

The structure of the effector domain of the influenza protein NS1, a validated antiviral drug target, has been solved in two space groups.
Keywords: influenza NS1, effector domain, dimer formation, protein–protein interactions
Abstract
The nonstructural protein NS1A from influenza virus is a multifunctional virulence factor and a potent inhibitor of host immunity. It has two functional domains: an N-terminal 73-amino-acid RNA-binding domain and a C-terminal effector domain. Here, the crystallographic structure of the NS1A effector domain of influenza A/Udorn/72 virus is presented. Structure comparison with the NS1 effector domain from mouse-adapted influenza A/Puerto Rico/8/34 (PR8) virus strain reveals a similar monomer conformation but a different dimer interface. Further analysis and evaluation shows that the dimer interface observed in the structure of the PR8 NS1 effector domain is likely to be a crystallographic packing effect. A hypothetical model of the intact NS1 dimer is presented.
1. Introduction
Influenza viruses are the causative agents of an infectious and debilitating disease commonly referred to as the flu. The virus has a complex replicative cycle (Lamb & Krug, 2001 ▶) and undergoes rapid evolutionary divergence (Webby & Webster, 2001 ▶). In its most aggressive form, the virus was responsible for one of the most devastating pandemics in human history: the notorious Spanish flu caused 20–40 million deaths worldwide in the early twentieth century. In 1997, a deadly influenza A virus strain H5N1 was transmitted from birds to humans in Hong Kong, resulting in six deaths among 18 infected persons. This H5N1 subtype also caused disease outbreaks in poultry in Asia, Europe and Africa during late 2003 and early 2004. The H5N1-subtype influenza A viruses have the potential to cause the next pandemic if they acquire an efficient ability to spread easily among human beings. There is currently great interest in defining influenza targets for the design of efficacious antiviral agents that can complement ongoing efforts towards vaccine development.
The genome of influenza A virus is composed of eight segments of single-stranded RNA. Segment 8 encodes a 26 kDa protein called NS1. NS1 is designated as a nonstructural protein because it is synthesized in infected cells but is not incorporated into virions; this makes it less subject to selective pressure than virus-coat proteins such as the hemagglutinin and neuraminidase proteins that give rise to the influenza serotype names. It has been proven that the NS1A protein can be targeted for the development of new antiviral drugs (Min & Krug, 2006 ▶; Twu et al., 2006 ▶).
The NS1 protein is a multifunctional protein that participates in both protein–protein and protein–RNA interactions (Krug et al., 2003 ▶). NS1 has two functional domains: the N-terminal 73-amino-acid RNA-binding domain (RBD) and the C-terminal effector domain. The RBD binds nonspecifically to double-stranded RNA (dsRNA) and protects the virus against the antiviral state induced by interferon α/β. It was subsequently discovered that this protection is achieved by blocking the activation of the 2′-5′-oligo(A) synthetase/RNase L pathway (Min & Krug, 2006 ▶). The RBD also overlaps the retinoic acid-inducible gene I (RIG-I) binding domain (Mibayashi et al., 2007 ▶). The RIG-I protein functions as an intracellular viral sensor which can be activated by influenza virus ssRNA. The binding of RIG-I to RBD of NS1 inhibits the viral detection ability of the cell.
The C-terminal effector domain has been found to interact with at least four different proteins: elongation initiation factor 4GI (eIF4GI), protein kinase R (PKR), poly(A)-binding protein II (PAB II) and the cleavage and 30 kDa subunit of the polyadenylation specificity factor (CPSF30). When the effector domain binds to CPSF30, it inhibits the maturation and export of host cellular antiviral mRNAs (Noah et al., 2003 ▶). The second and third zinc fingers (F2F3) of CPSF30 have been shown to mediate the binding of NS1A (Twu et al., 2006 ▶). The validity of the NS1 effector domain as a potential drug target has been attested to by a mutational analysis. NS1 mutations spanning the CPSF30-binding site (residues 184–188) reduce CPSF30 binding. Recombinant virus expressing these mutations induced a high level of host-cell interferon β (INF-β), while viral replication was attenuated 1000-fold (Noah et al., 2003 ▶). In addition, an engineered MDCK cell line which constitutively expresses epitope-tagged F2F3 in the nucleus effectively blocked the binding of endogenous CPSF30 to NS1A and thereby selectively inhibited influenza virus A replication (Twu et al., 2006 ▶). Taken together, these experiments suggest that the CPSF30 binding site can be targeted for the development of new antiviral drugs.
