

Abstract
High mortality rate for metastatic melanoma is related to its resistant to the current methods of therapy. Melanogenesis is a metabolic pathway characteristic for normal and malignant melanocytes that can affect the behavior of melanoma cells or its surrounding environment. Human melanoma cells in which production of melanin pigment is dependent on tyrosine levels in medium were used for experiments. Peripheral blood mononuclear cells were derived from the buffy coats purchased from Lifeblood Biological Services. Cell pigmentation was evaluated macroscopically, and tyrosinase activity was measured spectrophotometrically. Cell proliferation and viability were measured using lactate dehydrogenase release MTT, [3H]-thymidine incorporation and DNA content analyses, and gene expression was measured by real time RT-PCR. Pigmented melanoma cells were significantly less sensitive to cyclophosphamide and to killing action of IL-2-activated peripheral blood lymphocytes. The inhibition of melanogenesis by either blocking tyrosinase catalytic site or chelating copper ions sensitized melanoma cells towards cytotoxic action of cyclophosphamide, and amplified immunotoxic activities of IL-2 activated lymphocytes. Exogenous L-DOPA inhibited lymphocyte proliferation producing the cell cycle arrest in G1/0 and dramatically inhibited the production of IL-1beta, TNF-alpha, IL-6 and IL-10. Thus, the active melanogenesis could not only impair the cytotoxic action of cyclophosphamid but also has potent immunosuppressive properties. This resistance to a chemotherapeutic agent or immunotoxic activity of lymphocytes could be reverted by the action of tyrosinase inhibitors. Thus, the inhibition of melanogenesis might represent a valid therapeutic target for the management of advanced melanotic melanomas.
Keywords: melanogenesis, melanocyte, melanoma, therapy, tyrosinase
Cutaneous melanoma is a tumor derived from activated or genetically altered epidermal melanocytes, following complex interactions between genetic, constitutional and environmental factors.1–9 Melanoma represents the most rapidly increasing malignancy in the white population, and its mortality rate is surpassed only by lung cancer.1,3,5 The high mortality rate among melanoma patients is related to its progression to the therapy resistance in stages III and IV of the disease; clinical cure of melanomas is only possible by surgery at the radial growth phase (RGP) when the disease is still localized and restricted to the skin.1,3,10–12 The subsequent progression from RGP to the vertical growth phase (VGP), signals metastatic potential with important negative impact on survival; once the metastatic process has started, the tumor becomes resistant to current methods of therapy.1,11–15
In disseminated melanoma, treatment is usually palliative, addressed at improving the quality of life, although surgical excision of lone metastasis to the lung or brain can be associated with prolonged survival.3,12 Local relief for recurrent tumors or meta-static sites may follow radiation therapy, or localized intra-arterial limb perfusion with chemotherapeutic agents for advanced regional disease restricted to a limb.11 The response rate to chemotherapy is only 20–25%, rarely resulting in complete remission.13 Other agents tried, generally unsuccessfully, in advanced cases include retinoids, high-dosage chemotherapy combined autologous bone marrow transplantation and antibodies conjugated to isotopes, drugs and biological toxins, and most recently, immunotherapy with interleukin-2 and α-interferon.16,17
Therapeutic selectivity towards cells of melanocytic origin (and hence increase of local effectiveness) has likely focused on immunotargeting.10,11,13,18–27 Among the proposed antigens are the melanocyte related proteins (MRP) that include tyrosinase, tyrosinase related proteins type 1 and 2 (TRP-1, TRP-2) and melanosomal antigens HMB-45 and MART 1.25,26,28 Cell surface antigens that stimulate the humoral (antibody) response have also been proposed to target the plasma membrane gangliosides GD2, GD3, 0-Ac-GD3 or GM2.29–31 Although rational and logical, none of the above strategies has yet been established as a therapy.13,31
Interference with melanogenesis may be considered as an alternative, physiologically based approach.4,32–40 This metabolic pathway is characteristically expressed in normal and malignant melanocytes and involves the transformation of L-tyrosine to melanin pigment through a series of oxidoreduction reactions.35 Clinically the melanin synthesis pathway serves as a diagnostic tool, for example, differentiation marker that allows for the differentiation of melanomas from other tumors; biologically, the presence of melanogenesis affects the behavior of melanoma cells and their surrounding environment.8,35,41 More, specifically, melanogenesis generates an oxidative environment and some of its intermediates (quinones and semiquinones) are directly toxic and also mutagenic.35,42 Thus, the melanogenesis associated mutagenic environment may potentially lead to genetic instability. Furthermore, because melanogenesis intermediates can inhibit activity of immune cells an immunosuppressive environment may surround the tumor.42 Lastly, the final product, melanin (a light absorbing biopolymer with electromagnetic properties) can not only scavenge free radicals and reactive oxygen species (ROS) but also chelates chemotherapeutic agents and produces relatively hypoxic environment due to increased oxygen consumption.35,43 All of these properties can affect the efficacy of chemotherapy, radiotherapy or photodynamic therapy.
Because melanogenesis has the potential to affect the tumor behavior and thus melanoma therapy outcome, we have tested the effects of suppression of melanogenesis on the effectiveness of killing of melanoma cells by chemotherapeutic agent or by immune cells. We evaluated the inhibitors of tyrosinase activity D-penicillamine and N-phenylthiourea (PTU), and also tested the effect of the melanin precursor L-DOPA on lymphocyte viability and production of proinflammatory cytokines. We used human melanoma cell line whose amelanotic versus melanotic phenotype can be regulated by concentration of melanin precursors in culture medium.44
Methods
Cell culture
Human SKMEL-188 melanoma cells were cultured in either Ham’s F10, Dulbecco’s Modified Eagle’s Medium (DMEM) or DMEM:F10 at 1:1 ration supplemented with 5% fetal bovine serum (FBS) and 1% antibiotics (penicillin/streptomycin/amphotericin, Sigma-Aldrich, St. Louis, MO). Melanin content in melanoma cells is dependent on the L-tyrosine levels in medium, being ~10, 400 or 200 μM in F10, DMEM or F10:DMEM, respectively. The cells were cultured at 37°C in 5% CO2 and the media were changed every second day as described previously.44
Peripheral blood mononuclear cells (PBMC) were derived from the buffy coats (purchased from Lifeblood Biological Services, Memphis, TN), separated by the standard Ficoll method according to manufacturer’s protocol (Ficoll-Paque Plus, Amersham Biosciences, Uppsala, Sweden). PBMC were resuspended in medium RPMI 1640 with 10% FBS and antibiotics and incubated for 2 hr to let monocytes adhere to the surface of the culture dish. The lymphocytes remaining in suspension were transferred to a new bottle and rhIL-2 (Sigma, St. Louis, MI) was added to the concentration 200 U/ml. Alternatively, lymphocytes were activated with lipopolysaccharide (LPS; 1,000 ng/ml) and used for the subsequent experiments. Composition of lymphocytes suspensions was assessed with flow cytometry (CD3+: 73%, CD19+: 0.8%, CD3+/4+: 5%, CD3+/8+: 14%, CD3−/56+/16+: 3%).
