

The crystal structures of the three-component toluene 2,3-dioxygenase system provide a model for electron transfer among bacterial Rieske non-heme iron dioxygenases.
Keywords: toluene, dioxygenases, electron transfer, Rieske clusters, reductases, ferredoxins, NADH, FAD
Abstract
Bacterial Rieske non-heme iron oxygenases catalyze the initial hydroxylation of aromatic hydrocarbon substrates. The structures of all three components of one such system, the toluene 2,3-dioxygenase system, have now been determined. This system consists of a reductase, a ferredoxin and a terminal dioxygenase. The dioxygenase, which was cocrystallized with toluene, is a heterohexamer containing a catalytic and a structural subunit. The catalytic subunit contains a Rieske [2Fe–2S] cluster and mononuclear iron at the active site. This iron is not strongly bound and is easily removed during enzyme purification. The structures of the enzyme with and without mononuclear iron demonstrate that part of the structure is flexible in the absence of iron. The orientation of the toluene substrate in the active site is consistent with the regiospecificity of oxygen incorporation seen in the product formed. The ferredoxin is Rieske type and contains a [2Fe–2S] cluster close to the protein surface. The reductase belongs to the glutathione reductase family of flavoenzymes and consists of three domains: an FAD-binding domain, an NADH-binding domain and a C-terminal domain. A model for electron transfer from NADH via FAD in the reductase and the ferredoxin to the terminal active-site mononuclear iron of the dioxygenase is proposed.
1. Introduction
The extensive use of toluene as an industrial solvent and as a component of gasoline has led to the pollution of soil, groundwater and urban atmospheres (Greenberg, 1997 ▶). Rieske non-heme iron oxygenases (ROs) catalyze the initial step in microbial aerobic degradation of many aromatic compounds in which two hydroxyl groups are introduced into the aromatic ring, yielding cis-dihydrodiols as the main product. Pseudomonas putida F1 can grow with toluene as its sole source of carbon and energy; the initial degradation is catalyzed by the three-component toluene 2,3-dioxygenase (TDO) enzyme system (Supplementary Fig. 11; Gibson et al., 1968 ▶).
The TDO system is composed of a reductase (TDO-R; Subramanian et al., 1981 ▶), a Rieske [2Fe–2S] ferredoxin (TDO-F; Subramanian et al., 1985 ▶) and a terminal dioxygenase (TDO-O; Yeh et al., 1977 ▶). TDO-F shuttles electrons from NADH via a flavin in TDO-R to TDO-O, which catalyzes the enantioselective addition of dioxygen to the aromatic nucleus to form cis-(1R,2S)-dihydroxy-3-methylcyclohexa-3,5-diene (cis-toluene dihydrodiol; Gibson et al., 1970 ▶; Fig. 1 ▶).
Figure 1.

Schematic representation of electron transfer in the TDO enzyme system. Two electrons are transferred from NADH to FAD in the reductase (TDO-R). The ferredoxin (TDO-F; shown as a grey rhombus) shuttles one electron from FAD via the dioxygenase (TDO-O) [2Fe–2S] Rieske center to the mononuclear iron in the neighboring subunit, where the catalytic reaction take place. The S and Fe atoms in the Rieske clusters are represented as orange and magenta circles, respectively.
To date, the structures of several individual components of RO systems have been determined. A recent review has summarized the structural features of these enzymes (Ferraro et al., 2005 ▶). The oxygenase components naphthalene dioxygenase from Pseudomonas sp. (NDO-O; Kauppi et al., 1998 ▶) and Rhodococcus sp. (Gakhar et al., 2005 ▶), biphenyl dioxygenase from Rhodococcus sp. (BPDO-ORHA1; Furusawa et al., 2004 ▶) and Sphingobium yanoikuyae B1 (BPDO-OB1; Ferraro et al., 2007 ▶), nitrobenzene dioxygenase from Comamonas sp. (NBDO-O; Friemann et al., 2005 ▶), cumene dioxygenase from Pseudomonas fluorescens (Dong et al., 2005 ▶) and the ring-hydroxylating dioxygenase from Sphingomonas CHY-1 (Jakoncic et al., 2007 ▶) are all α3β3 heterohexamers of similar structure. The catalytic α-subunit contains a Rieske [2Fe–2S] center and a mononuclear Fe atom at the active site. Electrons are transferred from the ferredoxin via the dioxygenase Rieske [2Fe–2S] center of one α-subunit to the mononuclear iron in the neighboring subunit through a hydrogen-bonded system containing a conserved aspartic acid (Kauppi et al., 1998 ▶; Parales et al., 1999 ▶). The terminal oxygenase components of angular dioxygenase, carbazole 1,9a-dioxygenase from P. resinovornas (Nojiri et al., 2005 ▶) and 2-oxoquinoline monooxygenase from P. putida (Martins et al., 2005 ▶) are trimeric α3 structures in which the subunit structure and trimeric organization are similar to those of the α3 components of the hexameric ROs (Nojiri et al., 2005 ▶).
