

Abstract
The induction of the immediate early gene Arc is strongly implicated in synaptic plasticity. Although the role of ERK was demonstrated, the regulation of Arc expression is largely unknown. In this study, we investigated the major signaling pathways underlying brain-derived neurotrophic factor (BDNF)-mediated Arc transcription in cultured cortical neurons. The BDNF-stimulated Arc transcription was solely regulated by the Ras-Raf-MAPK signaling through ERK, but not by phosphoinositide 3-kinase (PI3K) and PLC-γ activities. Although it was demonstrated that BDNF might promote calcium entry through calcium channels and NMDA receptors, chelating extracellular calcium with EGTA failed to block Arc transcription. In contrast, chelating intracellular calcium ([Ca2+]i) by BAPTA-AM abolished BDNF-mediated Arc up-regulation. Surprisingly, BAPTA-AM did not block ERK activation, indicating that [Ca2+]i and Ras-Raf-MAPK are not coupled, and the activation of ERK alone is not sufficient to up-regulate Arc transcription. Moreover, we found that inhibition of calmodulin (CaM) by W13 blocked both Arc transcription and ERK activation, revealing a Ca2+-independent function of CaM. These data suggested novel functions of [Ca2+]i and CaM in BDNF signaling. Comparison of the Arc transcription profiles between Ca2+-stimulated and BDNF-stimulated neurons demonstrated that the regulatory mechanisms were distinctively tailored to the complex features of neuronal activity. Specifically, PI3K and CaM-dependent protein kinase (CaMK) activity were required for Ca2+-stimulated Arc transcription through regulating ERK signaling. Such cross-talks between PI3K, CaMK and ERK were absent in BDNF-stimulated neurons.
Keywords: neurotrophin, calmodulin-dependent protein kinase, phosphoinositide 3-kinase, MAPK, neuroplasticity
The immediate-early genes (IEG) are rapidly induced by various forms of neuronal stimulation. Among them, activity-regulated cytoskeleton-associated protein (ARC) serves as a sensitive marker for neuronal activity. Recent studies began to address the function of Arc in neurons, and revealed its involvement in regulating AMPA receptor trafficking, long-term potentiation (LTP) and the consolidation of long-term memories. For example, over-expression of Arc enhanced AMPA receptor endocytosis and reduced the surface expression of AMPA receptors (Chowdhury et al. 2006). Consistently, an increase in AMPA receptor surface expression and a decrease in AMPA receptor endocytosis were observed in Arc knock-out mice (Shepherd et al. 2006). Furthermore, inhibition of Arc expression by antisense oligonucleotides disrupted both the maintenance of LTP and the consolidation of spatial memory (Guzowski et al. 2000). Impaired late-phase LTP, long-term depression (LTD), and hippocampus-dependent memory were also observed in the Arc knock-out mice (Plath et al. 2006). These studies suggested that the activity-dependent Arc up-regulation might be of physiological relevance for certain neuronal functions.
The up-regulation of Arc expression was demonstrated during pentylenetrazole-induced seizures (Link et al. 1995), after the induction of LTP (Lyford et al. 1995), and in vivo after exploring a novel environment (Guzowski et al. 1999) or learning to escape from an aversively illuminated area (Montag-Sallaz and Montag 2003). Although the cellular behavior and induction profile of Arc are well documented, the regulatory mechanisms underlying the activity-dependent transcription remain largely unknown. Waltereit et al. observed that Arc transcription could be stimulated by either membrane depolarization with KCl or the activation of adenylyl cyclases with forskolin in PC12 cells (Waltereit et al. 2001). They further studied the molecular structure of the Arc promoter and found two SREs (serum response element) and two AP-1 consensus sequences, but failed to detect the cAMP responsive element (CRE) (Waltereit et al. 2001). However, the presence of SRE and AP-1 did not contribute to the cAMP-induced Arc transcription. Nevertheless, the forskolin-induced Arc expression required the activation of ERK, which regulates both SRE- and CRE-mediated transcription.
In addition to calcium and cAMP, Arc expression may also be up-regulated by neurotrophins, such as BDNF (Rao et al. 2006; Ying et al. 2002). The function of BNDF was initially implicated in cell survival, neuronal differentiation, and neurogenesis (Huang and Reichardt 2001; Lu et al. 2005). Recent investigations have strongly demonstrated its role in regulating synaptic plasticity (Schinder and Poo 2000). First, BDNF expression and release are tightly controlled by neuronal activities, and induced by NMDA activation, LTP and hippocampus-dependent learning (Ghosh et al. 1994; Hall et al. 2000; Patterson et al. 1992; Tao et al. 2002; West et al. 2001). Second, suppression of BDNF expression resulted in defective LTP and memory formation (Korte et al. 1995; Linnarsson et al. 1997; Ma et al. 1998; Mu et al. 1999). Theoretically, BDNF may regulate neuroplasticity by stimulating gene transcription, activating protein synthesis, promoting neuro-transmitter release, and modulating the activity and trafficking of post-synaptic receptors (Jovanovic et al. 2000; Kafitz et al. 1999; Nakata and Nakamura 2007; Poo 2001; Schinder and Poo 2000). Therefore, the BDNF-induced Arc transcription may be functionally relevant for the activity-dependent neuronal modifications.
The goal of this study is to investigate the molecular mechanisms for the BDNF-induced Arc transcription. We examined the function of the major signaling molecules in BDNF-stimulated neurons. Our results reveal that Arc transcription is regulated, in parallel, by ERK activity and the basal level of [Ca2+]i. Furthermore, by comparing the Arc transcription profiles between BDNF- and KCl (membrane depolarization)-stimulated neurons, distinct regulatory mechanisms were identified, suggesting that neuronal responses are tailored to specific stimulations.
MATERIALS AND METHODS
Antibodies and inhibitors
All antibodies were purchased from Cell Signaling and diluted to working concentrations with PBS-T (1% TritonX-100 and 1%NaF in PBS buffer) according to the manufacturer’s direction: phospho-ERK (P-ERK), 1:1000; total ERK (T-ERK), 1:1000; phospho-Akt (P-Akt), 1:1000; and total Akt (T-Akt), 1:1000. The concentrations of inhibitors used were: K252a (Sigma), 200nM; TrkB-IgG (R&D system) 200ug/ml; LY294002 (Calbiochem), 30uM; U0126 (Calbiochem), 10uM; U73122 (Calbiochem), 5uM; Actinomycin D (ACD, Sigma), 0.1ug/ml; EGTA (Sigma) 2.5mM; BAPTA-AM (Sigma), 33uM; APV (Calbiochem), 100uM; Nifedipine (Sigma), 10uM; W13(Sigma), 70uM; KN62 (Sigma), 10uM; and KN93 (Sigma), 5uM. The 1000X stock solution of TrkB-IgG was dissolved in PBS containing 1%BSA. EGTA (5mM, 2X) was dissolved in the cell culture media, pH was adjusted to 7.6. The stock solutions for W13 (35mM) and APV (100mM) were dissolved in H2O. The stock solutions (1000 X) for all other inhibitors/stimulators were made in DMSO, and diluted to working strength by adding directly to the cell culture media.