Cyclophosphamide, N-phenylthiourea (PTU) and D-penicillamine were purchased from Sigma (St. Louis, MI).
MTT assay
Cells were seeded at a density of 5,000 cells per well into 96-well plates in growth media. After 24 hr, media were changed to appropriate incubation media containing graded concentrations of cyclophosphamide and/or melanogenesis inhibitors. After incubation, 20 μl MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; 5 mg/ml in PBS; Sigma, St. Louis, MI) was added and the plates were incubated at 37°C for 4 hr in the presence of 5% CO2. At the end of the incubation period, media were discarded and 100 μl of acid (0.1 M hydrochloric acid) isopropanol was added before measuring optical density at 570 nm with a plate reader.
3[H]-thymidine incorporation
PBMC were seeded at a density of 100,000/well into 96-well plates and treated as described in Results and Discussion and Figure legends. [3H]thymidine (1 μCi/ml; Amersham Biosciences (Piscataway, NJ)) was then added for 6 hr (supernatant experiment) or 18 hr (L-DOPA experiment). Then, the media were discarded and the cells harvested on a glass fiber filter. Radioactivity proportional to [3H]thymidine incorporated into DNA was counted with Packard direct beta counter (Packard, Meriden, CA).
DNA content
Cell DNA content was measured by flow cytometry. PBMC were preincubated overnight in RPMI 1640 with 10% FBS with IL-2 (200 U/ml). Then the media were changed, and IL-2 and L-DOPA were added. After 24 hr, cells were washed with PBS and fixed with 70% ice-cold ethanol. Ethanol was removed by centrifugation and the pellets were washed twice with PBS before adding 0.5 ml of a solution of propidium iodide (50 μg/ml; Sigma (St. Louis, MI)) and ribonuclease IA (0.1 mg/ml Sigma; St. Louis, MI) in Ca2+- and Mg2+-free PBS. Samples were shaken for 30 min in 37°C and analyzed with a FACS Calibur cytometer (Beckton Dickinson, San Diego, CA). The DNA content was estimated with the ModFit 2.0 program (Verity Software House, Topsham, ME). Graph were prepared WinMDI2.8 (freeware from Joe Trotter, Scripps Institute, La Jolla, CA). Events were gated in the forward scatter/side scatter window to exclude debris, and in area/width FL2 windows to exclude doublets.
Cytotoxicity assay (lactate dehydrogenase release)
Nonpigmented and pigmented melanoma cells were cultured as described above. Some of the pigmented SKMEL-188 cells were cultured in DMEM:F10 medium in the presence or absence of 10−3 M of N-phenylthiourea or D-penicillamine for 3 days. These cells were then coincubated with IL-2-activated lymphocytes (preincubated with IL-2 200 U/ml for 24 hr) at target:effector ratio 1:5; 5,000 melanoma cells per well in Ham’s F10 medium with 5% FBS in U-bottom microwell plates. After 4 hr, the PBL-mediated cytotoxicity was measured with the Cytotox96® nonradioactive cytotoxicity assay (Promega, Madison, WI). Briefly, plates were centrifuged at 250g for 4 min, and 50 μl aliquots of supernatants were taken and analyzed. The LDH amount released from target cells was measured using Promega kit reagents according to the manufacturer’s protocol. Specific cytotoxicity was calculated according to the formula:
% cytotoxicity=100 × [(PBL and melanoma cells LDH release –spontaneous PBL LDH release –spontaneous melanoma cells release/(maximal melanoma cells LDH release –melanoma spontaneous LDH release)]. Maximal release was obtained after lysis of the cells with a control solution provided by the manufacturer. Culture medium background was subtracted from all values.
Melanin content and tyrosinase activity
Cell pigmentation was evaluated macroscopically as described previously.44,45 Briefly, the cells cultured in Ham’s F10, DMEM or DMEM supplemented with inhibitors of melanogenesis N-phenylthiourea (PTU) or D-penicillamine were harvested, the equal amounts of the cells were centrifuged into pellets and then photographed. Tyrosinase activity assay (DOPA oxidase) was assayed in cell extracts as described previously.46 The values are presented either as fold change in comparison to the control (Fig. 2) or in nmols of dopachrome formed per 1 mg protein during 1 hr of incubation (Fig. 2 legend); calculations were done using molar coefficient of dopachrome at 475 nm (3,700).45
Figure 2.

D-penicillamine and N-phenylthiourea inhibit melanogenesis and tyrosinase activity in SKMEL-188 melanoma cells. SKMEL-188 were incubated in either Ham’s F10 (containing 10 μM tyrosine) or DMEM:F10 (containing 200 μM tyrosine) medium with 5% FBS and antibiotics in the presence or absence of D-penicillamine or N-phenylthiourea for 5 days (media were changed and fresh compounds added every 2nd day). After harvesting and centrifugation the pellets were photographed (a) and tyrosinase activity measured as described46 (b). Data are presented as means ± SEM (n = 3). #p < 0.0005 versus F10,*p < 0.0005. **p < 0.00005 versus DMEM control. Tyrosinase activity for the cells cultured in F10 or DMEM was 10.8 ± 2.48 and 36.0 ± 0.7 nmols/mg/hr, respectively.
Real-time RT-PCR
Levels of proinflammatory cytokines mRNAs were measured at 1 hr, because modulation of cytokine production has been previously reported at this time frame point.47 RNA was extracted using Absolutely RNA RT-PCR Miniprep Kit (Stratagene, La Jolla, CA). Reverse transcription was performed using High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). Transcripts were quantitated using applied biosystems primer/probe gene-specific master mixes as described in Table I. The reaction was performed with Taqman® Universal PCR Master Mix; data collected on ABI Prism 7700 and analyzed on Sequence Detector 1.9.1. Specific mRNA amounts were calculated in relation to 18SrRNA using the comparative ΔΔCT method.
TABLE I.
PROBES USED FOR REAL TIME PCR (APPLIED BIOSYSTEMS)
| Gene name | Genebank accession number | FAM™ dye-labeled probe sequence | Applied biosystems catalog number |
|---|---|---|---|
| 18SrRNA | X03205.1 | GGAGGGCAAGTCTGGTGCCAGCAGC | Hs99999901_s1 |
| TNF-α | NM_000594.2 | CCCATGTTGTAGCAAACCCTCAAGC | Hs00174128_m1 |
| IL-1β | NM_000576.2 | GGAGCAACAAGTGGTGTTCTCCATG | Hs00174097_m1 |
| IL-6 | NM_000600.1 | TTCAATGAGGAGACTTGCCTGGTGA | Hs00174131_m1 |
| IL-10 | NM_000572.2 | CGGCGCTGTCATCGATTTCTTCCCT | Hs00174086_m1 |
Statistical analysis
Differences between means were tested for significance by one way ANOVA or Student’s test (depending on the assay); p < 0.05 was considered as statistically significant using GraphPad Prism 4 (GraphPad Software, San Diego, CA).