The RO reductases belong to two distinct families: the ferredoxin-NADP reductase (FNR) family and the glutathione reductase (GR) family. The structures of phthalate dioxygenase reductase (Correll et al., 1992 ▶) and benzoate 1,2-dioxygenase reductase (Karlsson et al., 2002 ▶) belonging to the FNR family have been determined. Both these reductases contain an FAD-binding domain, an NADH-binding domain and a domain containing a plant-type [2Fe–2S] center. Electrons are transferred from NAD(P)H via FMN/FAD to the iron–sulfur cluster. Biphenyl dioxygenase reductase (BPDO-RKKS102; Senda et al., 2000 ▶) is currently the only known RO reductase structure of the GR type. It is a three-domain protein consisting of an NADH-binding domain, an FAD-binding domain and a C-terminal domain corresponding to the interface domain of GR.
The ferredoxins in three-component RO systems serve to shuttle electrons from the reductase to the terminal dioxygenase. These proteins contain a Rieske-type [2Fe–2S] center. To date, the structures of biphenyl dioxygenase ferredoxin (BPDO-FLB400; Colbert et al., 2000 ▶), carbazole 1,9a-dioxygenase ferredoxin (Nam et al., 2005 ▶), toluene 4-monooxygenase ferredoxin (Moe et al., 2006 ▶) and naphthalene dioxygenase ferredoxin (NDO-F9816–4; Brown et al., 2008 ▶) have been determined.
In order to gain insight into the structural basis of electron transfer in a three-component RO system, we have determined the structures of all three components of the TDO system of P. putida F1. We have used these structures of TDO-R, TDO-F and TDO-O to trace a path for electron transfer from NADH of TDO-R to the mononuclear iron at the active site of TDO-O. In addition, the structure of TDO-O without mononuclear iron has been determined (apo-TDO-O), showing that part of the structure becomes flexible.
2. Material and methods
2.1. Expression, purification, crystallization, data collection and processing
TDO-F, TDO-R, TDO-O and apo-TDO-O were overexpressed in Escherichia coli, purified and crystallized as previously described (Lee et al., 2005 ▶). Crystallization was performed by the hanging-drop or sitting-drop vapour-diffusion method. TDO-R crystallized in 1.4 M ammonium sulfate, 0.1 M HEPES pH 7.7. TDO-F crystallized in 38% PEG 8000, 0.1 M MES pH 6.1 or 37% PEG 8000, 0.1 M MES pH 5.8. Apo-TDO-O crystallized in 40%(w/v) PEG 600, 0.1 M sodium citrate pH 5.8. Cocrystallization experiments with Fe(NH4)2(SO4)2 and toluene were performed under strict anaerobic conditions in 40%(w/v) PEG 600, 0.1 M sodium citrate pH 6.1, 20–50 mM Fe(NH4)2(SO4)2 and 20 mM toluene.
X-ray diffraction data were collected on beamline X6A (TDO-R) at Brookhaven National Laboratories (BNL), Upton, New York, USA, beamlines ID14-1 (TDO-F) and ID14-2 (TDO-O) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France and beamline 17-ID (IMCA-CAT; apo-TDO-O) at the Advanced Photon Source (APS), Argonne, Illinois, USA (Lee et al., 2005 ▶).
TDO-R diffracted to 1.8 Å resolution and belongs to space group P41212, with unit-cell parameters a = b = 77.1, c = 156.3 Å. TDO-F diffracted to 1.2 Å resolution and belongs to space group P21, with unit-cell parameters a = 30.5, b = 52.0, c = 30.95 Å, β = 113.7°. Apo- and holo-TDO-O both diffracted to 3.2 Å resolution and belongs to the cubic space group P4332, with unit-cell parameters 235.9 and 234.5 Å, respectively.