Neuronal culture and treatments
Cortices obtained from neonatal (postnatal day 0) Sprague Dawley rats were used for primary neuronal cultures as described (Chan et al. 1998). Briefly, dorsal regions of the frontal cortex were dissected in Hibernate A (BrainBits LLC), and chopped into small dices (about 1mm3). The tissues were then treated with papain (10 units/ml, Worthington, Freehold, NJ) and DNase I (Roche, 100 units/ml) for 30–40min at 37°C. After washing (3 times) with Neurobasal A (Invitrogen, Carlsbad, CA), the digestion was triturated and plated on poly-D-lysine (50 ug/ml, Sigma, St. Louis, MO)-coated plates at a density of 0.25 to 0.5 million cells/cm2. One third of the growth media (Neurobasal A with B27 supplement, 0.5mM glutamine and 1X penicillin and streptomycin) was replenished once every 3 days.
Bath incubation with KCl (50mM), forskolin (50uM) or recombinant human BDNF (Calbiochem) was used to stimulate cultured cortical neurons on 5 to 7 DIV (days in vitro).
RNA extraction and semi-quantitative RT-PCR
Neurons were stimulated with KCl, BDNF, or forskolin on 5–7 DIV (days in vitro) for 1h. Total RNA was extracted with TRIzol (Invitrogen) following manufacturer’s protocol. One ug RNA was reverse transcribed to cDNA using SuperScript III kit (Invitrogen). A 420-base pair-long product for Arc was amplified by semi-quantitative PCR with specific primers 5′-AGACACAGCAGATCCAGCTG-3′ (forward) and 5′-TGGCTTGTCTTCACCTTCAG-3′ (reverse). The housekeeping gene GAPDH was used as an internal reference, and amplified with the forward primer 5′-TCCATGACAACTTTGGCATTGTGG-3′ and the reverse primer 5′-GTTGCTGTTGAAGTCG CAGGAGAC-3′. The annealing temperature for both genes was 55°C. Unless specified for certain experiments, the number of thermo cycles is 20 for GAPDH and 26 for Arc. PCR products were separated on 1.2% agarose gels, documented by digitalimaging, and quantified by the Scion Image software (Scion Corp. Frederick, Maryland).
Western blot
Fifteen minutes after stimulation, the medium was quickly aspirated. The treated neurons were harvested in hot SDS loading buffer (10mM Tris-HCl buffer pH 6.8, 10% glycerol, 2% sodium dodecyl sulfate, 0.01% bromophenol blue and 5% β-mercaptoethanol). After sonication, proteins from 0.1 million cells were separated by 10% SDS-PAGE, and the level of p-ERK1/2, p-Akt, T-ERK1/2, and T-Akt was analyzed by Western blot analysis. The incubation with primary antibodies was overnight at 4ºC, in PBST with 0.1% triton X-100 (PBST) and 5% non-fat milk. After extensive wash with PBS-T, the blots were incubated with horseradish peroxidase-conjugated goat-anti-rabbit antibodies (1:5000, Pierce, Rockford, IL) for 1hr at room temperature. The ECL system (SuperSignal® West Pico, Pierce, Rockford, IL) was used for signal detection. The exposed films were scanned by an EPSON flat-bed scanner, and the signal intensity was quantified by the Scion Image software (Scion Corp. Frederick, Maryland).
Immunocytochemistry
Fifteen minutes after stimulation, the neurons were fixed in PBS with 6% paraformaldehyde at room temperature for 20min, and then permeabilized in PBST. After extensive wash with PBST, the neurons were incubated with anti-NeuN (mouse monoclonal antibody) (1:500, Chemicon) and anti-p-ERK1/2 (rabbit polyclonal antibody) (1:300, Cell Signaling) followed by incubation with Alexa-488-conjugated (1:200, Invitrogen) goat-anti-rabbit and Alexa-594-conjugated goat-anti-mouse antibodies (1:200, Invitrogen). All antibody dilutions were done in PBST/3% BSA/3% goat serum. The stained cells were mounted on slides with GEL/MOUNT™ (Biomeda), and examined by a Nikon fluorescent microscope using the Q-capture program.
Data analysis
After quantification, the value of Arc mRNA level was normalized to GAPDH. The value of p-ERK1/2 and p-Akt was normalized to T-ERK and T-Akt. The quantification data were expressed as average +/− SEM (standard error of mean). One-way ANOVA and Student’s t-test was used to determine the statistical significance (with p-value less than 0.05).
RESULTS
Sub-nanomolar BDNF activates Arc transcription and ERK through the tyrosine kinase receptor TrkB
Previous studies have demonstrated that BDNF, at 100ng/ml, stimulated Arc transcription through the tyrosine kinase receptor TrkB (Yasuda et al. 2007). To obtain a dose-responsive curve, we incubated cortical neurons with BDNF at different concentrations. Compared to nontreated control neurons, a robust elevation of Arc mRNA was observed in neurons treated with 5ng/ml (equivalent to 0.2nM for the BDNF dimer) BDNF (Fig. 1A). No further induction of Ar transcription was observed with higher BDNF concentration. Moreover, comparable induction of Arc transcription was achieved by 1ng/ml, 2ng/ml and 5ng/ml BNDF treatment (Supplementary Fig. 1A). A significant smaller, but detectable, induction was observed with 0.5ng/ml BDNF (Supplementary Fig. 1A). As a control, the mRNA level of GAPDH remained constant after BDNF treatment (Fig. 1A).
Fig. 1.