Results and discussion
Inhibition of melanogenesis increases melanoma sensitivity to killing action of cyclophosphamide
To compare the effects of cyclophosphamide on the viability of amelanotic or melanotic human melanoma cells, SKMEL-188 cells were propagated in the Ham’s F10 to maintain nonpigmented phenotype, or in Hams’ F10:DMEM to induce melanin synthesis.44,45 Pigmented and nonpigmented cells were seeded into 96-well plates and incubated with serial dilutions of cyclophosphamide. After 24 hr, the viability of the cells was assayed with the MTT test as described.48 As shown on Figure 1, the viability of pigmented melanoma cells is not affected by cyclophosphamide at the concentrations tested (p > 0.05). In contrast, nonpigmented SKMEL-188 cells were killed by cyclophosphamide in a dose-dependent manner (Fig. 1). The effect was visible already at cyclophosphamide concentration of 10−6 M (p = 0.033) and reached the maximum at 10−4 M (p < 0.0005).
Figure 1.
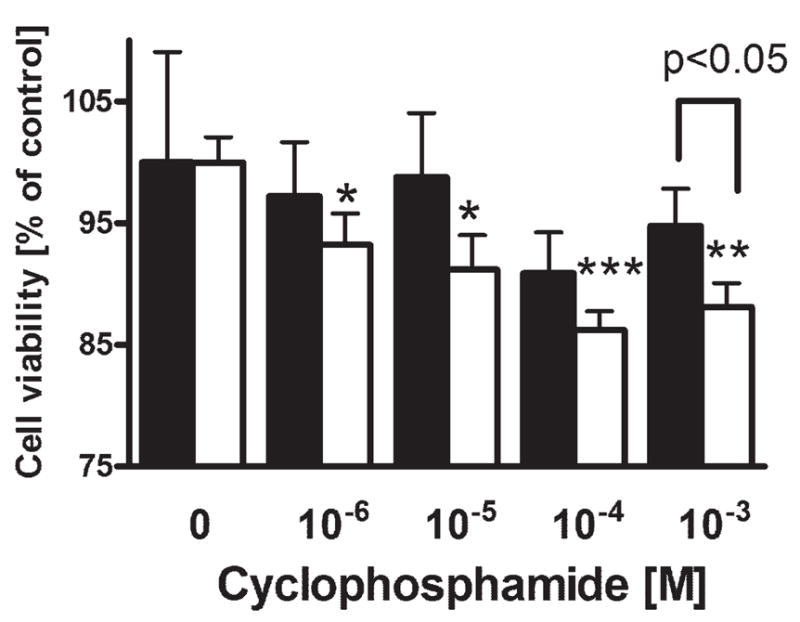
The absence of melanin pigment sensitizes SKMEL-188 melanoma cells against cyclophosphamide. SKMEL-188 were grown in the medium containing low tyrosine levels (10 μM, F10, nonpigmented) or high tyrosine (200 μM, DMEM:F10, pigmented). Pigmented (black bars) and nonpigmented (white bars) cells were then seeded into 96-well plates and incubated with serial dilutions of cyclophosphamide. After 24 hr, the viability of cells was assayed with MTT test. Data is presented as means ± SEM for 6 independent measurements, *p < 0.05, **p < 0.005, ***p < 0.0005 versus control. The experiment was repeated with similar results.
Having observed that the presence of melanin significantly changes the response of melanoma cells to antineoplastic agent, we tested whether the inhibition of melanogenesis will sensitize melanoma cells to cyclophosphamide induced cytotoxicity. First, SKMEL-188 were cultured in either Ham’s F10 (containing 10 μM tyrosine) or F10:DMEM (containing 200 μM tyrosine) medium with 5% FBS and antibiotics in the presence or absence of D-penicillamine or PTU (inhibitors of tyrosinase35). As shown in Figure 2a either inhibitor at the concentration 10−3 M caused almost complete depigmentation of previously pigmented cells. To quantify the degree of melanogenesis inhibition, we measured tyrosinase activity as described previously.46 Although pigmented cells exhibited more than ~3-fold higher tyrosinase activity than nonpigmented cells, addition of melanization inhibitors decreased the tyrosinase activity to the level observed in nonpigmented cells (~1-fold, at 10−4 M)(Fig. 2b).
To test the effect of melanogenesis suppression on cyclophosphamide toxicity, we used pigmented SKMEL-188 cells or the cells that were depigmented by the action of PTU or D-penicillamine (as described in Fig. 2). Cells were incubated in Ham’s F10 medium (low in tyrosine) with 5% FBS and antibiotics with serial dilutions of cyclophosphamide for 24 hr and the viability was assessed with the MTT test (Fig. 3). In this experiment, cyclophosphamide could exert a statistically significant effect on pigmented cells but only at the very high concentration (10−3 M) (~14%, p < 0.005)(Fig. 3a). In contrast, the cells depigmented by the melanogenesis inhibitors were killed by the cyclophosphamide already at 10−6 M (p = 0.040 and p = 0.002) and the effect reached the maximum level at 10−3 M for either inhibitor: PTU (p < 0.005) or D-penicillamine (p < 0.0005) (Fig. 3b and 3c).
Figure 3.

Inhibition of melanogenesis by N-phenylthiourea and D-penicillamine sensitizes KMEL-188 melanoma cells against cyclophosphamide. Pigmented melanoma cells (a) or cells depigmented by b10−4 M N-phenylthiourea (b) or 10−3 M D-penicillamine (c) were transferred to 96-well plates and incubated in Ham’s F10 medium with 5% FBS and serial dilutions of cyclophosphamide. After 24 hr, the viability of cells was assayed with MTT test as described.48 Data are presented as means ± SEM (n = 6), *p < 0.05, **p < 0.005, ***p < 0.0005.
These data clearly show that the melanotic phenotype attenuates cyclophosphamide toxicity and conversely, that inhibition of melanogenesis by tyrosinase inhibitors sensitizes melanoma cells to the chemotherapeutic action of the same agent. The results, therefore, are consistent with the known chemical properties of melanin being a strong detoxifying agent and ROS scavenger.34,35,43,49,50 Moreover, the induced sensitization of melanoma cells to chemotherapy by inhibition of melanogenesis suggests a new therapeutic arena for the control (adjuvant strategy) of this intractable disease in its advanced stages.