Although the structure of full-length NS1A is not available, the structures of the two functional domains have been solved independently. NMR (Chien et al., 1997 ▶) and X-ray structures (Liu et al., 1997 ▶) of the 73-residue N-terminal domain of NS1A have been reported. They show that the protein is active as a dimer. The structural analysis revealed a unique six-helix structure for the dimer, which differs from that of the predominant class of dsRNA-binding proteins. The dimeric protein binds dsRNA as part of its function in the viral cycle.
The X-ray structure of the NS1 effector domain (residues 79–205) of the mouse-adapted influenza A/Puerto Rico/8/34 (PR8) virus strain has been solved recently (Bornholdt & Prasad, 2006 ▶). Like the N-terminal domain, the effector domain forms a dimer in solution. Each monomer consists of seven β-strands and three α-helices. The structure is believed to be a novel fold, which can be described as an α-helix β-crescent fold, as the β-strands form a crescent-like shape about the central α-helix. The NS1A from the PR8 virus strain does not bind CPSF30. It is thought that this arises from two mutations (F103S and M106I) in the F2F3-recognition site. As the CPSF30-binding site is crucial for virus replication and a proven target for new antiviral drug development, it is important to analyze the structure of an NS1 effector domain that does interact with CPSF30 (Twu et al., 2006 ▶). Recently, the X-ray structure of a complex between the influenza A/Udorn/72 NS1A effector domain and the F2F3 domain of CPSF30 has been published (Das et al., 2008 ▶). The key observation is that aromatic side chains from the F3 zinc finger (Phe97, Phe98 and Phe102) bind into a hydrophobic pocket in the effector domain. Here, we report the crystal structure of the 126-residue C-terminal effector domain (residues 79–205) of NS1A from the influenza A/Udorn/72 virus strain at 2.3 Å resolution and compare it with the structure of the PR8 virus strain NS1 effector domain, which has 89% sequence identity.
2. Materials and methods
2.1. Purification and expression
The gene expressing the influenza A/Udorn/72 NS1A effector domain, a polypeptide consisting of 126 amino acids (residues 79–205), was cloned by first amplifying the DNA with primers GACGACGACAAGATGACCATGGCCTCCACACC and GAGGAGAAGCCCGGTTTAGCTTCCCCAAGCGAATC. The product was cloned into pET-46 Ek/LIC (Novagen). The integrity of the cloned DNA was confirmed by DNA sequencing. Escherichia coli strain Rosetta 2 (DE3) (Novagen) containing the plasmid was grown overnight in broth medium [1%(w/v) tryptone, 0.5%(w/v) yeast extract, 0.5%(w/v) NaCl] containing ampicillin (100 µg ml−1) and chloramphenicol (34 µg ml−1) at 310 K. The cells were then diluted 1:100 in 2 l medium and grown to a cell density of approximately 4 × 108 per millilitre. IPTG was added to 1 mM and the cells were grown overnight at 298 K. The protein, residues 79–205 of the influenza A/Udorn/72 NS1A with a 14-residue His tag (MAHHHHHHVDDDDK) attached at the N-terminal end, was purified essentially according to a procedure developed by Novagen. Briefly, cells were collected by centrifugation, resuspended in 25 ml column buffer (CB; 50 mM HEPES pH 8.0, 300 mM NaCl, 20 mM imidazole) and disrupted in a French pressure cell. Cellular debris was pelleted by centrifugation at 5000g for 60 min at 277 K. The supernatant was applied onto a column containing Ni–NTA His-binding beads (2 ml bed volume, equilibrated with CB), washed with 100 ml CB and then eluted with 10 ml elution buffer (EB; 50 mM HEPES pH 8.0, 300 mM NaCl, 250 mM imidazole). The protein was concentrated to 1–2 ml using an Amicon bioseparator fitted with a 45 mm YM-3 membrane and applying 414–483 kPa N2 at 277 K. The concentrated sample then underwent size-exclusion chromatography with a 50 mM HEPES pH 8.0, 100 mM NaCl buffer.
