erythrocytes.
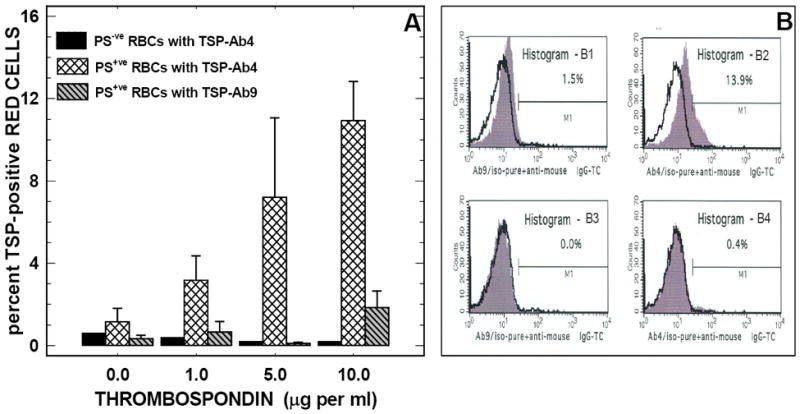
Panel A:Treatment of PS-positive erythrocytes with soluble TSP transforms PS-positive red cells into TSP-positive erythrocytes: Erythrocytes (1×108 cells/ml containing 12% PS-positive cells) were pre-incubated in the presence or absence of the indicated concentrations of soluble TSP, washed and then assayed for TSP-positivity using TSP antibodies: TSP-Ab4 (cross-hatched bars) and TSP-Ab9 (gray-hatched bars). TSP-positivity measured in parallel experiments with PS-negative control red cells using TSP-Ab4 is also shown (black solid bars) for comparison. Values presented are the means ± SD from 4 different experiments.
Panel B:Histogram profiles of TSP-positive erythrocytes: Erythrocytes (1×108 cells/ml containing 12% PS-positive cells) were pre-incubated in the presence (panels B1 and B2) or absence (panels B3 and B4) of soluble TSP (10 μg/ml) and then assayed for TSP positivity using anti-TSP-Ab4 (panels B2 and B4) or anti-TSP-Ab9 (panels B1 and B3). Histogram profiles of TC fluorescence from cells labeled with anti-TSP antibodies (gray histograms) and isotype-matched negative immunoglobulin control (solid black lines) are shown in each panel. The marker M1 is the positive histogram region defined using red cells labeled with the isotype-matched negative immunoglobulin control. Percent marker-positive cell, shown in each panel, is the difference in M1 between the cells labeled with anti-TSP and the isotype-matched negative immunoglobulin control. Results presented are from a representative experiment repeated four times with similar results. Note that the TSP-positivity was observed only with anti-TSP-Ab4 (an antibody that recognizes the collagen-binding domain on TSP). TSP positivity was almost undetectable when anti-TSP-Ab9 (an antibody that recognizes the heparin-binding domain on TSP) was used as the detecting agent, likely due to preoccupancy of the heparin binding domain on the TSP molecule by PS-positive erythrocytes. The TC-conjugated second antibody used in these experiments was labeled with PE-Cy5. This fluorochrome is excitated at 488 nm and emission detected at 667 nm. Its red fluorescence is detected in the FL3 channel on a FACScan flow cytometer.
Identification of Adhesion Molecules on Erythrocytes
Since multiple adhesion molecules are involved in mediating red cell adhesion to the components of sub-endothelial matrix, it was necessary to identify whether the other relevant adhesion molecules present on ionophore-activated erythrocytes were significantly altered. As depicted in Table-1, among the adhesion molecules evaluated only PS showed differences on the ionophore activated control erythrocytes. We also compared adhesion marker profiles between high PS and low PS expressing SCD groups and demonstrate that the differences in adhesion marker expression was confined only to PS-positivity (Table-1).