Inhibition of melanogenesis sensitizes melanoma cells against the action of IL-2-activated peripheral blood lymphocytes
Because melanoma-associated antigens may generate B cell-, T cell- and NK-mediated responses,11,29,31,51–53 we determined whether inhibition of pigmentation will make melanoma cells more susceptible to immunotherapy, specifically, to killing by human peripheral blood lymphocytes. For this experiment, we used four types of melanoma cells: nonpigmented (cultured in Ham’s F10 medium), pigmented (cultured in DMEM:F10 medium), depigmented (cultured in DMEM:F10 medium in the presence of N-phenylthiourea) for 3 days and depigmented (cultured in DMEM:F10 medium in the presence of D-penicillamine) for 3 days. Peripheral blood lymphocytes (obtained as above) were activated by preincubation with IL-2 200 U/ml for 24 hr, and the 4 types of SKMEL-188 cells were then coincubated with IL-2-activated peripheral blood lymphocytes (at target:effector ratio 1:5) in Ham’s F10 medium with 5% FBS. After 4 hr, release of LDH from melanoma cells killed by PBLs (PBL-mediated cytotoxicity) was measured. As shown in Figure 4, pigmented cells were almost completely resistant to PBLs (<0.1% killed). However, relatively significant proportion of nonpigmented cells were killed (~4%; p < 0.05). Most strikingly, the inhibition of melanogenesis by N-phenylthiourea resulted in the killing of ~50% of melanoma cells by PBLs (p < 0.000000005). Depigmented cells from treatment with D-penicillamine showed ~23% of killing by PBLs (p < 0.000005). Note, that neither N-phenylthiourea nor D-penicillamine had affected the viability of melanoma cells (Fig. 4b).
Figure 4.
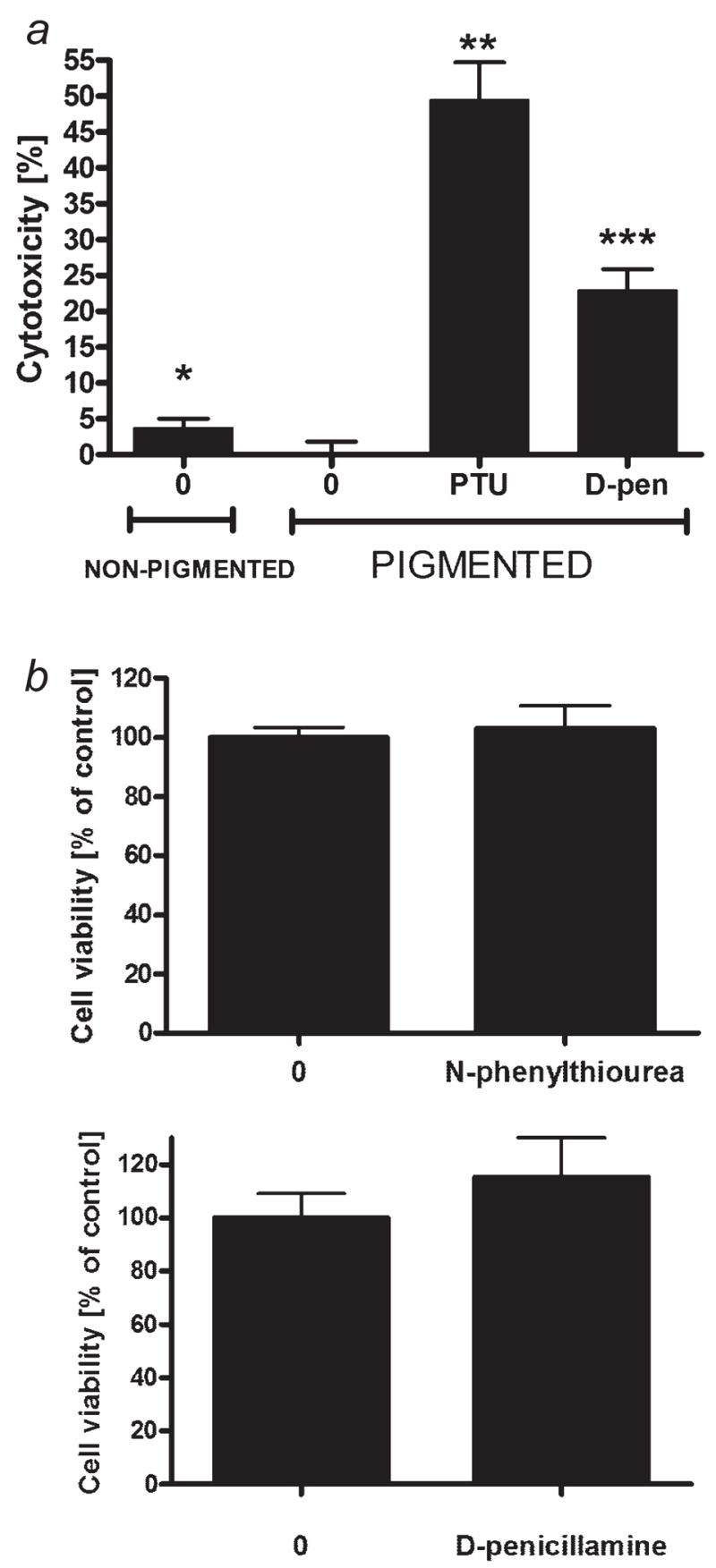
The absence of melanin or inhibition of melanogenesis by N-phenylthiourea or D-penicillamine sensitizes SKMEL-188 melanoma cells to killing action of IL-2-activated peripheral blood lymphocytes, whereas neither N-phenylthiourea nor D-penicillamine affect the viability of the melanoma cells. a: Nonpigmented SKMEL-188 cells were grown in Ham’s F10. Pigmented cells were grown in DMEM:F10 medium; the depigmentation was achieved by addition of either PTU or D-penicillamine (at 10−3 M) to the DMEM:F10 medium for 3 days. These cells were coincubated with IL-2-activated (preincubated with IL-2 200 U/ml for 24 hr) peripheral blood lymphocytes (at ratio 1:5) in Ham’s F10 medium with 5% FBS. After 4 hr, the PBL-mediated cytotoxicity was measured with Cytotox96® nonradioactive cytotoxicity assay (Promega, Madison, WI). Data are presented as means ± SEM (n = 12), *p < 0.05, **p < 0.000005, ***p < 0.000000005 versus pigmented control cells. b: Pigmented SKMEL-188 were grown in the medium containing high tyrosine (200 μM, DMEM:F10) and were seeded into 96-well plates and incubated with N-phenylthiourea or D-penicillamine at 10−3. After 24 hr, the viability of cells was assayed with MTT test. Data are presented as means ± SEM (n = 6).
Therefore, the melanotic phenotype attenuates or abrogates the killing action of IL-2 activated immune cells. This finding is important within the context of an ongoing effort to develop effective immunotherapies against melanoma, including the use of IL-2.52,54 Of great interest is striking stimulation of immunotoxicity against melanoma cells by tyrosinase inhibitors suggesting that the inhibition of generation of immunosuppressive intermediates of melanogenesis is allowing to mount an efficient immune response as proposed by us previously.34,55 However, an additional effect of N-phenylthiourea nor D-penicillamine (cation chelator) independent of melanogenesis inhibition cannot be ruled out.