2.2. Crystallization
One crystal form (I) of the influenza A/Udorn/72 NS1A effector domain was grown at room temperature using the batch method by mixing 5 µl 10 mg ml−1 NS1A effector domain in 50 mM HEPES pH 8.0, 0.1 M NaCl with 5 µl 20%(w/v) PEG 400, 0.1 M sodium acetate pH 5.5, 0.1 M MgSO4. This condition was identified from screens carried out in the high-throughput crystallization screening laboratory at the Hauptman–Woodward Medical Research Institute (Luft et al., 2003 ▶). A second crystal form (II) was grown at 277 K using the sitting-drop method. 5 µl protein solution (10 mg ml−1 in 50 mM HEPES pH 8.0, 0.1 M NaCl) was mixed with 5 µl reservoir solution and equilibrated against a reservoir containing 800 µl 12%(w/v) PEG 3350 and 4%(v/v) Tacsimate pH 6.0. This condition was found using the Hampton Research PEG/Ion HT Screen kit.
2.3. Data collection and processing
Crystal form I of the influenza A/Udorn/72 NS1A effector domain was cryoprotected by dipping it into an artificial mother liquor containing 30% PEG 400. Crystal form II was similarly cryoprotected with an artificial mother liquor containing 20% PEG 3350. Crystals mounted in a cryoloop (Hampton Research) were flash-frozen by dipping them into liquid nitrogen and were placed in the cold stream on the goniostat. Diffraction data were collected at 100 K on a MAR 345 image-plate detector (MAR Research) with X-rays generated by a Rigaku Micromax007 rotating-anode generator (Rigaku, The Woodlands, Texas, USA) operated at 40 mV and 30 mA. Diffraction data were collected using a crystal-to-detector distance of 200 mm. Diffraction data were integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶). Crystal form I (grown at pH 5.5 and room temperature) was determined to belong to space group P212121, with unit-cell parameters a = 47.9, b = 61.5, c = 132.1 Å. The second crystal form, grown at pH 6.0 and 277 K, belonged to space group C2221, with unit-cell parameters a = 62.8, b = 74.0, c = 121.9 Å.
2.4. Structure determination and refinement
Molecular-replacement solutions for both crystal forms were obtained using the influenza A/Puerto Rico/8/34 NS1 protein as a model (PDB code 2gx9; Bornholdt & Prasad, 2006 ▶); the searches were conducted using the program MOLREP (Vagin & Teplyakov, 1997 ▶). Rigid-body refinement of the molecular-replacement solutions was performed using CNS (Brünger et al., 1998 ▶). Subsequent refinement of atomic positions and isotropic B factors was performed using CNS and REFMAC (Collaborative Computational Project, Number 4, 1994 ▶; Murshudov et al., 1997 ▶). Molecular visualization and rebuilding were performed using Coot (Emsley & Cowtan, 2004 ▶). Water and sulfate molecules were identified using Coot based on an F o − F c difference map. 5% of the diffraction data were set aside throughout refinement for cross-validation (Brünger, 1993 ▶). PROCHECK was used to produce the Ramachandran plots (Laskowski et al., 1993 ▶). Model pictures were produced using PyMOL (DeLano Scientific, San Carlos, California, USA).
3. Results and discussion
3.1. X-ray structure determination
Recombinant His-tagged influenza A/Udorn/72 NS1A effector domain (residues 79–205) was expressed in good yield from E. coli (∼10 mg per litre of cell culture). A screen of crystallization conditions produced two useful forms. The best crystals of the NS1A effector domain grew at room temperature at pH 5.5 and were found to belong to space group P212121, with unit-cell parameters a = 47.9, b = 61.5, c = 132.1 Å. In this crystal form there are two molecules per asymmetric unit, giving a V M value of 3.04 Å3 Da−1 (Matthews, 1968 ▶). A second useful form grew at 277 K at pH 6.0. These crystals belonged to space group C2221, with unit-cell parameters a = 62.8, b = 74.0, c = 121.9 Å. These crystals also contained two molecules in the asymmetric unit, giving a V M value of 2.21 Å3 Da−1. X-ray data and refinement statistics for the two structures are shown in Table 1 ▶. A section of the final 2F o − F c electron-density map used for model construction from the pH 5.5 crystal is shown in Fig. 1 ▶.