DISCUSSION
Previous studies from our laboratories and others have documented a role for PS-positive red cells in erythrocyte-endothelial adhesion with data suggesting that adhesion was in part due to the PS-positive erythrocyte-TSP interaction. The PS-binding site on TSP has not been characterized to date. In this study using a dynamic flow adhesion assay and artificially generated PS-positive erythrocytes, we demonstrate that PS-positive erythrocytes bind to both immobilized and soluble thrombospondin via its heparin-binding domain located at the amino-terminus of the polypeptide. We show that the binding is PS specific and requires the presence of both divalent cations Ca2+ and Mg2+. Significant binding to TSP of erythrocytes from patients with SCD, who demonstrated high levels of percent PS-positive red cells, also occurred at the heparin-binding domain. In addition, both heparin and its low-molecular weight derivative enoxaparin inhibit this interaction.
TSP is a homotrimeric 450-kDa protein, and as shown in Figure-1, each TSP subunit contains five distinct cell-binding domains, which interact with select protein(s), adhesion marker(s) or receptor(s).9-13 The heparin-binding domain which is located at the amino-terminus of the polypeptide interacts with heparin, heparansulfate proteoglycans, sulfatides, and β1-integrins.10-12 The WSPWS sequence, present within the type-I repeats, also interacts with heparansulfate proteoglycans.10,11 The CSVTCG motif that occurs twice within the type-I repeats binds to sulfatides, and CD36.10,11 The integrin-binding motif, RGDS, found in the last type-III repeat interacts with the fibrinogen receptor αIIbβ3, and the vitronectin receptor αVβ3.10,11,13 The RFYVVM sequence located at the carboxy-terminal of the polypeptide binds to CD47.10,11 While TSP can interact with a variety of cells via multiple cell-binding domains, the PS binding site has not been identified to date. PS-mediated cell binding may have potential implications not only in tethering of PS expressing apoptotic cells to phagocytes, but also in red cell-endothelial adhesion. Our interest in characterization of PS-binding site on TSP is due to previously documented work including the findings from this laboratory demonstrating that the PS is involved in erythrocyte-endothelial adhesion4. Since heparin inhibits adhesion of sickle red cells to TSP,18,30,31 we hypothesized that PS-positive erythrocytes may bind to the heparin-binding domain on TSP. Using artificially generated PS-positive control erythrocytes, we demonstrate that PS-positive red cells bind to immobilized TSP via its heparin-binding domain. Our conclusion is based on the findings that heparin, the anti-TSP antibody recognizing the heparin-binding domain, and the specific TSP peptides containing the heparin-binding motif KKTRG inhibited PS-mediated erythrocyte adhesion to TSP (Figures 3B-C, 4A, and 5A). Anti-TSP antibodies that recognize the collagen- or the CD47-binding domain, or the TSP peptide lacking the heparin-binding motif did not affect the binding process (Figures 4A, and 5A). Additional findings, demonstrating that immobilized TSP peptides containing the KKTRG motif supported PS-mediated erythrocyte adhesion, confirmed that PS interacted with TSP’s heparin-binding domain (Figure-5B). Pathophysiologic relevance of this work was documented in SCD patients, since HbSS erythrocyte binding to immobilized TSP matrix was also significantly inhibited by the anti-TSP antibody recognizing the heparin-binding domain (Figure-4B). Magnitude of the inhibitory effects produced by the latter anti-TSP antibody with sickle erythrocytes (a mean inhibition of 38%) was, however, smaller compared to that noted with artificially generated PS-positive erythrocytes (a mean inhibition of ~80%), which could be due to the differences in adhesion markers expressed on these cells as shown in Table-1. In contrast to sickle erythrocytes which express multiple cell adhesion markers28,32-38 with capabilities of interacting with multiple cell-binding domains on TSP,9-13 PS is the sole functional adhesion marker present on the ionophore-activated red cell and its interaction with TSP may, therefore, be limited to TSP’s heparin-binding domain. Pre-treatment of immobilized TSP with an antibody against the heparin-binding domain may, therefore, produce greater inhibition with the ionophore-activated control erythrocytes.