Because peripheral blood lymphocytes exert a stronger cytotoxic effect against nonpigmented or depigmented cells versus pigmented cells, we decided to determine if pigmented melanoma cells release factors that inhibit lymphocyte proliferation. Pigmented and nonpigmented melanoma cells were incubated in serum free DMEM for 1, 6, 24 hr and then the media were collected. Conditioned media were added to the peripheral blood lymphocytes suspended in RPMI with FBS and [3H]thymidine. Lymphocytes were then incubated for 6 hr and incorporation of thymidine was measured. As shown in Figure 5a, proliferation of lymphocytes incubated with media derived from above pigmented cells is lower by 16 ± 5% (p < 0.05) than the proliferation of lymphocytes incubated with the media derived from above nonpigmented cells. This can be explained by the presence of increased concentrations of intermediates of melanogenesis in the medium conditioned by pigmented cells that inhibit proliferation and/or function of lymphocytes.
Figure 5.
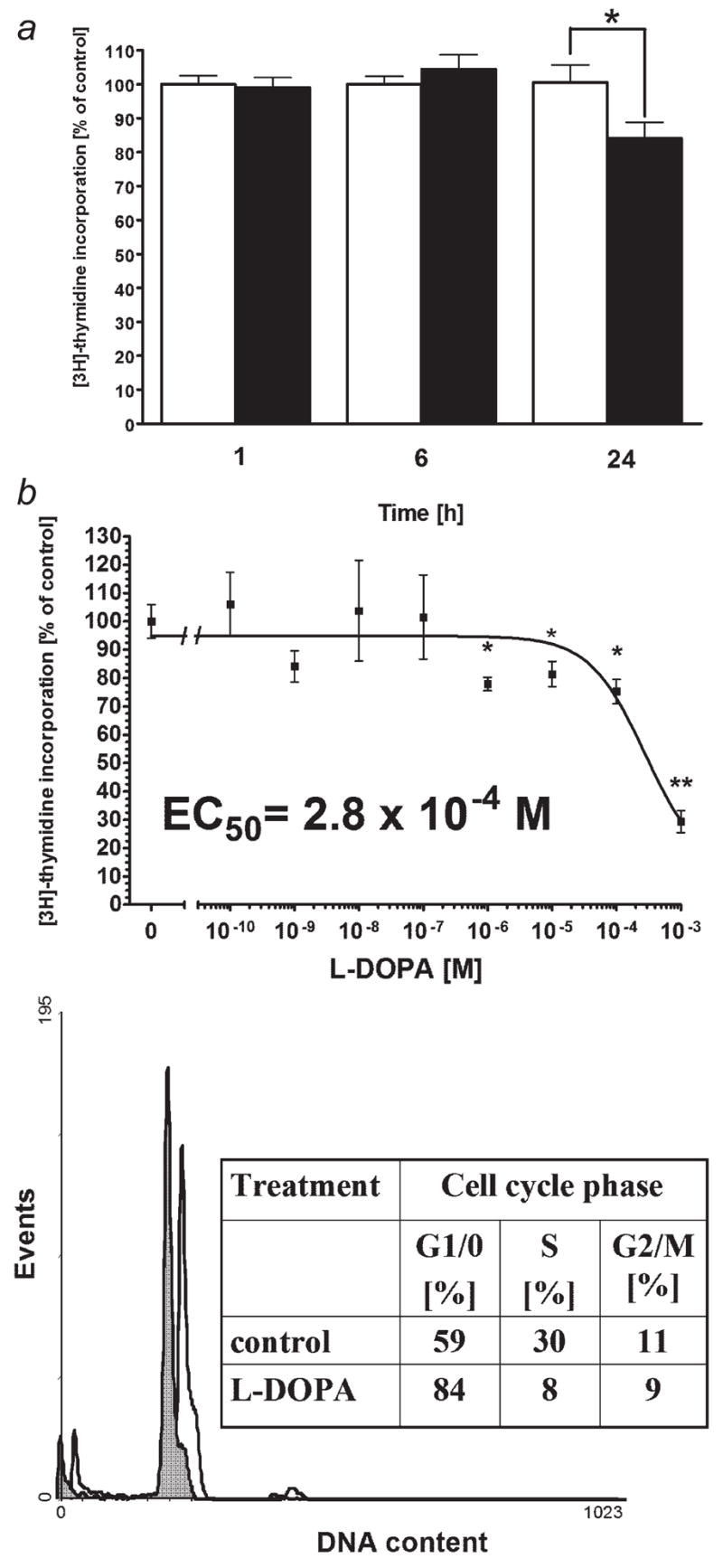
The supernatant from above pigmented melanoma cells and L-DOPA inhibits lymphocyte proliferation producing cell cycle arrest in G1/0. a: Pigmented and nonpigmented melanoma cells were incubated in serum free DMEM for 1, 6, 24 hr and then the media were collected. Conditioned media were added to the peripheral blood lymphocytes suspended in RPMI with FBS and [3H]thymidine. Lymphocytes were incubated for 6 hr and then incorporation of thymidine was measured. Data are presented as means ± SEM (n = 12). White columns: lymphocytes stimulated with supernantant from nonpigmented melanoma cells, black columns: lymphocytes stimulated with the supernatant from pigmented melanoma cells. Control: lymphocytes stimulated with the supernatant from nonpigmented melanoma cells in each time point. b: Peripheral blood lymphocytes were preincubated with LPS (1,000 ng/ml) for 24 hr and then incubated for 24 hr with serial dilutions of L-DOPA in medium RPMI 1640 containing 5% FBS and antibiotics. [3H]-thymidine was added for last 18 hr of incubation. Data are presented as means ± SEM (n = 6), *p < 0.05, **p < 0.0001. c: Peripheral blood lymphocytes were preincubated with IL-2 (200 U/ml) and then incubated for 24 hr with L-DOPA at 50 μM in medium RPMI 1640 containing 5% FBS and antibiotics. Cells were then fixed, stained and DNA content measured with flow cytometry. Data are representative of two separate experiments. White histogram: control cells, gray histogram: cells stimulated with L-DOPA.