Table 1. Data-collection and model-refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | ||
| Wavelength () | 1.5418 | 1.5418 |
| Space group | P212121 | C2221 |
| Unit-cell parameters () | a = 47.92, b = 61.46, c = 132.13 | a = 62.82, b = 74.03, c = 121.88 |
| Resolution () | 302.3 (2.382.30) | 302.8 (2.902.80) |
| R merge (%)† | 6.8 (26.3) | 6.1 (14.6) |
| I/(I) | 11.8 (2.7) | 16.0 (4.8) |
| Completeness | 98.1 (83.5) | 96.8 (79.0) |
| Redundancy | 6.4 (4.1) | 7.4 (3.1) |
| Refinement | ||
| No. of reflections | 17766 | 7261 |
| No. of protein atoms | 1909 | 1923 |
| No. of solvent atoms | 136 | 20 |
| No. of sulfate atoms | 10 | 0 |
| R working | 0.197 (0.234) | 0.197 (0.235) |
| R free | 0.232 (0.287) | 0.284 (0.316) |
| Average B factor for protein atoms (2) | 35.4 | 38.8 |
| R.m.s deviations from ideality | ||
| Bond lengths () | 0.011 | 0.014 |
| Bond angles () | 1.236 | 1.482 |
R
merge = 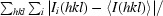
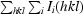 .
.
Figure 1.

Electron density for the influenza A/Udorn/72 NS1A effector domain. This is a section of a 2F o − F c map contoured at 1σ to show the interface of the effector-domain dimer.
Following refinement, a Ramachandran plot of the pH 5.5 crystal structure (space group P212121) shows 87.1% of residues to be in the most favorable region and 12.9% to be in additionally allowed space. The refined structure includes two sulfate ions and 136 solvent molecules. The pH 6.0 crystal structure (space group C2221) gives a Ramachandran plot with 87.3% of residues in the most favorable region and 12.7% in additionally allowed space. This refined structure includes 20 solvent molecules.
3.2. Structure of the influenza A/Udorn/72 NS1A effector domain
The structure of the influenza A/Udorn/72 NS1A effector domain shows an α-helix β-crescent fold that is generally similar to that of the PR8 NS1 effector domain (Bornholdt & Prasad, 2006 ▶) and that of the recently solved NS1A effector domain from an avian influenza virus (Hale et al., 2008 ▶). A ribbon drawing of the influenza A/Udorn/72 NS1A effector domain is shown in Fig. 2 ▶. There are seven β-strands and three α-helices in each monomer subunit. Six of the β-strands surround a long central α-helix and make an extensive network of hydrophobic interactions with it. It is these interactions around one side of the helix that give rise to the rough crescent shape.
Figure 2.

Ribbon drawing of the effector domain of influenza A/Udorn/72 NS1A. α-Helices and β-strands are labeled and the side chain of Trp187 is shown in order to mark the binding site for CPSF30.
As shown in Fig. 2 ▶, the first β-strand (a) lies on the convex side of the crescent and is connected by a short helix A to strand b. Five β-strands (b, c, d, e and f) then sequentially connect to each other to form an antiparallel twisted β-sheet that surrounds the central helix B. Helix B is the longest α-helix of the structure; it connects strand f to strand g, which lies adjacent and antiparallel to strand d. Strand g is followed by the last helix C, which lies at the the sharp end of the crescent. One sulfate ion is observed in each monomer and forms hydrogen bonds to the side chains of Gln199 and Arg193.
The influenza A/Udorn/72 NS1A effector domain and the PR8 NS1 effector domain share 89% sequence identity and consequently the overall folding of the two structures is quite similar. Superposition of the two structures, shown in Fig. 3 ▶, gives an r.m.s. distance between 81 equivalent Cα atoms of 0.29 Å. The most obvious differences between the two structures lie at the N-terminus and in two loop regions. Ser87 is near the N-terminus and marks that region; residues 135–143 (labeled Phe138) are shifted up to 5 Å towards strands a and d. The second loop region, residues 163–168 (labeled Pro167), is shifted towards strand b by ∼3 Å in the influenza A/Udorn/72 NS1A structure. Trp187, which is crucial to effector-domain function, is shown for reference.
Figure 3.

Superposition of the influenza A/Udorn/72 NS1A effector domain and the PR8 NS1 effector domain (PDB code 2gx9; Bornholdt & Prasad, 2006 ▶). The Cα trace of the influenza A/Udorn/72 NS1A effector domain is shown in blue bonds and that of the PR8 NS1 effector domain is shown in red bonds. The side chain of Trp187, indicating the area of the effector domain-binding pocket, is also shown.