Sub-nanomolar BDNF activates both Arc transcription and ERK phosphorylation through the TrkB tyrosine receptor. A) Cortical neurons were treated with (in ng/ml) 5, 10, 25, 50 and 100 BDNF for 1hr. Total RNA was extracted from control and BDNF-treated neurons. The mRNA level of Arc and GAPDH was determined by semi-quantitative RT-PCR using gene specific primers. B) Neurons were treated with or without a transcription inhibitor actinomycin (ACD) (0.1 ug/ml) for 30min followed by 1hr BDNF (5ng/ml) or KCl (50mM) incubation. Representative images are shown in the left panels, and quantification in the right panels (n = 3 from separate experiments). Relative intensity of Arc was normalized to GAPDH. C, D, and E) Sub-nanomolar BDNF activates ERK phosphorylation through the TrkB tyrosine receptor. Cortical neurons were treated with (in ng/ml) 5, 10, 25, 50 and 100 BDNF for 15min. The harvested samples were separated by 10% SDS-PAGE. The phosphorylation of ERK was determined by Western blot using an antibody against p-ERK1/2. To address the role of the receptor tyrosine kinase TrkB, Trk inhibitor K252a (0.2 uM) or TrkB-specific BDNF scavenger TrkB-IgG (0.4 ug/ml) were applied 30min before BDNF (5ng/ml) incubation. Samples were collected 15min after BDNF treatment. The level of p-ERK was determined by Western blot analysis. The activation of p-ERK in neurons was also examined by immunofluorescent microscopy (E). Control and BDNF-treated cortical neurons were double labeled with antibodies against p-ERK (upper panels) and the neuronal marker NeuN (lower panels).
We further confirmed that sub-nanomolar BDNF-mediated Arc transcription depended on TrkB activity, because the pre-treatment with Trk receptor inhibitor K252a or TrkB-IgG significantly blocked BDNF-induced Arc mRNA elevation (Supplementary Fig. 2).
Theoretically, the elevation of Arc mRNA by BDNF could be due to either enhanced transcription or increased mRNA stability. To rule out the function of BDNF on mRNA degradation or stability, we pre-treated the neurons with transcription inhibitor actinomycin D (ACD). As shown in Fig. 1B, BDNF had no effects on Arc mRNA level when transcription was blocked, indicating that BDNF does not regulate mRNA stability. Previous reports demonstrated that Arc transcription was up-regulated by KCl-mediated membrane depolarization in PC 12 cell (Waltereit et al. 2001). Here, we also examined KCl-stimulated neurons, and compared the regulatory mechanisms between BDNF- and Ca2+-mediated Arc transcription. As shown in Fig. 1B, KCl stimulation lead to a smaller, but significant increase of Arc transcription without affecting Arc mRNA stability.
The function of MAPK, PLC-γ and PI3 kinase activity on BDNF-induced Arc transcription
As demonstrated by previous studies, Ras-Raf-MAPK, PLC-γ and PI3K-Akt signaling are the three major pathways activated by BDNF through the TrkB (Reichardt 2006) (see illustration in Fig. 7A). Although it was demonstrated that BDNF (100ng/ml)-induced Arc elevation was significantly suppressed by MEK inhibition with U0126 (Ying et al. 2002), it is unknown whether these three signaling pathways differentially regulate Arc transcription. Therefore, we examined their function by pharmacological inhibitions. First, consistent with the Arc up-regulation profile, ERK was robustly activated by sub-nanomolar BDNF (Fig. 1C). Comparable elevation of p-ERK was observed for 1ng/ml, 2ng/ml and 5ng/ml BDNF (Supplementary Fig. 1B). A slightly lower, but detectable activation of p-ERK was achieved by 0.5ng/ml BDNF (Supplementary Fig. 1B). Western blot analysis and immuno-fluorescent staining demonstrated that the function of TrkB was required for p-ERK elevation in BDNF-stimulated neurons (Fig. 1D and E). We also found that the suppression of Ras-Raf-MAPK with U0126 signaling dampened Arc induction by BDNF (5ng/ml) stimulation (Fig. 2A). Because BDNF might result in calcium release from the internal storage (Huang and Reichardt 2001; Reichardt 2006) by activating PLC-γ, and the activity of MAPK/ERK could also be regulated by calcium, we examined the function of PLC-γ. However, as shown in Fig. 2A, blocking PLC-γ with U73122 had no effect on Arc up-regulation, suggesting PLC-γ is not required for BDNF-induced Arc expression. We next examined the effects of PI3K inhibition, and found that blocking PI3K activity by LY294002 did not affect BDNF-induced Arc mRNA elevation (Fig. 2A). These results demonstrated that the Trk B-dependent Arc transcription required only MAPK pathway, but not PLC and PI3K in BDNF-stimulated neurons.
Fig. 7.
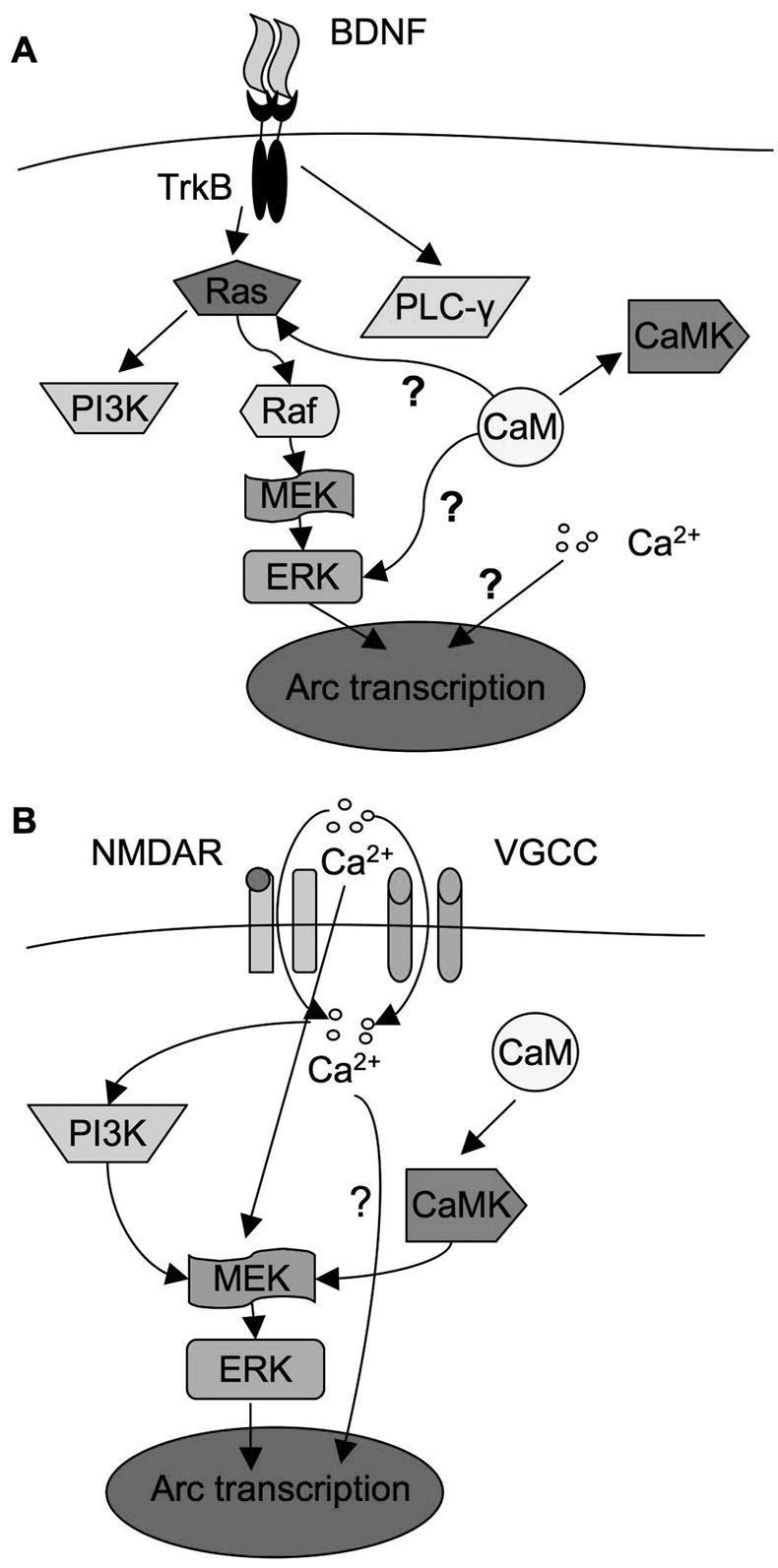