Because L-DOPA is a major product released by pigmented cells (benign or malignant),35,40,56–58 we tested its effects on immune cells (lymphocytes). We assessed L-DOPA effect on lymphocyte proliferation and production of inflammatory cytokines. Peripheral blood lymphocytes were activated with LPS and then incubated for 24 hr with serial dilutions of L-DOPA. As shown on Figure 5a, L-DOPA inhibits lymphocyte DNA synthesis with EC50=2.8 × 10−4 M. Peripheral blood lymphocytes activated with IL-2 were then incubated for 24 hr with L-DOPA at the 50 μM and the DNA content was measured with flow cytometry. L-DOPA inhibited the transition from G1/0 to S phase of the cell cycle (Fig. 5b), since, in the control sample, 59% of cells were in G1/0 phase of the cell cycle whereas L-DOPA treatment increased that proportion to 84%. Concordantly, there were correspondent decreases of cell numbers in S and G2/M phases. These data are in agreement with our previous findings on L-DOPA effects on murine splenocytes and human lymphocytes.55 LPS activated peripheral blood lymphocytes were also incubated with L-DOPA and the levels of inflammatory cytokines were measured with real-time PCR. Cells pretreated with LPS only showed ~7.5-fold increase in TNF-alpha mRNA, ~56.4-fold increase in IL-1beta mRNA, ~1,506-fold increase in IL-6 mRNA and 23.6-fold increase in IL-10 mRNA (Fig. 6). However, treatment of cells with L-DOPA at concentration as low as 5 μM resulted in decrease of cytokine mRNAs to the levels comparable or lower (p < 0.0005 at 1,000 μM) than these of unstimulated cells. Some authors have reported that L-DOPA could stimulate production of IL-6 and TNF-alpha on protein level in patients with Parkinson disease.59 However, because this effect has been moderate, random and observed only at selected concentrations of L-DOPA, it is likely that the cytokines accumulation in the media could be secondary to a release of proteins from lymphocytes killed by L-DOPA. This has further been substantiated by the reported significant inhibition of DNA synthesis in lymphocytes exposed to L-DOPA,59 which concurs with our findings, and indicates the lymphotoxic effect of this compound. Of note, the effect of L-DOPA on expression of cytokine mRNA in our model was not only inhibitory but also very robust (from 20 to more than 1,000-fold inhibition).
Figure 6.

L-DOPA inhibits cytokine production by peripheral blood lymphocytes. LPS (1,000 ng/ml) activated peripheral blood lymphocytes (for 24 hr) were incubated with graded concentrations of L-DOPA for 1 hr in RPMI 1640 containing 5% FBS and antibiotics. Cells were then lysed, total RNA extracted, and levels of IL-1beta, TNF-alpha, IL-6 and IL-10 mRNAs were measured after reverse transcription with real-time PCR. Data are presented as means ± SEM (n = 3), #p < 0.0001 versus control without LPS, *p < 0.05, **p < 0.005, ***p < 0.0005 versus control with LPS.
Thus, L-DOPA and/or quinone/semiquinone products of its auto-oxidation (L-DOPA can oxidize spontaneously starting slow nonenzymatic melanogenesis that can be accelerated by metal cations35) are now identified as both inhibitors of lymphocyte proliferation (through G1/0 arrest) and lymphocyte immune activity (through complete inhibition of tested cytokines genes transcription). These results further provide a mechanistic explanation for the preferential killing efficiency of IL-2 activated lymphocytes against amelanotic rather than melanotic melanoma cells, and for the striking amplification of the immune activity by the tyrosinase inhibitors PTU and D-penicillamine (Fig. 4). Hence, the production and release of melanogenesis intermediates that include L-DOPA would generate an immunosuppressive environment that enhances growth and unresponsiveness to therapy. Selective inhibition of tyrosinase, an enzyme catalyzing a rate limiting reactions of the pathway may prevent such outcome. Furthermore, the reported biologically active concentrations of L-DOPA can be routinely detected in the melanotic tumor environment57 or even detected in serum from patients with advanced melanotic melanomas.56,58
Conclusions
The data presented above have been generated in a well defined model, which clearly demonstrates that the presence of melanin pigment or of active melanogenesis impairs significantly the sensitivity of melanoma cells to chemo- or immunotherapy. Furthermore, we showed the expected sensitization of melanoma cells towards cytotoxic action of cyclophosphamide, and significant amplification of IL-2 activated lymphocytes immunotoxic activities after the inhibition of tyrosinase activity. Thus, these findings, in conjunction with the most recently reported radiosensitization of melanotic melanoma cells by inhibition of tyrosinase activity,60 support further effort (e.g., in vivo testing) for the development of novel therapies for melanotic melanoma based on the inhibition of melanogenesis. Such an approach could amplify the effectiveness of existing chemotherapy or immunotherapy, and directly contribute to inhibition of melanoma growth. Inhibition of melanogenesis may represent a dawn of novel adjuvant therapeutic modality; because it will utilize already FDA approved compounds used for treatment of Wilson disease and/or intoxication with heavy metals (chelation of copper and other metal chelatorscations would inhibit tyrosinase activity) in combination with existing chemotherapy or immunotherapy protocols.
In summary, we provide evidence obtained in vitro in support of the hypothesis that inhibition of melanogenesis should be explored as a valid therapeutic target for the management of advanced melanoma.
Acknowledgments
NIH; Grant number: AR052190; Grant sponsor: Department of Energy UTCI pilot grant.
Funding was provided by Department of Energy UTCI pilot grant and in part by NIH grant AR052190 to AS. We thank Dr. Jacobo Wortsman for his careful review of the manuscript, editorial help and valuable comments.
References
- 1.Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 2.Becker D, Mihm MC, Hewitt SM, Sondak VK, Fountain JW, Thurin M. Markers and tissue resources for melanoma: meeting report. Cancer Res. 2006;66:10652–7. doi: 10.1158/0008-5472.CAN-06-0921. [DOI] [PubMed] [Google Scholar]