2.2. Structure determination and refinement
The structure of TDO-R was solved by molecular replacement with MOLREP (Vagin & Teplyakov, 1997 ▶) using BPDO-RKKS102 (PDB code 1d7y; Senda et al., 2000 ▶) as a search model. Refinement was performed using REFMAC5 (Murshudov et al., 1997 ▶) and ARP/wARP (Perrakis et al., 1999 ▶).
The structure of TDO-F was determined with the molecular-replacement program MOLREP (Vagin & Teplyakov, 1997 ▶) using a polyalanine model generated from BPDO-FLB400 (PDB code 1fqt; Colbert et al., 2000 ▶) as a search model. Refinement was performed using REFMAC5 (Murshudov et al., 1997 ▶) and ARP/wARP (Perrakis et al., 1999 ▶).
The structures of apo-TDO-O and of TDO-O cocrystallized with toluene and Fe(NH4)2(SO4)2 were determined by molecular replacement with the program AMoRE (Navaza, 1994 ▶) using the αβ heterodimer of BPDO-ORHA1 (PDB code 1uli; Furusawa et al., 2004 ▶) as a search model. Refinement was carried out using CNS (Brünger et al., 1998 ▶) and REFMAC5 (Murshudov et al., 1997 ▶). Model building for all four structures was performed using O (Jones et al., 1991 ▶) and Coot (Emsley & Cowtan, 2004 ▶).
2.3. Modelling of binary complexes
Rigid-body dockings of the binary complexes TDO-R–TDO-F and TDO-F–TDO-O were performed using the program Z-DOCK (Chen et al., 2003 ▶). The large domain (residues 1–39 and 85–106) of TDO-F as well as the iron–sulfur clusters of TDO-F and TDO-O and the FAD in TDO-F were omitted from the docking procedure. The final ten top solutions were analyzed and adjusted using O (Jones et al., 1991 ▶).
The models that were closest in superposition to the determined structures of the biphenyl dioxygenase reductase–ferredoxin complex (Senda et al., 2007 ▶) and the carbozole dioxygenase oxygenase–ferredoxin complex (Ashikawa et al., 2006 ▶) were chosen for energy minimization. Energy minimization of selected docked protein–protein complexes was performed using GROMACS v.3.3.1 (Berendsen et al., 1995 ▶; Lindahl et al., 2001 ▶) and the GROMOS96 43a1 force field. Following protonation and solvation with approximately 52 000 rigid simple point-charge water molecules (Berendsen et al., 1981 ▶; Miyamoto & Kollman, 1992 ▶) and 77 sodium ions, 1000 steps of gradient-descent energy minimization were performed. Electrostatic effects were computed using the particle-mesh Ewald method (Essmann et al., 1995 ▶). The bonds of the Rieske iron–sulfur center were restrained at their crystallographic distances with a force constant of 100 000 kJ mol−1 nm−2 and the angles were restrained with a force constant of 1 000 kJ mol−1 rad−2.
3. Results and discussion
3.1. Overall structures
3.1.1. Toluene 2,3-dioxygenase reductase
TDO-R contains one molecule in the asymmetric unit, corresponding to a solvent content of 54%. The structure was refined at 1.8 Å resolution to a final R value of 20.3% (R free = 24.3%). Refinement statistics are summarized in Table 1 ▶. All residues except for the first two at the N-terminus and the last eight at the C-terminus could be fitted into the electron-density map. One residue (Lys48) which was modelled in well defined electron density lies in the generously allowed region of the Ramachandran plot (Laskowski et al., 1993 ▶).
Table 1. Refinement statistics.
| TDO-R | TDO-F | TDO-O | Apo-TDO-O | |
|---|---|---|---|---|
| Resolution (Å) | 20.0–1.8 | 28.4–1.2 | 20.0–3.2 | 48.2–3.2 |
| Space group | P41212 | P21 | P4332 | P4332 |
| Unit-cell parameters (Å, °) | a = b = 77.13, c = 156.38 | a = 30.54, b = 52.09, c = 30.95, β = 113.67 | a = b = c = 235.87 | a = b = c = 234.53 |
| R factor/Rfree† | 20.3/24.3 | 18.3/19.5 | 18.5/20.8 | 24.5/27.9 |
| R.m.s.d. bond lengths (Å) | 0.012 | 0.009 | 0.014 | 0.016 |
| R.m.s.d. bond angles (°) | 1.52 | 1.60 | 1.55 | 1.86 |
| No. of solvent atoms | 465 | 76 | n.a. | n.a. |
| Average B factor (Å2) | ||||
| Protein | 26.5 | 10.3 | 49.0 | 74.9 |
| NAD | 16.5 | n.a. | n.a. | n.a. |
| Rieske [2Fe–2S] | n.a. | 15.6 | 44.1 | 68.3 |
| Mononuclear Fe/toluene | n.a. | n.a. | 52.8/52.4 | n.a. |
| Solvent | 37.6 | 28.7 | 32.0 | n.a. |
| PDB code | 3ef6 | 3dqy | 3en1 | 3eqq |
R factor/R
free = 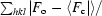
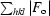 .