Regulation pathways of Arc transcription in BDNF- and Ca2+-mediated signaling. A). BDNF-mediated Arc expression is controlled through MAPK pathway and gated by intracellular calcium. Upon the activation of TrkB by BDNF, Ras-Raf-MAPK, PI3K, and PLC-γsignaling pathways are stimulated. It appeared that they are independent, no cross talk was identified among them in BDNF-stimulated neurons, and MAPK signaling is the sole regulator for Arc transcription. In addition, [Ca2+]i regulates Arc transcription in parallel to MAPK. Furthermore, CaM may regulate Arc transcription through Ras-Raf-MAPK, but is independent of [Ca2+]i and not through CaMKs. B) Ca2+-mediated Arc transcription requires MAPK, PI3K, extracellular Ca2+, intracellular Ca2+, CaM, and CaMK activity. It appeared that PI3K and CaMK regulated Arc transcription through ERK. Although a transient influx of extracellular Ca2+ is required for ERK activation, [Ca2+]i is not required for ERK activation. Even with the activation of ERK, chelation of [Ca2+]i blocks Arc up-regulation. This implicates that ERK and [Ca2+]i are also parallel pathways in KCl-stimulated neurons. “?” indicates that the mechanism is unknown.
Fig. 2.

Regulation of Arc transcription by MAPK, PLC-γ and PI3K signaling. Cortical neurons were stimulated by 5ng/ml BDNF (A, C) or KCl (50mM) (B and D). Total RNA was extracted 1h after the treatments and used for semi-quantitative RT-PCR (A to B). Samples for Western blots (C, D) were collected 15min after the treatments. All inhibitors were applied 30min before BDNF and KCl stimulation. U73122 (5uM), LY294002 (30uM), and U0126 (10uM) were used to inhibit PLC-γ, PI3K and MEK, respectively. The levels of Arc and GAPDH mRNA were measured by RT-PCR using gene specific primers. Representative images are shown in the left panels, and quantification in the right panels. The relative intensity of Arc was normalized to GAPDH. C) The BDNF-induced ERK activation was inhibited by MEK inhibitor U0126, but not by PI3K inhibitor LY294002 or PLC-γ inhibitor U73122. The activation of ERK and PI3K was determined by the level of p-ERK and p-Akt, respectively. D) PI3K activity is required for the phosphorylation of ERK induced by membrane depolarization. Quantification of p-ERK and p-Akt was calculated from 3 independent experiments after normalization to T-ERK and T-Akt. Representative Western blot images are shown in the left panels, and quantification in the right panels.
Previous study revealed that PI3K activity is required for the activation of MAPK pathway (Chen et al. 2005). Thus we wondered whether the crosstalk between PI3K pathway and MAPK pathway occurs in BDNF-stimulated neurons. We found that BDNF activation of ERK and PI3K was mutually independent. Specifically, inhibition of MEK by U0126 only suppressed BDNF-induced ERK phosphorylation, but not the phosphorylation of Akt (a downstream target of PI3K). Similarly, inhibition of PI3K by LY294002 significantly suppressed p-Akt, but not pERK in BDNF-stimulated neurons (Fig. 2C). The discrepancy between our data and those of Chen et al. (Chen et al. 2005) may be due to different features of the stimuli. In deed, the Ca2+-mediated Arc transcription required both MAPK and PI3K activity (Fig. 2B). Either U0126 or LY294002 significantly blocked KCl-stimulated Arc up-regulation (Fig. 2B). Although the cross-talk between MAPK and PI3K was absent in BDNF-stimulated neurons (Fig. 7A), it appeared that PI3K regulated Arc transcription through impinging on ERK in KCl-stimulated neurons (Fig. 2D, and Fig. 7B). LY294002 blocked both p-Akt and p-ERK, whereas U0126 blocked only p-ERK but not p–Akt (Fig. 2D). These data implicated that the regulation of both signal transduction and Arc transcription is tailored to specific neuronal stimulations (Fig. 7).
Arc transcription is differentially regulated by extracellular and intracellular calcium in BDNF-stimulated neurons
Calcium plays an essential role in regulating activity-dependent transcription of immediate early genes (West et al. 2001) (Fig. 7). As shown in Fig. 2, significant up-regulation of Arc transcription was observed in neurons stimulated with KCl, presumably by evoking calcium influx through voltage-gated calcium channels (VGCC) and NMDA receptors. Consistently, APV (NMDAR antagonist, 100uM) and nifedipine (L-VGCC antagonist, 10uM) blocked KCl-mediated Arc transcription (Supplementary Fig. 3A). Furthermore, chelating either extracellular calcium (with EGTA) or intracellular calcium (with BAPTA-AM) blocked KCl/depolarization-induced Arc up-regulation (Fig. 3B).
Fig. 3.

BDNF-induced Arc transcription requires intracellular, but not extracellular calcium. Cortical neurons were stimulated by 5ng/ml BDNF (A) or KCl (50mM) (B) for 1hr, after which total RNA was extracted and used for semi-quantitative RT-PCR. Extracellular calcium chelator (EGTA, 2.5mM), and intracellular calcium chelator (BAPTA-AM, 33uM) were applied 30min before BDNF and KCl stimulation. The levels of Arc and GAPDH mRNA were determined by RT-PCR using gene specific primers. Representative images are shown in the left panels, and quantification in the right panels. The relative intensity of Arc was normalized to GAPDH.