- 3.Carlson JA, Ross JS, Slominski A, Linette G, Mysliborski J, Hill J, Mihm M., Jr Molecular diagnostics in melanoma. J Am Acad Dermatol. 2005;52:743–75. doi: 10.1016/j.jaad.2004.08.034. quiz 75–8. [DOI] [PubMed] [Google Scholar]
- 4.Meyskens FL, Jr, Farmer PJ, Anton-Culver H. Etiologic pathogenesis of melanoma: a unifying hypothesis for the missing attributable risk. Clin Cancer Res. 2004;10:2581–3. doi: 10.1158/1078-0432.ccr-03-0638. [DOI] [PubMed] [Google Scholar]
- 5.Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bardia A, Vachon CM, Schild SE, McWilliams RR, Hand JL, Laman SD, Kottschade LA, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc. 2007;82:364–80. doi: 10.4065/82.3.364. [DOI] [PubMed] [Google Scholar]
- 6.Zabierowski SE, Herlyn M. Learning the ABCs of melanoma-initiating cells. Cancer Cell. 2008;13:185–7. doi: 10.1016/j.ccr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006;20:2149–82. doi: 10.1101/gad.1437206. [DOI] [PubMed] [Google Scholar]
- 8.Slominski A, Wortsman J, Carlson A, Matsuoka L, Balch CM, Mihm M. Malignant melanoma: an update. Arch Pathol Lab Med. 2001;125:1295–306. doi: 10.5858/2001-125-1295-MM. [DOI] [PubMed] [Google Scholar]
- 9.Lachiewicz AM, Berwick M, Wiggins CL, Thomas NE. Epidemiologic support for melanoma heterogeneity using the surveillance, epidemiology, and end results program. J Invest Dermatol. 2008;128:1340–2. doi: 10.1038/jid.2008.18. [DOI] [PubMed] [Google Scholar]
- 10.Cascinelli N, Heerlyn M, Schneeberger A, Kuwert C, Slominski A, Armstrong C, Belli F, Lukiewcz S, Maurer D, Ansel J, Stingl G, Saida T. What is the most promising strategy for the treatment of metastasizing melanoma? Exp Dermatol. 2000;9:439–51. doi: 10.1034/j.1600-0625.2000.009006439.x. [DOI] [PubMed] [Google Scholar]
- 11.Tawbi HA, Kirkwood JM. Management of metastatic melanoma. Semin Oncol. 2007;34:532–45. doi: 10.1053/j.seminoncol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Linette GP, Carlson JA, Slominski A, Mihm MC, Ross JS. Biomarkers in melanoma: stage III and IV disease. Expert Rev Mol Diagn. 2005;5:65–74. doi: 10.1586/14737159.5.1.65. [DOI] [PubMed] [Google Scholar]
- 13.Eggermont AM, Gore M. Randomized adjuvant therapy trials in melanoma: surgical and systemic. Semin Oncol. 2007;34:509–15. doi: 10.1053/j.seminoncol.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Eggermont AM, Gore M. European approach to adjuvant treatment of intermediate- and high-risk malignant melanoma. Semin Oncol. 2002;29:382–8. doi: 10.1053/sonc.2002.34117. [DOI] [PubMed] [Google Scholar]
- 15.Herlyn M, Halaban R, Ronai Z, Schuchter L, Berwick M, Pinkel D. Roadmap for new opportunities in melanoma research. Semin Oncol. 2007;34:566–76. doi: 10.1053/j.seminoncol.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Eggermont AM, Suciu S, MacKie R, Ruka W, Testori A, Kruit W, Punt CJ, Delauney M, Sales F, Groenewegen G, Ruiter DJ, Jagiello I, et al. Post-surgery adjuvant therapy with intermediate doses of interferon alfa 2b versus observation in patients with stage IIb/III melanoma (EORTC 18952): randomised controlled trial. Lancet. 2005;366:1189–96. doi: 10.1016/S0140-6736(05)67482-X. [DOI] [PubMed] [Google Scholar]
- 17.Johnson TM, Smith JW, II, Nelson BR, Chang A. Current therapy for cutaneous melanoma. J Am Acad Dermatol. 1995;32:689–707. doi: 10.1016/0190-9622(95)91443-9. quiz 08–9. [DOI] [PubMed] [Google Scholar]
- 18.Demierre MF, Koh HK. Adjuvant therapy for cutaneous malignant melanoma. J Am Acad Dermatol. 1997;36:747–64. doi: 10.1016/s0190-9622(97)80329-5. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberg SA. The immunotherapy of solid cancers based on cloning the genes encoding tumor-rejection antigens. Annu Rev Med. 1996;47:481–91. doi: 10.1146/annurev.med.47.1.481. [DOI] [PubMed] [Google Scholar]
- 20.Scott AM, Cebon J. Clinical promise of tumour immunology. Lancet. 1997;349(Suppl 2):SII19–SII22. doi: 10.1016/s0140-6736(97)90016-7. [DOI] [PubMed] [Google Scholar]
- 21.Curiel-Lewandrowski C, Atkins MB. Immunotherapeutic approaches for the treatment of malignant melanoma. Curr Opin Investig Drugs. 2001;2:1553–63. [PubMed] [Google Scholar]
- 22.Bonnekoh B, Bickenbach JR, Roop DR. Immunological gene therapy approaches for malignant melanoma. 2 Preclinical studies and clinical strategies. Skin Pharmacol. 1997;10:105–25. doi: 10.1159/000211476. [DOI] [PubMed] [Google Scholar]
- 23.Shurin MR, Kirkwood JM, Esche C. Cytokine-based therapy for melanoma: pre-clinical studies. Forum (Genova) 2000;10:204–26. [PubMed] [Google Scholar]
- 24.Ravindranath MH, Morton DL. Immunogenicity of membrane-bound gangliosides in viable whole-cell vaccines. Cancer Invest. 1997;15:491–9. doi: 10.3109/07357909709047588. [DOI] [PubMed] [Google Scholar]
- 25.Dufour E, Carcelain G, Gaudin C, Flament C, Avril MF, Faure F. Diversity of the cytotoxic melanoma-specific immune response: some CTL clones recognize autologous fresh tumor cells and not tumor cell lines. J Immunol. 1997;158:3787–95. [PubMed] [Google Scholar]
- 26.Hearing VJ. Biochemical control of melanogenesis and melanosomal organization. J Investig Dermatol Symp Proc. 1999;4:24–8. doi: 10.1038/sj.jidsp.5640176. [DOI] [PubMed] [Google Scholar]
- 27.Cochran AJ, Ohsie SJ, Binder SW. Pathobiology of the sentinel node. Curr Opin Oncol. 2008;20:190–5. doi: 10.1097/CCO.0b013e3282f46d70. [DOI] [PubMed] [Google Scholar]
- 28.Eggert AO, Becker JC, Ammon M, McLellan AD, Renner G, Merkel A, Brocker EB, Kampgen E. Specific peptide-mediated immunity against established melanoma tumors with dendritic cells requires IL-2 and fetal calf serum-free cell culture. Eur J Immunol. 2002;32:122–7. doi: 10.1002/1521-4141(200201)32:1<122::AID-IMMU122>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 29.Chapman PB. Melanoma vaccines. Semin Oncol. 2007;34:516–23. doi: 10.1053/j.seminoncol.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Hakomori S. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. 2001;491:369–402. doi: 10.1007/978-1-4615-1267-7_24. [DOI] [PubMed] [Google Scholar]
- 31.Shah GD, Chapman PB. Adjuvant therapy of melanoma. Cancer J. 2007;13:217–22. doi: 10.1097/PPO.0b013e318074dfd4. [DOI] [PubMed] [Google Scholar]
- 32.Meyskens FL, Jr, Farmer PJ, Yang S, Anton-Culver H. New perspectives on melanoma pathogenesis and chemoprevention. Recent Results Cancer Res. 2007;174:191–5. doi: 10.1007/978-3-540-37696-5_16. [DOI] [PubMed] [Google Scholar]
