

The native oxygen-carrier haemoglobins complex (HbII–III) is composed of haemoglobin II (HbII) and haemoglobin III (HbIII), which are found in the ctenidia tissue of the bivalve mollusc Lucina pectinata. This protein complex was isolated and purified from its natural source and crystallized using the vapour-diffusion and capillary counter-diffusion methods.
Keywords: haemoglobins complex, Lucina pectinata
Abstract
The native oxygen-carrier haemoglobins complex (HbII–III) is composed of haemoglobin II (HbII) and haemoglobin III (HbIII), which are found in the ctenidia tissue of the bivalve mollusc Lucina pectinata. This protein complex was isolated and purified from its natural source and crystallized using the vapour-diffusion and capillary counter-diffusion methods. Oxy and cyano derivatives of the complex crystallized using several conditions, but the best crystals in terms of quality and size were obtained from sodium formate pH 5 using the counter-diffusion method in a single capillary. Crystals of the oxy and cyano complexes, which showed a ruby-red colour and nonsingular prismatic shapes, scattered X-rays to resolution limits of 2.15 and 2.20 Å, respectively, using a 0.886 Å synchrotron-radiation source. The crystals belonged to the tetragonal system, space group P42212, with unit-cell parameters a = b = 74.07, c = 152.07 and a = b = 73.83, c = 152.49 Å for the oxy and cyano complexes, respectively. The asymmetric unit of both crystals is composed of a single copy of the heterodimer, with Matthew coefficients (V M) of 3.08 and 3.06 Å3 Da−1 for the oxy and cyano complexes, respectively, which correspond to a solvent content of approximately 60.0% by volume.
1. Introduction
The clam Lucina pectinata is found in the sulfide-rich southwest coastal sediments of Puerto Rico in the Caribbean Sea. This clam has specific biological attributes that make it an excellent biotic model. The clam has developed a chemoautotrophic symbiotic relation with the bacteria within it, with varying levels of interaction via intracellular and intercellular pathways in a sulfide-rich environment (Kraus & Wittenberg, 1990 ▶). This symbiotic scenario has raised questions regarding the biology of the clam. One of the most interesting interaction paths with the bacteria takes place through the family of haemoproteins HbI, HbII and HbIII (Kraus & Wittenberg, 1990 ▶). These three proteins and the bacteria coexist in the ctenidia tissue of the clam, in which the interchange of hydrogen sulfide and oxygen occurs. HbI, with 142 amino acids (Antommattei et al., 1999 ▶), is in charge of providing hydrogen sulfide to the bacteria, while HbII and HbIII, with 150 and 152 amino acids, respectively (Torres-Mercado et al., 2003 ▶; Hockenhull-Johnson et al., 1993 ▶), have been identified as transporting oxygen to the clam. The protein complex HbII–III, which has been isolated from the ctenidia tissue, also acts as an oxygen carrier (Kraus & Wittenberg, 1990 ▶). The ligand-selection and discrimination mechanism of the proteins and the protein complex in this biotic scenario remain unclear.
The structures of HbI in its aquomet, sulfide and cyanide forms (Rizzi et al., 1994 ▶; Bolognesi et al., 1999 ▶) and the oxy form of HbII (Gavira et al., 2008 ▶) have been solved and initial crystallization studies of the oxygen-carrier haemoglobins complex (HbII–III) were published in 1991 (Kemling et al., 1991 ▶). However, the structure of HbII–III has remained unsolved to date. Previous structural studies of HbI and HbII showed correlations with the proposed functions of these proteins. To our knowledge, the only crystallographic study of the HbII–III protein complex is that published by Kemling and coworkers, who reported its crystallization and preliminary crystallographic data and analysis (Kemling et al., 1991 ▶). The crystal was grown from a 65 mg ml−1 equimolar mixture of HbII and HbIII by the hanging-drop vapour-diffusion technique, using 10% saturated ammonium sulfate, 0.05 M Tris–HCl pH 7.0 as the precipitating agent. Prismatic crystals with dimensions of 0.25 × 0.1 × 0.05 mm diffracted to 3.1 Å resolution and were assigned to the tetragonal space group P42212. Unfortunately, no further crystallographic analysis has been reported since this study. The structure of the HbII–III complex is necessary in order to obtain information on the protein–protein interactions and the ligand-selection and discrimination mechanism and to correlate the structure with the proposed protein function and properties such as (i) the existence of inhibitory mechanisms of NO oxidation as a possible blood substitute (De Jesús-Bonilla et al., 2007 ▶), (ii) amino-acid modulation of the ligand-binding activity (Pietri et al., 2006 ▶) and (iii) the cooperative or noncooperative behaviour related to the measured Hill coefficient of the complex (Kraus & Wittenberg, 1990 ▶).