3.3. Effector-domain dimerization
The NS1 effector domain behaves as a dimer in solution based on its chromatographic properties (Nemeroff et al., 1995 ▶). Not surprisingly, the asymmetric unit in both of our crystal forms of the influenza A/Udorn/72 NS1A is a dimer. In both of our crystal structures, the main stabilizing interaction for noncrystallographic dimer formation appears to be the pseudo-symmetrical insertion of the Trp187 indole ring into the hydrophobic F2F3-binding pocket of its dimer partner; the dimer interface is shown in Fig. 4 ▶. Fig. 4 ▶(a) shows a space-filling model of a monomer subunit, with the Trp187 side chain of the other subunit fitting into it; Fig. 4 ▶(b) shows further details of the interactions of the buried Trp187 side chain with the hydrophobic binding pocket. The pocket is formed by main-chain residues Gly183–Asn188 together with the side chains of residues Lys108, Lys110, Ile117, Gln121 and Val180. The hydrophobic pocket is located at the base of the central long helix, with the side chain of Trp187 pointing outwards from the periphery. The recently obtained X-ray structure of the influenza A/Udorn/72 NS1A effector domain bound to F2F3 (Das et al., 2008 ▶) shows that this hydrophobic pocket is indeed the CPSF30-binding pocket. This is consistent with the observation that residues Gly184–Asn188 are crucial for binding to the CPSF30 subunit and are highly conserved among human influenza A viruses, including the influenza A/Hongkong/493/1997 virus and the influenza A/Vietnam/1203/2004 virus (Twu et al., 2007 ▶).
Figure 4.

The dimer interface for the influenza A/Udorn/72 NS1A effector domain. (a) A space-filling model of monomer subunit A reveals a distinct largely hydrophobic pocket that has evolved to bind aromatic residues from the F2F3 domain of CPSF30. Here, Trp187 from effector domain monomer subunit B (shown in blue) binds in that pocket. (b) A detailed view of the interactions of Trp187 with monomer subunit A binding-cleft residues (shown in green).
The X-ray structure of an avian virus NS1 effector domain (Hale et al., 2008 ▶) also exhibited a noncrystallographic dimer very similar to that seen in our two influenza A/Udorn/72 NS1A structures. The authors also realised the importance of the interaction of Trp187 with the CPSF30-binding site. They mutated three residues near the dimeric interface to alanines, M106A, Q121A and W187A, but only the tryptophan alteration caused the protein to act as a monomer in solution.
The PR8 NS1 effector domain also forms a noncrystallographic dimer and has the same kind of interface as described here (Bornholdt & Prasad, 2006 ▶). The authors of this study focused their attention not on the Trp187 contact but on the formation of a segment of antiparallel β-sheet between the N-terminal strands of the neighboring molecules. This β-sheet pairing is not seen in either of our structures nor in the avian virus structure and appears to result from adventitious crystal-packing forces that are unique to the PR8 NS1 crystal. The pseudo-dimeric interaction of Trp187 with the F2F3-binding site is common to all four structures and is clearly the main interaction driving effector-domain dimerization, as confirmed by the recent mutagenic studies.
Fig. 5 ▶ shows the relative orientations of the noncrystallographic dimers from our two crystal forms of the influenza A/Udorn/72 NS1A effector domain. One monomer subunit from each structure superimposes in a least-squares sense with an r.m.s. deviation of 0.28 Å for Cα atoms. In each case its dimeric partner makes interactions similar to those described above, burying the Trp187 side chain in the neighboring binding site (the side chains are shown and labeled). However, subtle differences at the interface generate rather large differences for remote regions of the molecule. To superpose the second monomer subunit from the two systems requires a rotation of 44°. The central B helices of the monomers are shown as cartoons in Fig. 5 ▶ in order to facilitate the comparison. The orientation of the PR8 NS1 effector-domain dimer is similar but not identical to that of the space group C2221 influenza A/Udorn/72 NS1A crystal (red in Fig. 5 ▶). Again, the dimer interface is basically the same in all the crystal structures; however, subtle differences at the interface still allow larger differences in the crystal packing of the entire dimer.
Figure 5.