.
TDO-R has a glutathione reductase (GR) fold (Karplus & Schulz, 1989 ▶; Schulz et al., 1978 ▶) and can be divided into three domains: an FAD-binding domain (residues 1–108 and 239–317), an NADH-binding domain (residues 109–238) and a C-terminal domain (residues 318–410) (Fig. 2 ▶ a). Both the FAD-binding and NADH-binding domains are of the α/β-fold type characteristic of members of the GR family. Both the FAD-binding and NADH-binding domains have a central parallel β-sheet. In both cases there are three α-helices on one side of the sheet and a three-stranded β-meander on the other side (Rice et al., 1984 ▶; Wierenga et al., 1983 ▶). The C-terminal domain consists of a five-stranded antiparallel β-sheet with two α-helices and one 310-helix on the C-terminal side of the sheet.
Figure 2.

Overall structures of the TDO enzyme system. (a) TDO-R. The FAD-binding domain, NADH-binding domain and C-terminal domain are colored blue, orange and green, respectively. FAD (red) and NADH (blue) are shown in stick representation and as a transparent surface representation. NADH was modeled in the NADH-binding pocket based on the structure of BPDO-RKKS102. (b) TDO-F. The large and cluster-binding domains are colored dark and light gray, respectively. The Rieske center and coordinating side-chain residues are shown in stick representation, with Fe, S and N atoms colored magenta, orange and blue, respectively. (c) Side view of the mushroom-shaped TDO-O α3β3 hexamer. The α-subunits are colored red, green and yellow and the β-subunits pink, light green and gray. The Rieske [2Fe–2S] cluster and the mononuclear iron are shown in CPK model representation, with Fe and S atoms colored magenta and orange, respectively. (d) Top view of TDO-O using the same color representations as in (c). This figure was produced using PyMOL (http://www.pymol.org).
Interestingly, TDO-R not only shares overall similarity with the glutathione reductase family enzymes (Chen et al., 1994 ▶; Karplus & Schulz, 1989 ▶; Lantwin et al., 1994 ▶; Mattevi et al., 1992 ▶; Schierbeek et al., 1989 ▶; Schiering et al., 1991 ▶; Schulz et al., 1978 ▶) but also with the mammalian apoptosis-inducing factor (Mate et al., 2002 ▶; Ye et al., 2002 ▶). Among the reductase structures determined to date, TDO-R shares the highest structural similarity with BPDO-RKKS102 from Acidovorax sp. strain KKS102 (Senda et al., 2000 ▶), which also belongs to the toluene/biphenyl dioxygenase family (Gibson & Parales, 2000 ▶). The proteins share 33% sequence identity and their structures superimpose with an r.m.s. deviation of 1.3 Å for 375 Cα atoms. The structural differences between the proteins are all located on loops that connect secondary-structural elements in solvent-exposed surface regions of the protein. These regions also have significantly higher temperature factors than other regions in both structures, suggesting that they are flexible. None of these regions are close to the ferredoxin-binding site (described in §3.4.1).
Gel-filtration experiments on BPDO-RKKS102 (Senda et al., 2000 ▶) suggested that the protein forms a dimer in solution. The crystal structure of BPDO-RKKS102 revealed two possible dimer forms, both with small contact areas. None of these crystal contact areas could be seen in the TDO-R structure. Packing interactions with neighboring molecules in the TDO-R crystals are few, suggesting that TDO-R is a monomer, which is consistent with biochemical observations (Subramanian et al., 1981 ▶).