Besides PS, other relevant adhesion markers expressed on pathologic erythrocytes that are involved in red cell adhesion to endothelial cells and the sub-endothelial matrix components include CD36,32,33 VLA4,33,34 CD47,35,36 BCAM/LU37,38 and sulfatides.28 To exclude an ancillary role for these latter adhesion molecules in PS-TSP adhesion, we first elected to use un-activated and ionophore-activated control HbAA erythrocytes where, as shown in Table-1, ionophore treatment did not alter the expression of other adhesion molecules except causing PS-positivity i.e. the ideal condition for our proposed experiments related to PS-TSP adhesion. In addition, pretreatment of TSP with an antibody that recognizes the CD47-binding domain on TSP had no effect on ionophore-activated erythrocyte adhesion (Figure-4A). Further, while it is not known whether erythrocyte activation with A23187 perturbs membrane sulfatide composition, pretreatment of immobilized TSP with bovine brain sulfatide had no effect on A23187-treated red cell binding (Figure-3C). These results demonstrate that the erythrocyte adhesion molecules including CD47, BCAM/LU and sulfatides do not appear to be involved in mediating ionophore-activated erythrocyte binding to the heparin-binding domain on TSP.
Protein conformational changes may affect the cell-binding characteristics of TSP.39 For example, studies have demonstrated that chelation of Ca2+ unfolds the globular cell-binding domain of TSP, destabilizes disulfide bonds and results in extensive thiol-disulfide exchange leading to altered cell-binding characteristics.40-42 We therefore evaluated the effects of divalent cations including Ca2+ and Mg2+, and found a 12-fold increase in PS-mediated erythrocyte binding to TSP in the presence of Ca2+, although this effect also required the presence of Mg2+. Additionally, both Ca2+ and Mg2+, by decreasing molecular motion of the PS head groups and narrowing the distance between the neighboring PS head groups,43 may maximize binding of PS-positive erythrocytes to TSP. Protein conformation of immobilized matrix TSP also appears to be different from that of soluble TSP as previously documented by differential adhesion of sickle erythrocytes to soluble verses immobilized matrix TSP.28,32,44 While soluble TSP-mediated sickle red cell adhesion to cultured endothelial cells was blocked by both OKM-5 (a murine monoclonal antibody against CD36) and by a TSP peptide containing the CD36-binding motif CSVTCG,32 these blocking agents had no effect on sickle erythrocyte adhesion to immobilized matrix TSP.28,44 In light of these documented differential effects of blocking agents on sickle erythrocyte adhesion to TSP, we elected to also evaluate soluble TSP interactions with PS-positive cells. We found that soluble TSP, like immobilized TSP, appears to interact with PS-positive erythrocytes via its heparin-binding domain. We demonstrate that erythrocytes acquire TSP-positivity following incubation of PS-positive red cells with soluble TSP. No TSP positivity was found on the surface of erythrocytes that were incubated in the absence of TSP, or PS-negative red cells incubated with soluble TSP (Figure-6). In addition, TSP positivity was noted when an antibody recognizing the collagen-binding domain, but not an antibody recognizing the heparin-binding domain, was used as the detecting agent, likely due to preoccupancy of the heparin-binding domain, the binding site for anti-TSP-Ab9, on the TSP molecule by PS-positive erythrocytes. Since TSP is one of the components of the sub-endothelial matrix, in its immobilized form it facilitates adhesion of PS-positive red cells to sub-endothelium. In its soluble form TSP transforms a PS-positive red cell into a TSP-positive red cell which can interact not only with multiple adhesion receptors including CD36, CD47, α4β1, αvβ3 and αIIbβ3, but may also, potentially, play a role in the formation of hetero-cellular aggregates by facilitating adhesive interactions with other circulating cellular elements of blood including platelets and monocytes, as well as vascular endothelial cells. In this context it is interesting to note that previous studies in patients with SCD documented the presence of platelet-erythrocyte and neutrophil-erythrocyte aggreagates.45-47