Because the activation of Ras-MAPK signaling is sensitive to calcium, we hypothesized that extra- and intracellular calcium may regulate BDNF-mediated Arc transcription through ERK. Although blocking PLC-γ had no effects on ERK-dependent Arc transcription, BDNF-stimulated calcium influx might affect ERK activity and hence regulate Arc transcription. It has been demonstrated that BDNF could directly potentiate NMDA receptor activity, enhance glutamate release, and evoke calcium influx through both NMDA receptors and VGCC by TrkB-dependent opening of sodium channels (Nav 1.9) (Rose et al. 2004). To examine the function of NMDA receptors, we pre-treated neurons with APV (100uM) before BDNF stimulation. Similarly, the role of L-type VGCC was examined by pre-treating the neurons with antagonist nifedipine (10uM). Consistent with a previous finding (Yasuda et al. 2007), blocking NMDA receptors and L-VGCC did not affect BDNF-induced Arc up-regulation (Supplementary Fig. 3B). The BDNF-induced ERK phosphorylation was not blocked by APV and nifedipine either (Supplementary Fig. 3C). To totally rule out the function of extracellular calcium, we used calcium chelator EGTA (2.5mM). Pretreatment of neurons with EGTA did not block BDNF-mediated Arc up-regulation (Fig. 3A). In contrast, chelating intracellular calcium with BAPTA-AM (33uM) abolished BDNF-induced Arc transcription (Fig. 3A), indicating the functional relevance of intracellular, rather than extracellular calcium, in BDNF-stimulated signaling.
The activation of ERK does not depend on intracellular calcium
Because ERK belongs to Ca2+-stimulated protein kinases (Della Rocca et al. 1999; Improta-Brears et al. 1999), we reasoned that the effects of BAPTA-AM on Arc transcription might be mediated through the inhibition of ERK. However, neither BAPTA-AM nor EGTA inhibited BDNF-induced ERK phosphorylation (Fig. 4C and 4D). Furthermore, although the Ca2+-mediated ERK phosphorylation (in KCl-stimulated neurons) was abolished by EGTA, BAPTA-AM did not inhibit KCl-induced ERK phosphorylation, indicating that a transient influx of extracellular calcium is sufficient to activate ERK (Fig. 4A and B). Taken together, our data suggest that the [Ca2+]i and ERK signaling pathways in BDNF-stimulated neurons are parallel. [Ca2+]i regulation on Arc transcription is not mediated through ERK, and the activation of ERK alone is not sufficient for Arc transcription.
Fig. 4.

Activity-dependent ERK phosphorylation does not depend on intracellular calcium. Cortical neurons were stimulated by KCl (50mM) or 5ng/ml BDNF. Pre-treatments with EGTA (2.5mM) or BAPTA-AM (30uM) were applied 30min before stimulation. Samples were collected 15min after stimulation. The ERK phosphorylation was analyzed by Western blot (A and C) and immunofluorescent microscopy (B and D). For quantification, the relative intensity of p-ERK was normalized to total ERK. Representative Western blots are shown in the left panels, and quantification (n = 3 for each treatment group) is shown in the right panels.
Calmodulin (CaM) regulates BDNF-mediated Arc Transcription
To further investigate how intracellular calcium regulates BDNF-dependent Arc transcription, we examined the function of calmodulin (CaM), whose activity depends on calcium and regulates many aspects of neuroplasticity (Xia and Storm 2005). As shown in Fig. 5A, pre-treatment of neurons with a CaM antagonist W13 significantly inhibited the BDNF-induced Arc up-regulation. Because CaM-dependent kinase II and IV (CaMKII and CaMKIV) were implicated in regulating other immediate early genes (such as c-fos) (Finkbeiner et al. 1997; Ho et al. 2000; Wang et al. 2003), we pre-treated neurons with KN93 (an inhibitor of CaMKI, II, and IV) (Fig. 5A) before BDNF application. However, KN93 failed to block BDNF-induced Arc up-regulation (Fig. 5A), indicating that the regulatory function of CaM is independent of CaM KI, II and IV. Another CaMK inhibitor KN62 also failed to block BDNF-induced Arc up-regulation (data not shown).
Fig. 5.

The activity-dependent Arc transcription and ERK phosphorylation depend on CaM activity. DIV6 Cortical neurons were pre-incubated with a CaM inhibitor W-13 (70uM) or CaMK inhibitor KN93 (5uM) for 30min before neuronal stimulation with BDNF (5ng/ml) or KCl (50mM). The samples for total RNA extraction were collected 1hr after stimulation. The samples for Western blot were collected 15min after the stimulations. The mRNA level of Arc and GAPDH was determined by semi-quantitative RT-PCR (A and C). ERK phosphorylation was determined by Western blots using phospho-specific antibodies (B and D). Representative RT-PCR and Western blot results are shown in the left panels, and quantification in the right panels (n = 3 for each treatment group). The signals of Arc were normalized to GAPDH. The signal of p-ERK was normalized to T-ERK.
Although an earlier study by Quinn et al. (Quinn et al. 2002) demonstrated an inhibition effect of W13 on 5-HT-mediated p-ERK in PC 12 cells, how CaM modulates BDNF-mediated activation of Ras-Raf-MAPK is unknown. Because BAPTA-AM suppressed BDNF-mediated Arc up-regulation in an ERK-independent manner, we assumed that blocking CaM by W13 would not affect ERK phosphorylation. However, we found that W13 significantly blocked ERK phosphorylation in BDNF-stimulated neurons (Fig. 5B), indicating a Ca2+-independent function of CaM.
Our earlier results implicated that BDNF- and Ca2+-mediated Arc transcription was differentially regulated (Fig. 2, 3, and supplementary Fig. 3). Here, we further investigated how CaM and CaMKs regulated KCl-stimulated Arc transcription. Similarly, W13 blocked both Arc up-regulation and p-ERK in KCl-stimulated neurons (Fig. 5C and D). In contrast, KN93 significantly blocked KCl-induced Arc up-regulation (Fig. 5C). Although KN93 failed to inhibit BDNF-induced ERK phosphorylation, it significantly suppressed KCl-induced p-ERK (Fig. 5B and D). These data demonstrated that CaMKs might impinge on MAPK signaling and regulate Ca2+-mediated Arc transcription, and further implicated that the cross talk between CaMK and ERK is absent in BDNF-mediated signaling (Fig. 7).