In this work, we present the isolation, purification, crystallization and preliminary crystallographic data of oxy and cyano forms of the HbII–III complex. In addition to the intrinsic interest of the oxy HbII–III protein complex, the cyano form will provide us with insight into the haem-iron oxidation-state effect in the HbI, HbII and HbIII ligand-selection mechanism.
2. Materials and methods
2.1. Protein isolation, purification and concentration
The HbII–III complex was extracted from L. pectinata clams collected at La Parguera bay in Lajas, Puerto Rico. The HbII–III complex was isolated from the ctenidia tissue as described previously by Kraus & Wittenberg (1990 ▶) with slight modifications (Pietri et al., 2006 ▶). In brief, we employed a fast protein liquid-chromatography system (FPLC) with a HiLoad 26/60 Superdex 200 grade (ÄKTA, Amersham Bioscience) size-exclusion chromatography (SEC) column as the last step. The column was equilibrated with 660 ml 0.5 mM EDTA and 50 mM sodium phosphate buffer pH 7.5 at a flow rate of 1.0 ml min−1. The HbII–III protein complex was separated into HbII and HbIII by an anionic ion-exchange chromatography (IEC) step using an IEC HiPrep 16/10 Q FF column (ÄKTA FPLC, Amersham Bioscience). A HiTrap Q FF (5 ml) pre-column was used to assure the integrity and efficiency of purification. The separation was carried out with a gradient of NaCl from 0 to 180 mM at a flow rate of 5.0 ml min−1 and was followed by monitoring the absorbance at 280 nm. The concentrations of HbII, HbIII and the HbII–III complex were determined spectrophotometrically using the reported Soret and Q-band values (Kraus & Wittenberg, 1990 ▶). The protein purity was validated by employing SDS–PAGE (14%) at 0.1 mg ml−1 protein concentration (Fig. 1 ▶).
Figure 1.

14% SDS–PAGE. Lane 1, empty; lanes 2 and 6, molecular-weight markers; lane 3, HbII; lane 4, HbII–III; lane 5, HbIII.
The oxy HbII–III complex was prepared by reduction of the sample with 0.1 mM sodium dithionate (Na2S2O4) followed by exposure to a constant flow of pure oxygen at 101 kPa. The oxy complex is very stable owing to its high oxygen affinity and very slow release of bound oxygen (Kraus & Wittenberg, 1990 ▶). The cyano HbII–III complex was prepared by following the protocol previously outlined by Kraus & Wittenberg (1990 ▶). Ferric Hb was obtained by adding a 10% excess of potassium ferricyanide [K3Fe(CN)6] to a solution of the oxy form in water, which was followed by the addition of 50 mM KCl/20 mM KCN prepared at pH 7 to generate the cyano complex. All HbII–III complexes were confirmed spectrophotometrically using the reported Soret and Q-band values (Kraus & Wittenberg, 1990 ▶). As a final step, the samples were concentrated in pure water using a YM-10 Centricon (Millipore Co.).