Previous studies have evaluated the effects of several anionic polysaccharides including heparin, CSA and HDS on sickle erythrocyte adhesion to both immobilized matrix and soluble TSP and found differential inhibitory effects,18,28,31,44 in contrast to inhibition of a similar magnitude noted in this study with artificially generated PS-positive erythrocytes. These differences could be due to polysaccharides’ ability to recognize multiple GAG binding domains on TSP, as previously documented with CSA, which in addition to its interaction with the heparin binding domain, can also bind to regions within the type-I repeats and also to regions in the carboxy-terminal cell binding domain.48 Heparin has been used clinically as an anti-coagulant and anti-thrombotic agent.49 Major clinical concerns of prolonged heparin therapy, however, include heparin-induced thrombocytopenia, and abnormal bleeding. To overcome these adverse clinical effects of unfractionated heparin, several low molecular weight heparin derivatives have been developed, which have numerous advantages over unfractionated preparations including clinical safety and efficacy.50 In the present study we tested the effects on adhesion of heparin and enoxaparin, a low molecular weight heparin derivative, and demonstrate that both these polysaccharides inhibited PS-mediated erythrocyte adhesion to immobilized TSP not only at pharmacologic concentrations (5 to 50 U/ml) but also at levels (0.5 U/ml) achievable clinically in patients.49,51 Other investigators have demonstrated that heparin can inhibit soluble TSP-,18,31 and P-selectin-30 mediated sickle erythrocyte adhesion to cultured human endothelial cells. In addition, a previous study has suggested that prophylactic heparin therapy reduced the frequency of vaso-occlusive pain in patients with SCD.52 Our results taken together with those previously published demonstrate that heparin can modulate multiple red cell adhesion pathways in patients with SCD and suggests that heparin and its low molecular weight derivatives may be useful as potential therapeutic agents targeting the enhanced pro-adhesive state in this patient group.
In summary, we have demonstrated that PS-positive erythrocytes bind to both immobilized and soluble thrombospondin via its heparin-binding domain located at the amino-terminus of the polypeptide. We have shown that soluble TSP by binding to PS-positive red cells can potentially generate a sub-population of red cells which are TSP-positive with potentially enhanced pro-adhesive capabilities. In addition, we have demonstrated that heparin and its low molecular weight derivative enoxaparin inhibit PS-mediated erythrocyte-TSP interaction. The therapeutic potential of these observations are germane not only to sickle cell disease pathogenesis, but to other disorders in which a proadhesive propensity due to cellular PS-thrombospondin interactions exacerbates disease pathophysiology.
Acknowledgments
We thank Dr Marie Stuart for her continued support and her critical review of our manuscript. We also thank Mrs. Dottie Shields and Surekha Kulkarni for their assistance in data acquisition in some flow cytometry experiments related to PS analysis, Dr Carlton Dampier and the staff members of the division of Hematology, St Christopher’s Hospital for Children, Drexel University, for obtaining blood samples from patients with sickle cell disease, and Miss Priyanka Setty for providing secretarial assistance and preparing illustrations.
This work was supported by grants U54 HL70585 and R01 HL73944 from the National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, MD.