Because BAPTA-AM and W13 suppressed both Ca2+-mediated and BDNF-mediated Arc transcription, we examined whether they are universal regulators. We stimulated cAMP pathway by treating neurons with adenylyl cyclase activator forskolin, and observed robust up-regulation of Arc transcription (Fig. 6A). Chelation of intracellular calcium by BAPTA-AM abolished cAMP-mediated Arc up-regulation (Fig. 6A). Interestingly, inhibition of CaM by W13 had no effects on Arc transcription in forskolin-stimulated neurons (Fig. 6A). Western blot analysis demonstrated that the forskolin-induced ERK phosphorylation was blocked by W13, but not by BAPTA-AM (Fig. 6B). It is surprising that the forskolin-induced Arc transcription does not require ERK activation, and the basal activity of ERK may be permissive for cAMP-mediated signaling.
Fig. 6.

Regulation of cAMP-mediated Arc transcription and ERK phosphorylation by intracellular Ca2+ and CaM. Cortical neurons were pre-treated with BAPTA-AM (33uM) or W13 (70uM) for 30min before forskolin (50uM) stimulation. Samples were collected 1h after stimulation for RT-PCR, and 15min after stimulation for Western blots. A). Forskolin-induced Arc up-regulation was blocked by BAPTA-AM, but not by W13. B). Forskolin-induced ERK phosphorylation was inhibited by W13, but not by BAPTA-AM. Representative images are shown in the left panels, and quantification in the right panels.
DISCUSSION
The up-regulation of Arc transcription is stimulated by neuronal activity both in vitro and in vivo (Guzowski et al. 1999; Link et al. 1995; Montag-Sallaz et al. 1999; Steward et al. 1998). Previous studies using PC12 cells and cultured neurons demonstrated ERK as the major regulator for Arc transcription (Waltereit et al. 2001). However, other neuronal regulators are not identified. It is also not clear whether ERK activation is sufficient or necessary. The present work took advantage of cultured primary neurons and thoroughly examined the role of the major signaling molecules. Our results revealed that the regulatory mechanisms are tailored to specific neuronal stimulations, such as BDNF vs. membrane depolarization. The activity of CaMK and PI3K may converge on ERK, and regulates Ca2+-mediated Arc transcription (Fig. 7B). In contrast, ERK, but not PI3K and CaMK, was required for BDNF-mediated Arc transcription (Fig. 7A). We further demonstrated that the intracellular calcium is a parallel pathway to ERK, and CaM may regulate both ERK phosphorylation and Arc transcription in a calcium independent manner.
It has been demonstrated that the primary cultured neurons are better representations than PC12 cells and other neuronal cell lines. Synaptic specialization and functional axons and dendrites are readily developed after several days of culturing. Moreover, HFS-induced LTP in cultured neurons shares many common features with LTP induced in brain slices and live brains (Lonart et al. 2003; Malenka and Bear 2004; Man et al. 2003; Tong et al. 1996). Compared to the in vivo situations, precise incubation time, stimulant/inhibitor concentration and penetration are easily controlled with primary neurons. We chose to study Arc up-regulation by BDNF for the following reasons. First, the activity-dependent BDNF release plays an obligatory role for the maintenance of late phase LTP. Secondly, infusion of BDNF into the dentate gyrus results in LTP without high frequency stimulation (Ying et al. 2002). Because gene transcription is required for LTP and memory formation, the BDNF-induced mRNA synthesis may be functionally relevant. Previous studies have demonstrated that the mRNA level of the plasticity-related gene Arc is rapidly elevated by BDNF stimulation in the dentate gyrus and in cultured neurons (Rao et al. 2006; Ying et al. 2002).
First, we found that robust Arc up-regulation was stimulated by BDNF at 5ng/ml, a much lower concentration than the dose (100ng/ml or 1mg/ml) used in the previous studies (Rao et al. 2006; Ying et al. 2002). The concentration of total BNDF is around 80ng/ml in the rat brain (Szapacs et al. 2004). Although it is difficult to determine the BDNF concentration at the synapses, 5ng/ml may be easier to achieve than 100ng/ml. In addition, we found that the BDNF function depended on its concentration. For example, 100ng/ml, but not 5ng/ml, BDNF attenuated glutamate-induced neuronal death (Zheng et al. 2008), suggesting that the effects of BDNF at high concentration may be more relevant in pathological situations.
Second, we examined the role of Ras/Raf/MAPK, PI3K and PLC-γ, which are the three major signaling pathways stimulated by BDNF-TrkB. Previous studies showed that Arc transcription was induced by calcium when neurons or PC12 cells were stimulated by KCl (Waltereit et al. 2001), HFS (Bramham 2007), and bicuculline (Rao et al. 2006). However, our results show that the PLC-γ-mediated calcium release from internal storage, if there is any, is not required for Arc up-regulation. We confirmed that ERK activity is required for both Ca2+- and BDNF-mediated Arc transcription. The role of PI3K is intriguing. Although blocking PI3K activity had no effects on BDNF-induced Arc transcription, PI3K activity is required for both Ca2+-induced Arc transcription and ERK phosphorylation, indicating the differential role of calcium and neurotrophins in synaptic plasticity. Similarly, the cross talk between CaMK and MAPK was stimulus-specific. KN93 blocked Ca2+-mediated, but not BDNF-mediate ERK phosphorylation and Arc transcription. It was well documented that CaMK and PI3K were required for HFS-induced LTP. It would be interesting to investigate whether CaMK and PI3K are required for BDNF-induced LTP (Ying et al. 2002).
Third, we examined the role of calcium, since BDNF might elevate [Ca2+]i through routes independent of PLC-γ (Rose et al. 2004; Schinder and Poo 2000). For example, it was demonstrated that BDNF might promote calcium influx through NMDA receptors and VGCC. BDNF also promotes pre-synaptic glutamate release (Schinder and Poo 2000). However, our data demonstrated that blocking NMDA receptor, L-VGCC and chelating extra-cellular calcium had no effects on BDNF-induced Arc transcription. In contrast, [Ca2+]i is required, because pre-treatment with BAPTA-AM abolished Arc up-regulation. To our surprise, we did not detect any measurable elevation of [Ca2+]i in BDNF-stimulated neurons (Zheng et al. 2008). Although it is generally agreed that neurotrophins induce mild PLC-γ-dependent calcium release from the internal storage, another study demonstrated no significant increase of [Ca2+]i in the rat visual cortex slices and cultured neurons when stimulated by BDNF (Pizzorusso et al. 2000). The discrepancy may be due to the types of neurons involved (hippocampus vs. cortex), culturing conditions (DMEM vs. Neurobasal as media), BDNF concentration (5ng/ml vs. 100ng/ml), and the methods of calcium imaging (fura-2 vs. fluo-3) (Numakawa et al. 2002; Pizzorusso et al. 2000; Yagasaki et al. 2006). Because the [Ca2+]i remained unchanged and blocking PLC-γ had no effects, our data suggest that the basal level of [Ca2+]i is required for gating the BDNF-induced Arc transcription. Interestingly, BAPTA-AM did not block c-fos expression in BDNF-stimulated neurons in the visual cortex (Pizzorusso et al. 2000), implicating that the induction of IEGs might be differentially regulated.