2.2. Crystallization
Initial searches for crystallization conditions were performed in parallel using the vapour-diffusion method (2:2 µl hanging drops and 1 ml reservoir) with the Crystal Screen crystallization kit (HR2-110; Hampton Research) and by counter-diffusion (García-Ruiz, 2003 ▶; Ng et al., 2003 ▶) using a pre-filled GCB-CSK (24-condition Crystallization Screening Kit; Triana Science & Technology) in capillaries of 0.2 mm inner diameter (1.57 µl). Optimization experiments were performed with pre-filled ammonium sulfate (KIT-AS-49), sodium chloride (KIT-SC-49), sodium formate (KIT-SF-49) and polyethylene glycol mixture (KIT-PEG448-49) Capillary Counter-diffusion Kits (Triana Science & Technology) in the pH range 4–9. The experiments were performed using capillaries of 0.2–0.5 mm inner diameter for the Capillary Counter-diffusion Kits and of 0.7 mm inner diameter for the three-layer configuration (Gavira et al., 2006 ▶). In order to avoid convection and sedimentation, 0.1%(w/v) agarose was added to the protein solution for the capillaries with 0.5 and 0.7 mm inner diameter (Gavira et al., 2006 ▶). All screenings and crystallization experiments were set up using a 30 mg ml−1 initial protein concentration at 293 K.
2.3. Data collection
X-ray diffraction data sets were collected on beamline BM-16 of the European Synchrotron Radiation Facility (ESRF). Scattering intensities were collected with an ADSC Quantum 4R detector using a wavelength of 0.886 Å. The crystals grown in the GCB capillaries were extracted into and equilibrated in 50 µl cryoprotectant solution as described previously (Gavira et al., 2006 ▶). Briefly, a number of crystals (1–3) were removed from the capillary by increasing the pressure in the upper part: a piece of rubber tube was attached through which air was blown. If the crystals were stuck to the capillary wall, they were gently moved with the help of a cat whisker. A cryoprotectant solution containing 15%(v/v) glycerol was placed in a small plastic Petri dish and the crystals were equilibrated for a minimum of 30 s. After equilibration, one crystal was selected with a loop and flash-cooled in a 100 K liquid-nitrogen stream produced by an Oxford 600 Cryosystem. For the oxy HbII–III crystal, a total of 275 frames were obtained using a crystal-to-detector distance of 199.64 mm and a 11.0 s exposure time with 1° oscillations (Fig. 2 ▶ a). For the cyano HbII–III crystal, 275 frames were collected with a crystal-to-detector distance of 195.9 mm and a 10.93 s exposure time with 1° oscillations (Fig. 2 ▶ b). All the collected data were indexed, integrated and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). The method employed to obtain the crystallographic model for both proteins was molecular replacement using the coordinates of HbII chain A (PDB code 2olp; Gavira et al., 2008 ▶) from which the ligand and water molecules had been removed. The solution was found with the program MOLREP (Vagin & Teplyakov, 1997 ▶) and refinement is in progress using REFMAC5 (Murshudov et al., 1997 ▶); both programs were run in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
Figure 2.

(a) X-ray diffraction frame of oxy HbII–III obtained at 100 K. The circle shows the approximate resolution limit of 2.0 Å. The insert shows diffraction spots below 2.2 Å resolution. (b) X-ray diffraction frame of cyano HbII–III obtained at 100 K. The circle shows the approximate resolution limit of 2.0 Å. The insert shows diffraction spots below 2.15 Å resolution.