- 33.Meyskens FL, Jr, Berwick M. UV or Not UV: metals are the answer. Cancer Epidemiol Biomarkers Prev. 2008;17:268–70. doi: 10.1158/1055-9965.EPI-07-0653. [DOI] [PubMed] [Google Scholar]
- 34.Slominski A, Paus R, Mihm MC. Inhibition of melanogenesis as an adjuvant strategy in the treatment of melanotic melanomas: selective review and hypothesis. Anticancer Res. 1998;18:3709–15. [PubMed] [Google Scholar]
- 35.Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155–228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- 36.Riley PA. Melanogenesis and melanoma. Pigment Cell Res. 2003;16:548–52. doi: 10.1034/j.1600-0749.2003.00069.x. [DOI] [PubMed] [Google Scholar]
- 37.Prota G, d’Ischia M, Mascagna D. Melanogenesis as a targeting strategy against metastatic melanoma: a reassessment. Melanoma Res. 1994;4:351–8. doi: 10.1097/00008390-199412000-00002. [DOI] [PubMed] [Google Scholar]
- 38.Slominski AT, Paus R. Inhibition of melanogenesis for melanoma therapy? J Invest Dermatol. 1994;103:742. doi: 10.1111/1523-1747.ep12398639. [DOI] [PubMed] [Google Scholar]
- 39.Pawelek J, Korner A, Bergstrom A, Bologna J. New regulators of melanin biosynthesis and the autodestruction of melanoma cells. Nature. 1980;286:617–9. doi: 10.1038/286617a0. [DOI] [PubMed] [Google Scholar]
- 40.Pawelek JM. Studies on the Cloudman melanoma cell line as a model for the action of MSH. Yale J Biol Med. 1985;58:571–8. [PMC free article] [PubMed] [Google Scholar]
- 41.Slominski A. Coming of age of melanogenesis-related proteins. Arch Pathol Lab Med. 2002;126:775–7. doi: 10.5858/2002-126-0775-COAOMR. [DOI] [PubMed] [Google Scholar]
- 42.Slominski A, Paus R, Mihm M. Inhibition of melanogenesis as an adjuvant strategy in the treatment of melanotic melanomas: selective review and hypothesis. Anticancer Res. 1998;18:3709–16. [PubMed] [Google Scholar]
- 43.Wood JM, Jimbow K, Boissy RE, Slominski A, Plonka PM, Slawinski J, Wortsman J, Tosk J. What’s the use of generating melanin? Exp Dermatol. 1999;8:153–64. doi: 10.1111/j.1600-0625.1999.tb00365.x. [DOI] [PubMed] [Google Scholar]
- 44.Slominski A, Ermak G, Wortsman J. Modification of melanogenesis in cultured human melanoma cells. In Vitro Cell Dev Biol. 1999;35:564–65. doi: 10.1007/s11626-999-0093-6. [DOI] [PubMed] [Google Scholar]
- 45.Slominski A, Moellman G, Kuklinska E, Bomirski A, Pawelek J. Positive regulation of melanin pigmentation by two key substrates of the melanogenic pathway: L-tyrosine and L-dopa. J Cell Sci. 1988;89:287–96. doi: 10.1242/jcs.89.3.287. [DOI] [PubMed] [Google Scholar]
- 46.Slominski A, Scislowski PW, Bomirski A. Tyrosinase activity in primary cell culture of amelanotic melanoma cells. Biosci Rep. 1983;3:1027–34. doi: 10.1007/BF01121029. [DOI] [PubMed] [Google Scholar]
- 47.Tsuboi I, Tanaka H, Nakao M, Shichijo S, Itoh K. Nonsteroidal anti-inflammatory drugs differentially regulate cytokine production in human lymphocytes: up-regulation of TNF, IFN-gamma and IL-2, in contrast to down-regulation of IL-6 production. Cytokine. 1995;7:372–9. doi: 10.1006/cyto.1995.0047. [DOI] [PubMed] [Google Scholar]
- 48.Slominski A, Pisarchik A, Zbytek B, Tobin DJ, Kauser S, Wortsman J. Functional activity of serotoninergic and melatoninergic systems expressed in the skin. J Cell Physiol. 2003;196:144–53. doi: 10.1002/jcp.10287. [DOI] [PubMed] [Google Scholar]
- 49.Slominski A, Paus R, Schanderdorf D. Melanocytes as sensory and regulatory cells in the epidermis. J Theor Biol. 1993;164:103–20. doi: 10.1006/jtbi.1993.1142. [DOI] [PubMed] [Google Scholar]
- 50.Hoogduijn MJ, Cemeli E, Ross K, Anderson D, Thody AJ, Wood JM. Melanin protects melanocytes and keratinocytes against H2O2-induced DNA strand breaks through its ability to bind Ca2+ Exp Cell Res. 2004;294:60–7. doi: 10.1016/j.yexcr.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Parmiani G, Castelli C, Rivoltini L, Casati C, Tully GA, Novellino L, Patuzzo A, Tosi D, Anichini A, Santinami M. Immunotherapy of melanoma. Semin Cancer Biol. 2003;13:391–400. doi: 10.1016/j.semcancer.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Dudley ME, Rosenberg SA. Adoptive cell transfer therapy. Semin Oncol. 2007;34:524–31. doi: 10.1053/j.seminoncol.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paulos CM, Kaiser A, Wrzesinski C, Hinrichs CS, Cassard L, Boni A, Muranski P, Sanchez-Perez L, Palmer DC, Yu Z, Antony PA, Gattinoni L, et al. Toll-like receptors in tumor immunotherapy. Clin Cancer Res. 2007;13:5280–9. doi: 10.1158/1078-0432.CCR-07-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J, Kerstann KW, Ahmadzadeh M, Li YF, El-Gamil M, Rosenberg SA, Robbins PF. Modulation by IL-2 of CD70 and CD27 expression on CD8+ T cells: importance for the therapeutic effectiveness of cell transfer immunotherapy. J Immunol. 2006;176:7726–35. doi: 10.4049/jimmunol.176.12.7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slominski A, Goodman-Snitkoff GG. Dopa inhibits induced proliferative activity of murine and human lymphocytes. Anticancer Res. 1992;12:753–6. [PubMed] [Google Scholar]
- 56.Yamada K, Walsh N, Hara H, Jimbow K, Chen H, Ito S. Measurement of eumelanin precursor metabolites in the urine as a new marker for melanoma metastases. Arch Dermatol. 1992;128:491–4. [PubMed] [Google Scholar]
- 57.Jimbow K, Miyake Y, Homma K, Yasuda K, Izumi Y, Tsutsumi A, Ito S. Characterization of melanogenesis and morphogenesis of melanosomes by physicochemical properties of melanin and melanosomes in malignant melanoma. Cancer Res. 1984;44:1128–34. [PubMed] [Google Scholar]
- 58.Wakamatsu K, Kageshita T, Furue M, Hatta N, Kiyohara Y, Nakayama J, Ono T, Saida T, Takata M, Tsuchida T, Uhara H, Yamamoto A, et al. Evaluation of 5-S-cysteinyldopa as a marker of melanoma progression: 10 years’ experience. Melanoma Res. 2002;12:245–53. doi: 10.1097/00008390-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M. IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mono-nuclear cells from patients with Parkinson’s disease. Biomed Pharmacother. 1999;53:141–5. doi: 10.1016/S0753-3322(99)80079-1. [DOI] [PubMed] [Google Scholar]
- 60.Brozyna A, VanMiddlesworth L, Slominski A. Inhibition of melanogenesis as a radiation sensitizer for melanoma therapy. Int J Cancer. 2008;123:1448–56. doi: 10.1002/ijc.23664. [DOI] [PubMed] [Google Scholar]