Another interesting result was that BAPTA-AM had no effects on either Ca2+-mediated or BDNF-mediated ERK phosphorylation. This suggests that intracellular calcium and MAPK activation is un-coupled, and the activation of MAPK alone is not sufficient to induce Arc transcription. We also assume that blocking ERK has no effects on [Ca2+]i level, because p-Akt was suppressed by BAPTA-AM but not by U0126 in BDNF-stimulated neurons (Zheng et al. 2008). If U0126 decreased [Ca2+]i, then it would have suppressed p-Akt. These data suggest that regulatory pathways of intracellular calcium and ERK are parallel and independent of each other.
How does [Ca2+]i regulate Arc expression? It was demonstrated that BDNF stimulated the transcription of IEG c-fos through both MAPK and CaMK IV pathway (Finkbeiner et al. 1997). However, the application of CaMK inhibitor KN93 or KN62, which inhibit the activity of CaMK I, II and IV, failed to suppress the induction of Arc in BDNF-stimulated neurons. In contrast, the CaM antagonist W13 remarkably blocked BDNF-induced Arc up-regulation. It would be interesting to identify the functional molecules downstream of CaM. Intriguingly, our results demonstrated that CaM was not a downstream target of intracellular calcium in the BDNF-stimulated neurons. While BAPTA-AM had no suppression on ERK phosphorylation, W13 significantly blocked ERK phosphorylation. CaM itself may be directly required for the activity of the transcription factors responsible for Arc expression in a calcium-independent manner. Such possibility was suggested by the crucial role of CaM binding to estrogen receptors (ER). The transcriptional activity of ER-alpha was lost when CaM binding was disrupted (Li et al. 2005). We also found that the regulatory function of CaM depended on the types of stimulation. Although W13 inhibited ERK phosphorylation, it did not block Arc transcription in forskolin-stimulated neurons. It was reported that inhibition of MEK by PD098059 blocked forskolin-induced Arc transcription (Waltereit et al. 2001), our results implicated that ERK activation may not be not necessary for cAMP-mediated Arc transcription (Fig. 6).
In summary, our results demonstrated an interesting interaction between intracellular calcium and BDNF, the two important signaling molecules in the central nervous system. We suggest novel mechanisms for the BDNF-dependent activation of Arc transcription. Certain aspects of neurotrophin-mediated synaptic plasticity may be regulated, in parallel, by basal [Ca2+]i and the activation of ERK. In addition to pharmacological interventions, these regulatory pathways will be examined by genetic approaches in the future.
Supplementary Material
Supplementary Fig. 1. Treatment of cortical neurons with 1ng/ml BDNF stimulate significant transcription up-regulation of Arc. Cortical neurons were treated with 0.2, 0.5, 1, 2, and 5 (in ng/ml) BDNF. Total RNA was extracted 1hr after stimulation for RT-PCR analysis (A). Samples were collected for Western blots 15min after treatment (B).
Supplementary Fig. 2. BDNF-mediated Arc transcription depends on the TrkB tyrosine kinase receptor. Trk inhibitor K252a (0.2 uM) or TrkB-specific BDNF scavenger TrkB-IgG (0.4 ug/ml) were applied 30min before BDNF (5ng/ml) incubation. One hour after BDNF treatment, samples were collected, and RT-PCR products for Arc and GAPDH were synthesized from total RNA.
Supplementary Fig. 3. BDNF-induced Arc transcription and ERK phosphorylation does not require NMDAR and L-VGCC. Cortical neurons were stimulated by KCl (50mM) or 5ng/ml BDNF. Total RNA was extracted 1 h after the stimulation, and used for semi-quantitative RT-PCR (A and B). Protein samples for Western blot (C) were harvested 15min after the stimulation. NMDA receptor antagonist (APV, 100uM) and L-VGCC antagonist (nifedipine, 10uM) were applied 30min before KCl or BDNF stimulation. The levels of Arc and GAPDH mRNA were determined by RT-PCR using gene specific primers. ERK phosphorylation was determined by Western blot using anti-phospho-ERK antibody. Representative images are shown in the left panels, and quantification in the right panels.
Acknowledgments
We thank Dr. Xianju Zhou for the help with the neuronal cultures. This study is supported by NIH grant (MH076906 to H.W.).