ABBREVIATIONS
- BCAM/LU
basal cell adhesion molecule/Lutheran protein
- CSA
chondroitin sulfate A
- FITC
fluorescein isothiocyanate
- GAGs
glycosaminoglycans
- HDS
High molecular weight dextran sulfate
- IAP
integrin-associated protein
- PS
phosphatidylserine
- SCD
sickle cell disease
- TC
Tri-Color
- TSP
thrombospondin
- VLA4
very late activation antigen-4
Footnotes
Brief Statement of Key Findings: Besides serving as a signal for phagocytic recognition and removal of apoptotic cells, cell surface PS can also function as an adhesion ligand. PS-dependent erythrocyte adhesion to endothelium and sub-endothelial matrix components is mediated in part via TSP. We demonstrate that PS-positive erythrocytes bind to TSP via its heparin-binding domain and that both heparin and enoxaparin, at clinically relevant concentrations, block this interaction. Our results taken together with previously documented heparin inhibitory effects on P-selectin- and soluble-TSP-mediated sickle erythrocyte-endothelial adhesion suggest that heparin and its low-molecular-weight derivatives may prove beneficial as anti-adhesive therapeutics targeting various pathways in the erythrocyte adhesion-cascade.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuypers FA. Phospholipid asymmetry in health and disease. Current Opinion in Hematology. 1998;5:122–131. doi: 10.1097/00062752-199803000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 3.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. TRENDS in Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Setty BNY, Kulkarni S, Stuart MJ. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood. 2002;99:1564–1571. doi: 10.1182/blood.v99.5.1564. [DOI] [PubMed] [Google Scholar]
- 5.Eda S, Sherman IW. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell Physiol Biochem. 2002;12:373–384. doi: 10.1159/000067908. [DOI] [PubMed] [Google Scholar]
- 6.Bonomini M, Sirolli V, Gizzi F, Di Stante S, Grilli A, Felaco M. Enhanced adherence of human uremic erythrocytes to vascular endothelium: role of phosphatidylserine exposure. Kidney Intl. 2002;62:1358–1363. doi: 10.1111/j.1523-1755.2002.kid560.x. [DOI] [PubMed] [Google Scholar]
- 7.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endotheliumin sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302:992–995. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 8.Manodori AB, Barabino GA, Lubin BH, Kuypers FA. Adherence of phosphatidylserine-exposing erythrocytes to endothelial matrix thrombospondin. Blood. 2000;95:1293–1300. [PubMed] [Google Scholar]
- 9.Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130:503–506. doi: 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 11.Adams JC. Thrombospondins: Multifunctional regulators of cell interactions. Ann Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 12.Elzie CA, Murphy-Ullrich JE. The N-terminus of thrombospondin: the domain stands apart. Int J Biochem Cell Biol. 2004;36:1090–1101. doi: 10.1016/j.biocel.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Frazier WA. Thrombospondins. Curr Opin Cell Biol. 1991;3:792–799. doi: 10.1016/0955-0674(91)90052-z. [DOI] [PubMed] [Google Scholar]
- 14.McPherson J, Sage H, Bornstein P. Isolation and characterization of a glycoprotein secreted by aortic endothelial cells in culture: apparent identity with platelet thrombospondin. J Biol Chem. 1981;256:11330–11336. [PubMed] [Google Scholar]
- 15.Mosher DF, Doyle MJ, Jaffe EA. Synthesis and secretion of thrombospondin by cultured human endothelial cells. J Cell Biol. 1982;93:343–348. doi: 10.1083/jcb.93.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffe EA, Ruggiero JT, Leung LL, Doyle MJ, McKeown-Longo PJ, Mosher DF. Cultured human fibroblasts synthesize and secrete thrombospondin and incorporate it into extracellular matrix. Proc Natl Acad Sci USA. 1983;80:998–1002. doi: 10.1073/pnas.80.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manodori AB, Matsui NM, Chen JY, Embury SH. Enhanced adherence of sickle erythrocytes to thrombin-treated endothelial cells involves interendothelial cell gap formation. Blood. 1998;92:3445–3454. [PubMed] [Google Scholar]