3. Results and discussions
Similar crystallization conditions were found for the oxy and cyano forms of the HbII–III complex after screening using vapour diffusion and capillary counter-diffusion methods. The crystals selected for diffraction were those that had a good size and quality from visual inspection for both protein complexes. After two weeks, the GCB-CSK yielded the best crystals using the following precipitating agents: 1.5 M ammonium sulfate, 0.1 M sodium citrate pH 5.6 (condition No. 14), 30% PEG 400, 0.1 M HEPES–NaOH pH 7.5, 0.2 M calcium chloride (condition No. 16), 2% PEG 4000, 0.1 M sodium acetate pH 4.6, 0.2 M ammonium sulfate (condition No. 18), 20% PEG 8000, 0.05 M potassium phosphate (condition No. 23), 2.0 M sodium formate, 0.1 M sodium acetate pH 4.6 (condition No. 22) and 6.0 M sodium formate (condition No. 21). In contrast, vapour-diffusion screening using Crystal Screen (HR2-110; Hampton Research) produced crystals under several crystallization conditions: 1.4 M sodium acetate, 0.1 M sodium cacodylate pH 6.5 (condition No. 7), 30% PEG 400, 0.1 M Na HEPES pH 7.5, 0.2 M magnesium chloride (condition No. 23), 1.6 M potassium sodium tartrate, 0.1 M Na HEPES pH 7.5 (condition No. 29) and 4.0 M sodium formate (condition No. 33). This sparse-matrix sampling (Jancarik & Kim, 1991 ▶) generated similar crystallization conditions to those obtained with the GCB-CSK, based on the work of Kimber et al. (2003 ▶), but with a greater consumption of protein and smaller crystal sizes. No significant differences were found between the oxy and cyano complexes in terms of the crystallization profile. Optimization experiments were performed by the counter-diffusion method, screening the pH range 4–9 under the selected hit condition 5.0 M sodium formate, 2.0 M sodium potassium tartrate, 3.0 M ammonium sulfate. The best crystals according to visual inspection and preliminary X-ray diffraction experiments at a home source were obtained using 5.0 M sodium formate pH 5. We did not observe any difference in crystal quality between the two setups (Capillary Counter-diffusion Kits and the three-layer configuration) in the presence of agarose (Fig. 3 ▶).
Figure 3.

Crystals of (a) oxy HbII–III and (b) cyano HbII–III obtained in capillaries of 0.5 and 0.7 mm inner diameter, respectively.
The oxy and cyano complex derivatives are isomorphous with HbII, HbIII and the HbII–III complex (Kemling et al., 1991 ▶) and belong to the tetragonal space group P42212. The unit cell is composed of a heterodimer, with V M values of 3.08 Å3 Da−1 for the oxy form and 3.06 Å3 Da−1 for the cyano form, which are within the range of expected values (Matthews, 1968 ▶). Both the oxy and cyano crystals showed rounded rather than facetted faces (Fig. 3 ▶). The average dimensions of the oxy and cyano forms were 0.21 × 0.18 × 0.18 and 0.08 × 0.09 × 0.09 mm, respectively. Despite their difference in size, the two crystals showed similar diffraction properties in terms of resolution limit and mosaicity (Table 1 ▶).
Table 1. X-ray data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Oxy HbII–III | Cyano HbII–III | |
|---|---|---|
| Wavelength (Å) | 0.886 | 0.886 |
| Space group | P42212 | P42212 |
| Unit-cell parameters (Å) | a = b = 74.07, c = 152.07 | a = b = 73.83, c = 152.49 |
| Resolution range (Å) | 20.0–2.15 (2.23–2.15) | 20.0–2.20 (2.28–2.20) |
| Observed reflections | 253034 | 165962 |
| Independent reflections | 23941 | 22319 |
| Data completeness (%) | 99.4 (100.0) | 92.2 (89.2) |
| Rmerge† (%) | 7.0 (36.7) | 7.7 (32.9) |
| Average I/σ(I) | 27.8 (6.9) | 20.9 (5.9) |
| Redundancy | 10.7 (10.9) | 8.1 (7.4) |
| Mosaicity | 0.45 | 0.69 |
| Molecules per ASU | 2 | 2 |
| Matthews coefficient (Å3 Da−1) | 3.08 | 3.06 |
| Solvent content (%) | 60.01 | 59.85 |
R
merge = 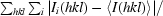
 , where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements.
, where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements.