References
- Bramham CR. Control of synaptic consolidation in the dentate gyrus: mechanisms, functions, and therapeutic implications. Prog Brain Res. 2007;163:453–471. doi: 10.1016/S0079-6123(07)63025-8. [DOI] [PubMed] [Google Scholar]
- Chan GC, Hinds TR, Impey S, Storm DR. Hippocampal neurotoxicity of Delta9-tetrahydrocannabinol. J Neurosci. 1998;18(14):5322–5332. doi: 10.1523/JNEUROSCI.18-14-05322.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8(7):925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52(3):445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Rocca GJ, Mukhin YV, Garnovskaya MN, Daaka Y, Clark GJ, Luttrell LM, Lefkowitz RJ, Raymond JR. Serotonin 5-HT1A receptor-mediated Erk activation requires calcium/calmodulin-dependent receptor endocytosis. J Biol Chem. 1999;274(8):4749–4753. doi: 10.1074/jbc.274.8.4749. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19(5):1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263(5153):1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20(11):3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2(12):1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3(6):533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci. 2000;20(17):6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci U S A. 1999;96(8):4686–4691. doi: 10.1073/pnas.96.8.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3(4):323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401(6756):918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92(19):8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li Z, Sacks DB. The transcriptional activity of estrogen receptor-alpha is dependent on Ca2+/calmodulin. J Biol Chem. 2005;280(13):13097–13104. doi: 10.1074/jbc.M410642200. [DOI] [PubMed] [Google Scholar]
- Link W, Konietzko U, Kauselmann G, Krug M, Schwanke B, Frey U, Kuhl D. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc Natl Acad Sci U S A. 1995;92(12):5734–5738. doi: 10.1073/pnas.92.12.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9(12):2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115(1):49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6(8):603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14(2):433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Ma YL, Wang HL, Wu HC, Wei CL, Lee EH. Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience. 1998;82(4):957–967. doi: 10.1016/s0306-4522(97)00325-4. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D’Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38(4):611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Montag-Sallaz M, Montag D. Learning-induced arg 3.1/arc mRNA expression in the mouse brain. Learn Mem. 2003;10(2):99–107. doi: 10.1101/lm.53403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag-Sallaz M, Welzl H, Kuhl D, Montag D, Schachner M. Novelty-induced increased expression of immediate-early genes c-fos and arg 3.1 in the mouse brain. J Neurobiol. 1999;38(2):234–246. [PubMed] [Google Scholar]
- Mu JS, Li WP, Yao ZB, Zhou XF. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res. 1999;835(2):259–265. doi: 10.1016/s0006-8993(99)01592-9. [DOI] [PubMed] [Google Scholar]
- Nakata H, Nakamura S. Brain-derived neurotrophic factor regulates AMPA receptor trafficking to post-synaptic densities via IP3R and TRPC calcium signaling. FEBS Lett. 2007;581(10):2047–2054. doi: 10.1016/j.febslet.2007.04.041. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Yamagishi S, Adachi N, Matsumoto T, Yokomaku D, Yamada M, Hatanaka H. Brain-derived neurotrophic factor-induced potentiation of Ca(2+) oscillations in developing cortical neurons. J Biol Chem. 2002;277(8):6520–6529. doi: 10.1074/jbc.M109139200. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9(6):1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Ratto GM, Putignano E, Maffei L. Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. J Neurosci. 2000;20(8):2809–2816. doi: 10.1523/JNEUROSCI.20-08-02809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, Kobalz U, Stawrakakis A, Fernandez E, Waltereit R, Bick-Sander A, Therstappen E, Cooke SF, Blanquet V, Wurst W, Salmen B, Bosl MR, Lipp HP, Grant SG, Bliss TV, Wolfer DP, Kuhl D. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron. 2006;52(3):437–444. doi: 10.1016/j.neuron.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2(1):24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Quinn JC, Johnson-Farley NN, Yoon J, Cowen DS. Activation of extracellular-regulated kinase by 5-hydroxytryptamine(2A) receptors in PC12 cells is protein kinase C-independent and requires calmodulin and tyrosine kinases. J Pharmacol Exp Ther. 2002;303(2):746–752. doi: 10.1124/jpet.102.038083. [DOI] [PubMed] [Google Scholar]
- Rao VR, Pintchovski SA, Chin J, Peebles CL, Mitra S, Finkbeiner S. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9(7):887–895. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361(1473):1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Blum R, Kafitz KW, Kovalchuk Y, Konnerth A. From modulator to mediator: rapid effects of BDNF on ion channels. Bioessays. 2004;26(11):1185–1194. doi: 10.1002/bies.20118. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Poo M. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci. 2000;23(12):639–645. doi: 10.1016/s0166-2236(00)01672-6. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21(4):741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Szapacs ME, Mathews TA, Tessarollo L, Ernest Lyons W, Mamounas LA, Andrews AM. Exploring the relationship between serotonin and brain-derived neurotrophic factor: analysis of BDNF protein and extraneuronal 5-HT in mice with reduced serotonin transporter or BDNF expression. J Neurosci Methods. 2004;140(1–2):81–92. doi: 10.1016/j.jneumeth.2004.03.026. [DOI] [PubMed] [Google Scholar]
- Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33(3):383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16(6):1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- Waltereit R, Dammermann B, Wulff P, Scafidi J, Staubli U, Kauselmann G, Bundman M, Kuhl D. Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen-activated protein kinase/extracellular regulated kinase activation. J Neurosci. 2001;21(15):5484–5493. doi: 10.1523/JNEUROSCI.21-15-05484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mishra R, Simonson MS. Ca2+/calmodulin-dependent protein kinase II stimulates c-fos transcription and DNA synthesis by a Src-based mechanism in glomerular mesangial cells. J Am Soc Nephrol. 2003;14(1):28–36. doi: 10.1097/01.asn.0000043180.18456.47. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98(20):11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6(4):267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Yagasaki Y, Numakawa T, Kumamaru E, Hayashi T, Su TP, Kunugi H. Chronic antidepressants potentiate via sigma-1 receptors the brain-derived neurotrophic factor-induced signaling for glutamate release. J Biol Chem. 2006;281(18):12941–12949. doi: 10.1074/jbc.M508157200. [DOI] [PubMed] [Google Scholar]
- Yasuda M, Fukuchi M, Tabuchi A, Kawahara M, Tsuneki H, Azuma Y, Chiba Y, Tsuda M. Robust stimulation of TrkB induces delayed increases in BDNF and Arc mRNA expressions in cultured rat cortical neurons via distinct mechanisms. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04851.x. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22(5):1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng F, Soellner D, Nunez J, Wang H. The basal level of intracellular calcium gates the activation of phosphoinositide 3-kinase -Akt signaling by brain-derived neurotrophic factor in cortical neurons. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05478.x. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Treatment of cortical neurons with 1ng/ml BDNF stimulate significant transcription up-regulation of Arc. Cortical neurons were treated with 0.2, 0.5, 1, 2, and 5 (in ng/ml) BDNF. Total RNA was extracted 1hr after stimulation for RT-PCR analysis (A). Samples were collected for Western blots 15min after treatment (B).
Supplementary Fig. 2. BDNF-mediated Arc transcription depends on the TrkB tyrosine kinase receptor. Trk inhibitor K252a (0.2 uM) or TrkB-specific BDNF scavenger TrkB-IgG (0.4 ug/ml) were applied 30min before BDNF (5ng/ml) incubation. One hour after BDNF treatment, samples were collected, and RT-PCR products for Arc and GAPDH were synthesized from total RNA.
Supplementary Fig. 3. BDNF-induced Arc transcription and ERK phosphorylation does not require NMDAR and L-VGCC. Cortical neurons were stimulated by KCl (50mM) or 5ng/ml BDNF. Total RNA was extracted 1 h after the stimulation, and used for semi-quantitative RT-PCR (A and B). Protein samples for Western blot (C) were harvested 15min after the stimulation. NMDA receptor antagonist (APV, 100uM) and L-VGCC antagonist (nifedipine, 10uM) were applied 30min before KCl or BDNF stimulation. The levels of Arc and GAPDH mRNA were determined by RT-PCR using gene specific primers. ERK phosphorylation was determined by Western blot using anti-phospho-ERK antibody. Representative images are shown in the left panels, and quantification in the right panels.