- 18.Gupta K, Gupta P, Solovey A, Hebbel RP. Mechanism of interaction of thrombospondin with human endothelium and inhibition of sickle erythrocyte adhesion to human endothelial cells by heparin. Biochim Biophys Acta. 1999;1453:63–73. doi: 10.1016/s0925-4439(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 19.Dixit VM, Galvin NJ, O’Rourke KM, Frazier WA. Monoclonal antibodies that recognize calcium-dependent structures of human thrombospondin: characterization and mapping of their epitopes. J Biol Chem. 1986;261:1962–1968. [PubMed] [Google Scholar]
- 20.Dixit VM, Haverstick DM, O’Rourke KM, Hennessy SW, Grant GA, Santoro SA, Frazier WA. A monoclonal antibody against human thrombospondin inhibits platelet aggregation. Proc Natl Acad Sci USA. 1985;82:3472–3476. doi: 10.1073/pnas.82.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosfeld MD, Pavlopoulos TV, Frazier WA. Cell attachment activity of the carboxyl-terminal domain of human thrombospondin expressed in Escherichia coli. J Biol Chem. 1991;266:24257–24259. [PubMed] [Google Scholar]
- 22.Merle B, Malaval L, Lawler J, Delmas P, Clezardin P. Decorin inhibits cell attachment to thrombospondin-1 by binding to a KKTR-dependent cell adhesive site present within the N-terminal domain of thrombospondin-1. J Cellular Biochem. 1997;67:75–83. [PubMed] [Google Scholar]
- 23.Clezardin P, Lawler J, Amiral J, Quentin G, Delmas P. Identification of cell adhesive active sites in the N-terminal domain of thrombospondin-1. Biochem J. 1997;321:819–827. doi: 10.1042/bj3210819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuypers FA, Lewis RA, Hua M, Schott MA, Discher D, Ernst JD, Lubin BH. Detection of altered membrane phospholipids asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood. 1996;87:1179–1187. [PubMed] [Google Scholar]
- 25.Setty BNY, Gayen Betal S. Microvascular endothelial cells express a phosphatidylserine receptor: a functionally active receptor for phosphatidylserine-positive erythrocytes. Blood. 2008;111:905–914. doi: 10.1182/blood-2007-07-099465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setty BNY, Rao AK, Stuart MJ. Thrombophilia in sickle cell disease: the red cell connection. Blood. 2001;98:3228–3233. doi: 10.1182/blood.v98.12.3228. [DOI] [PubMed] [Google Scholar]
- 27.Setty BNY, Kulkarni S, Dampier CD, Stuart MJ. Fetal hemoglobin in sickle cell anemia: relationship to erythrocyte adhesion markers and adhesion. Blood. 2001;97:2568–2573. doi: 10.1182/blood.v97.9.2568. [DOI] [PubMed] [Google Scholar]
- 28.Hillery CA, Du MC, Montgomery RR, Scott JP. Increased adhesion of erythrocytes to components of the extracellular matrix: isolation and characterization of a red blood cell lipid that binds thrombospondin and laminin. Blood. 1996;87:4879–4886. [PubMed] [Google Scholar]
- 29.Raynal P, Pollard HB. Annexins: The problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 30.Matsui NM, Varki A, Embury SH. Heparin inhibits the flow adhesion of sickle red blood cells to P-selectin. Blood. 2002;100:3790–3796. doi: 10.1182/blood-2002-02-0626. [DOI] [PubMed] [Google Scholar]
- 31.Barabino GA, Liu XD, Ewenstein BM, Kaul DK. Anionic polysaccharides inhibit adhesion of sickle erythrocytes to the vascular endothelium and result in improved hemodynamic behavior. Blood. 1999;93:1422–1429. [PubMed] [Google Scholar]
- 32.Sugihara K, Sugihara T, Mohandas N, Hebbel RP. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;80:2634–2642. [PubMed] [Google Scholar]
- 33.Joneckis CC, Ackley RL, Orringer EP, Wayner EA, Parise LV. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating endothelial reticulocytes in sickle cell anemia. Blood. 1993;82:3548–3555. [PubMed] [Google Scholar]
- 34.Swerlick RA, Eckman JR, Kumar A, Jeitler M, Wick TM. Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood. 1993;82:1891–1899. [PubMed] [Google Scholar]