The isomorphous structure of HbII (PDB code 2olp) was used as a search model. After removing water molecules and ligand, the coordinates of polypeptide chain A were used in MOLREP (Vagin & Teplyakov, 1997 ▶) together with reflections in the resolution range 10.0–4.0 Å. As expected, MOLREP was able to position two polypeptide chains in both structures with a final contrast and R factor of 2.68 and 38.0%, respectively, for the oxy complex and 2.63 and 39.4%, respectively, for the cyano complex. After ten cycles of restrained refinement with REFMAC (Murshudov et al., 1997 ▶), the values of R and R free were 24% and 29%, respectively, for the cyano complex and 24% and 27%, respectively, for the oxy complex. An inspection with Coot (Emsley & Cowtan, 2004 ▶) already shows clear displacement of the Tyr side chain at the B10 position of the cyano HbII–III complex of approximately 1 Å in both polypeptide chains.
Acknowledgments
This work was supported in part by grants NSF-MCB-0544250 (JLG) and NIH–NIGMS MBRS-SCORE 2 S06GM08103-34 (JLG) and the University of Puerto Rico at Mayagüez (JLG) and at Aguadilla (CRM, RAEA) Campuses. Other support includes grant No. RNM-1344 from the Andalusian Regional Government, Spain and the OptiCryst project of the Sixth Framework of the European Union. This is a product of the ‘Factoría Española de Cristalización’, Consolider-Ingenio 2010 project (MEC). The authors wish to thank the staff at BM-16 (ESRF) for their assistance during data collection.
References
- Antommattei, F. M., Rosado, T., Cadilla, C. L. & López-Garriga, J. (1999). J. Protein Chem.18, 831–836. [DOI] [PubMed]
- Bolognesi, M., Rosano, C., Losso, R., Boraddi, A., Rizzi, M., Wittenberg, J. B., Boffi, A. & Ascenzi, P. (1999). J. Biophys.77, 1093–1099. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- De Jesús-Bonilla, W., Jia, Y., Alayash, A. I. & López-Garriga, J. (2007). Biochemistry, 46, 10451–10460. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- García-Ruiz, J. M. (2003). Methods Enzymol.368, 130–154. [DOI] [PubMed]
- Gavira, J. A., De Jesús, W., Camara-Artigas, A., López-Garriga, J. & García-Ruiz, J. M. (2006). Acta Cryst. F62, 196–199. [DOI] [PMC free article] [PubMed]
- Gavira, J. A., De Jesús, W., Camara-Artigas, A., López-Garriga, J. & García-Ruiz, J. M. (2008). J. Biol. Chem.283, 9414–9423. [DOI] [PMC free article] [PubMed]
- Hockenhull-Johnson, J. D., Stern, M. S., Wittenberg, J. B., Vinogradov, S. N., Kapp, O. H. & Walz, D. A. (1993). J. Protein Chem.12, 261–277. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- Kemling, N., Kraus, D. W., Hockenhull-Johnson, J. D., Wittenberg, J. D., Vinogradov, S. N., Walz, D. A., Edwars, B. F. P. & Martin, P. (1991). J. Mol. Biol.222, 463–464. [DOI] [PubMed]
- Kimber, M. S., Vallee, F., Houston, S., Nečakov, A., Skarina, T., Evdokimova, E., Beasley, S., Christendat, D., Savchenko, A., Arrowsmith, C. H., Vedadi, M., Gerstein, M. & Edwards, A. M. (2003). Proteins, 51, 562–568. [DOI] [PubMed]
- Kraus, D. W. & Wittenberg, J. B. (1990). J. Biol. Chem.265, 16043–16053. [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Ng, J., Gavira, J. & García-Ruiz, J. M. (2003). J. Struct. Biol.142, 218–231. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pietri, R., León, R. G., Kiger, L., Marden, M. C., Granell, L. B., Cadilla, C. L. & López-Garriga, J. (2006). Biochim. Biophys. Acta, 1764, 758–765. [DOI] [PubMed]
- Rizzi, M., Wittenberg, J. B., Coda, A., Fosano, M., Ascenzi, P. & Bolognesi, M. (1994). J. Mol. Biol.244, 86–99. [DOI] [PubMed]
- Torres-Mercado, E., Renta, J. Y., Rodríguez, Y., López-Garriga, J. & Cadilla, C. L. (2003). J. Protein Chem.22, 683–690. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.