Superposition of the influenza A/Udorn/72 NS1A effector domain (pH 5.5 structure; blue) and the PR8 NS1 effector domain (red).
In discussing the F2F3-binding site of the NS1 effector domain, it is interesting to note that the pocket residues Lys108, Lys110, Ile117, Gln121, Val180 and Gly183–Trp187 are conserved among almost all influenza A viruses, including those having an intrinsic defect in binding CPSF30. It appears that proteins such as PR8 NS1 that are defective in CPSF30 binding lack the consensus amino acids Phe103 and Met106. The recent structure of the influenza A/Udorn/72 NS1A effector domain with the F2F3 fragment of CPSF30 bound reveals that Phe103 and Met106 are not part of the CPSF30-binding pocket, but are involved in intermolecular interactions that stabilize the complex at a site remote from the hydrophobic pocket. Phe102 and Met106 are not involved in the dimerization of the influenza A/Udorn/72 NS1A effector domain either, as shown in Fig. 4 ▶(b).
3.4. Model of intact NS1
As described in §1, NS1 is a functional dimer with two major domains (Nemeroff et al., 1995 ▶). We recently solved the structure of the NS1A N-terminal domain, residues 1–79 (unpublished data), but found that it was the same as that described previously for residues 1–73 (Liu et al., 1997 ▶). In our structure we note that residues beyond 73 are not observed, presumably because they are disordered. In the C-terminal effector domain reported here, residues 79–83 are not observed in either crystalline form, again because they are likely to be disordered. This suggests that there is a flexible linker region of about ten amino acids (residues 74–83) between the two domains.
The NS1 dimer is maintained by dimerization of the N-terminal domain (Wang et al., 1999 ▶) and it may be that the effector domains of NS1 also participate in NS1 dimerization (Wang et al., 2002 ▶). It is likely that in the NS1 dimer the effector domains dimerize in a manner similar to that which we and others have observed crystallographically. To help understand the action of the NS1 effector-domain dimers, in which the biologically important CPSF30-binding sites are buried, we constructed a hypothetical model of the intact NS1 dimer. This model is illustrated in Fig. 6 ▶. To build the model, we aligned the twofold axes of the N-terminal and C-terminal dimers and rotated them so as to bring the C-terminal residues of the N-terminal domain near the N-terminal residues of the C-terminal domain. We found that this was very straightforward and allowed the domains to be linked plausibly by a ten-residue linker. We minimized the energy of the model using CNS (Brünger et al., 1998 ▶). In Fig. 6 ▶, the N-terminal domains are shades of blue for chain 1 and cyan for chain 2. The effector domains are shades of red for chain 1 and orange for chain 2. The ten-residue linkers are colored yellow. The Trp187 side chains are shown as van der Waals structures and chain 1 is labeled; the indole ring binds deep in the pocket of chain 2. We have also added a plausible binding site for dsRNA to the NS1 model. RNA is thought to bind in the prominent channel between symmetrical helices 2 of the N-terminal dimer. The RNA binding is stabilized by a number of ionic interactions (Wang et al., 1999 ▶). This notion has been given strong support from site-directed mutations which show, among other things, that Arg38 is essential to dsRNA binding. This key side chain is shown as stick bonds pointing to the dsRNA; Arg38 on chain 1 is labeled and also serves to identify helix 2 which forms the binding channel.
Figure 6.

Hypothetical model of intact NS1. One chain has the N-terminal domain colored blue and the effector domain colored red; the second chain is colored cyan and orange, respectively. A ten-residue linker (yellow) joins the domains which have been observed crystallographically.
In the NS1 dimer, it is likely that the effector domains dimerize such that for each subunit the Trp187 side chain is buried in the hydrophobic pocket of its dimer partner. The binding of CPSF30 is likely to require a large quaternary structural change to the effector-domain dimer. Initially, the CPSF30-binding pocket is probably blocked by the Trp187 side chain of its dimeric partner, as seen in Fig. 6 ▶. In order to bind CPSF30, the Trp187 side chain must be displaced and the effector domains rotate to accommodate CPSF30 phenylalanines 97, 98 and 103. It is clear from our model that the long unstructured linkers would allow the effector domains to rotate freely into the solvent from the stable N-terminal domain platform. Presumably, there is an equilibrium between the dimerized state that we observe in the crystal structure and a more open conformation. If that open form interacts with the F2F3 domain of CPSF30, those interactions are presumably much stronger than the internal dimer. The effector domains of NS1 proteins that do not bind CPSF30, such as the PR8 NS1, also form dimers with Trp187 pointing into hydrophobic pocket of an adjacent monomer and presumably also have some open conformation. However, without Phe at position 103 and Met at position 106 to help stabilize the F2F3 interactions, the internal effector-domain dimer may be more stable. This would explain why all influenza viruses have this conserved hydrophobic pocket, but only viruses with Phe at position 103 and Met at position 106 are able to break their internal dimer and bind CPSF30 (Twu et al., 2007 ▶).