- 35.Brittain JE, Milnar KJ, Anderson CS, Orringer EP, Parise LV. Integrin-associated protein is an adhesion receptor on sickle red blood cells for immobilized thrombospondin. Blood. 2001;97:2159–2164. doi: 10.1182/blood.v97.7.2159. [DOI] [PubMed] [Google Scholar]
- 36.Brittain JE, Milnar KJ, Anderson CS, Orringer EP, Parise LV. Activation of sickle red blood cell adhesion via integrin-associated protein/CD47-induced signal transduction. J Clin Invest. 2001;107:1555–1562. doi: 10.1172/JCI10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zen Q, Cottman M, Truskey G, Fraser R, Telen MJ. Critical factors in basal cell adhesion molecule/Lutheran-mediated adhesion to laminin. J Biol Chem. 1999;274:728–734. doi: 10.1074/jbc.274.2.728. [DOI] [PubMed] [Google Scholar]
- 38.Udani M, Zen Q, Cottman M, Leonard N, Jefferson S, Daymont C, Truskey G, Tellen MJ. Basal cell adhesion molecule/Lutheran protein. The receptor critical for sickle cell adhesion to laminin. J Clin Invest. 1998;101:2550–2558. doi: 10.1172/JCI1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ugarova T, Agbanyo FR, Plow EF. Conformational changes in adhesive proteins modulate their adhesive function. Thromb Haem. 1995;74:253–257. [PubMed] [Google Scholar]
- 40.Lawler J, Simons ER. Cooperative binding of calcium to thrombospondin: The effect of calcium on the circular dichroism and limited trytpic digestion of thrombospondin. J Biol Chem. 1983;258:12098–12101. [PubMed] [Google Scholar]
- 41.Slane JMK, Mosher DF, Lai CS. Conformational change in thrombospondin induced by removal of bound Ca2+: a spin label approach. FEBS Letters. 1988;229:363–366. doi: 10.1016/0014-5793(88)81157-8. [DOI] [PubMed] [Google Scholar]
- 42.Speziale MV, Detwiler TC. Free Thiols of platelet thrombospondin: evidence for disulfide isomerization. J Biol Chem. 1990;265:17859–17867. [PubMed] [Google Scholar]
- 43.Araiso T, Kawada S-I, Yoshida K, Hasebe K, Koyama T. Effects of Ca2+ and Mg2+ on dynamics of the polar head group of phosphatidylserine bilayers. Jap J Physiol. 1995;45:369–380. doi: 10.2170/jjphysiol.45.369. [DOI] [PubMed] [Google Scholar]
- 44.Joneckis CC, Shock DD, Cunningham ML, Orringer EP, Parise LV. Glycoprotein IV-independent adhesion of sickle red blood cells to immobilized thrombospondin under flow conditions. Blood. 1996;87:4862–4870. [PubMed] [Google Scholar]
- 45.Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregation in patients with sickle cell anemia. J Lab Clin Med. 1997;129:507–516. doi: 10.1016/s0022-2143(97)90005-6. [DOI] [PubMed] [Google Scholar]
- 46.Wun T, Paglieroni T, Field CL. Platelet-erythrocyte adhesion in sickle cell disease. J Investig Med. 1999;47:121–127. [PubMed] [Google Scholar]
- 47.Hofstra TC, Karla VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996;87:4440–4447. [PubMed] [Google Scholar]
- 48.Adams JC, Lawler J. Cell-type specific adhesive interactions of skeletal myoblasts with thrombospondin-1. Mol Biol Cell. 1994;5:423. doi: 10.1091/mbc.5.4.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirsh J, Raschke R, Warkentin TE, Dalen JE, Deykin D, Poller L. Heparin: mechanism of action, pharmacokinetics, dosing considerations, monitoring, efficacy, and safety. Chest. 1995;108:258S–275S. doi: 10.1378/chest.108.4_supplement.258s. [DOI] [PubMed] [Google Scholar]
- 50.Hirsh J, Warkentin TE, Shaughnessy SG, Anand SS, Helperin JL, Raschke R, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, effeicacy, and safety. Chest. 2001;119(1 Suppl):64S–94S. doi: 10.1378/chest.119.1_suppl.64s. [DOI] [PubMed] [Google Scholar]
- 51.Ger-Jan CM, Sanderink G-JCM, Guimart CG, Ozoux M-L. Pharmacokinetics and pharmacodynamics of the prophylactic dose of enoxaparin once daily over 4 days in patients with renal impairement. Thrombosis Res. 2002;105:225–231. doi: 10.1016/s0049-3848(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 52.Chaplin H, Jr, Monroe MC, Malecek AC, Morgan LK, Michael J, Murphy WA. Preliminary trial of minidose heparin prophylaxis for painful sickle cell crises. East Afri Med J. 1989;66:574–584. [PubMed] [Google Scholar]