Supplementary Material
PDB reference: NS1A effector domain, P212121 crystals, 3ee9, r3ee9sf
PDB reference: C2221 crystals, 3ee8, r3ee8sf
Acknowledgments
This work was supported by NIH grants AI 074497 and AI 075509, by the Robert A. Welch Foundation and by support to the Center for Structural Biology from the College of Natural Sciences.
References
- Bornholdt, Z. A. & Prasad, B. V. (2006). Nature Struct. Mol. Biol. 13, 559–560. [DOI] [PubMed]
- Brünger, A. T. (1993). Acta Cryst. D49, 24–36. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chien, C. Y., Tejero, R., Huang, Y., Zimmerman, D. E., Rios, C. B., Krug, R. M. & Montelione, G. T. (1997). Nature Struct. Biol. 4, 891–895. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Das, K., Ma, L.-C., Xiao, R., Radvansky, B., Aramini, J., Zhao, L., Marklund, J., Kuo, R.-L., Twu, K. Y., Arnold, E., Krug, R. M. & Montelione, G. T. (2008). Proc. Natl Acad. Sci. USA, 105, 13093–13098. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Hale, B. G., Barclay, W. S., Randall, R. E. & Russell, R. J. (2008). Virology, 378, 1–5. [DOI] [PubMed]
- Krug, R. M., Yuan, W., Noah, D. L. & Latham, A. G. (2003). Virology, 309, 181–189. [DOI] [PubMed]
- Lamb, R. A. & Krug, R. M. (2001). Fields Virology, edited by D. M. Knipe & P. M. Howley, pp. 1487–1531. Philadephia: Lippincott, Williams & Wilkins.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Liu, J., Lynch, P. A., Chien, C. Y., Montelione, G. T., Krug, R. M. & Berman, H. M. (1997). Nature Struct. Biol. 4, 896–899. [DOI] [PubMed]
- Luft, J. R., Collins, R. J., Fehrman, N. A., Lauricella, A. M., Veatch, C. K. & DeTitta, G. T. (2003). J. Struct. Biol. 142, 170–179. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mibayashi, M., Martinez-Sobrido, L., Loo, Y. M., Cardenas, W. B., Gale, M. Jr & Garcia-Sastre, A. (2007). J. Virol. 81, 514–524. [DOI] [PMC free article] [PubMed]
- Min, J. Y. & Krug, R. M. (2006). Proc. Natl Acad. Sci. USA, 103, 7100–7105. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nemeroff, M. E., Qian, X. Y. & Krug, R. M. (1995). Virology, 212, 422–428. [DOI] [PubMed]
- Noah, D. L., Twu, K. Y. & Krug, R. M. (2003). Virology, 307, 386–395. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Twu, K. Y., Kuo, R. L., Marklund, J. & Krug, R. M. (2007). J. Virol. 81, 8112–8121. [DOI] [PMC free article] [PubMed]
- Twu, K. Y., Noah, D. L., Rao, P., Kuo, R. L. & Krug, R. M. (2006). J. Virol. 80, 3957–3965. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025.
- Wang, W., Riedel, K., Lynch, P., Chien, C. Y., Montelione, G. T. & Krug, R. M. (1999). RNA, 5, 195–205. [DOI] [PMC free article] [PubMed]
- Wang, X., Basler, C. F., Williams, B. R., Silverman, R. H., Palese, P. & Garcia-Sastre, A. (2002). J. Virol. 76, 12951–12962. [DOI] [PMC free article] [PubMed]
- Webby, R. J. & Webster, R. G. (2001). Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 1817–1828. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NS1A effector domain, P212121 crystals, 3ee9, r3ee9sf
PDB reference: C2221 crystals, 3ee8, r3ee8sf