The FAD molecule binds noncovalently in a cleft in the FAD-binding domain in an extended conformation similar to that observed in other members of the GR family. The isoalloxazine ring of FAD is completely buried in the central part of the TDO-R molecule. 11 residues and ten water molecules contribute to the extensive hydrogen bonding that holds the FAD in position. In front of the isoalloxazine ring of FAD there is a cavity that was found to be the NADH-binding site in BPDO-RKKS102. Cocrystallization and soaking experiments of TDO-R with NADH were unsuccessful; an ordered sulfate ion was located in the electron-density maps in the NADH-binding cavity (Supplementary Fig. 21). This was probably a consequence of the use of ammonium sulfate as a precipitant in the crystallization. A glycerol molecule from the cryoprotectant solution could also be located in the NADH-binding site. We were unable to obtain crystals with any other precipitant than ammonium sulfate. However, a comparison of the NADH-binding domains of BPDO-RKKS102 and TDO-R shows a very high structural similarity, making it possible to use NADH bound to BPDO-RKKS102 as a model for NADH binding to TDO-R. The carboxylate positioned at the adenosine-binding site (Glu173) probably determines the preference for NADH over NADPH (Scrutton et al., 1990 ▶).
3.1.2. Toluene 2,3-dioxygenase ferredoxin
TDO-F contains one molecule in the asymmetric unit, corresponding to a solvent content of 35%. The structure was refined to a final R value of 18.3% (R free = 19.5) at 1.2 Å resolution (Table 1 ▶). All residues could be located in the electron-density map and the model contained no Ramachandran outliers (89.4% of residues are in the most favored regions; Laskowski et al., 1993 ▶).
TDO-F is an elongated molecule with dimensions of approximately 40 × 30 × 20 Å (Fig. 2 ▶ b). Three antiparallel β-sheets dominate the structure, with a Rieske [2Fe–2S] cluster located at the tip of the molecule. TDO-F can be divided into two subdomains: a large domain (residues 1–39 and 85–106) and a cluster-binding domain (residues 40–85). The large domain has two β-sheets consisting of three β-strands each and two short 310-helices. The cluster-binding domain includes a four-stranded β-sheet, one 310-helix and a Rieske center.
Two histidines and two cysteines coordinate the Rieske iron–sulfur center. The first ligand pair is Cys42 and His44 in a CXH motif and the second ligand pair is Cys61 and His64 in a CXXH motif. The iron ions are coordinated with small deviations from tetrahedral geometry. The histidine ligands are exposed on the surface of the molecule. In the crystals, they form hydrogen bonds to carboxylates in symmetry-related molecules. The electron-density map shows the presence of a cysteine residue (Cys69) in the form of a cysteine sulfenic acid (Supplementary Fig. 31). This residue is located at the surface near the end of the third β-sheet of the cluster-binding domain.
TDO-F is very similar to BPDO-FLB400 of the biphenyl dioxygenase system from Burkholderia sp. strain LB400 (Colbert et al., 2000 ▶). TDO-F and BPDO-FLB400 both belong to the toluene/biphenyl dioxygenase family (Gibson & Parales, 2000 ▶) and their sequences are 54% identical. Their structures superimpose with an r.m.s. deviation of 0.97 Å for 106 Cα atoms. There are only minor differences between the proteins. BPDO-FLB400 has one additional residue in the N- and C-termini. The tight hairpin loop between β2 and β3 in the large domain of BPDO-FLB400 is replaced by an open loop in TDO-F which lacks the three outer hydrogen bonds of the BPDO-FLB400 hairpin. BPDO-FLB400 has an insertion at residue 54 of one extra glycine in an otherwise conserved part of the sequence located about 9 Å from the Rieske center. However, these two regions differ between the two molecules in the asymmetric unit of BPDO-FLB400.
TDO-F has a similar overall structure to other Rieske iron–sulfur domains: those that are soluble components of respiration and photosynthetic systems and those that are parts of the catalytic subunits of Rieske-type dioxygenases (Carrell et al., 1997 ▶; Dong et al., 2005 ▶; Ellis et al., 2001 ▶; Friemann et al., 2005 ▶; Furusawa et al., 2004 ▶; Hunsicker-Wang et al., 2003 ▶; Iwata et al., 1996 ▶; Kauppi et al., 1998 ▶). All Rieske domains have similar general structures with three β-sheets. However, Rieske domains that are part of larger proteins (including oxygenases) often have extra features compared with the small dioxygenase ferredoxins TDO-F and BPDO-FLB400. The iron–sulfur cluster and its ligands superimpose closely in all Rieske-center domains. The Rieske domains of the terminal dioxygenases in biphenyl dioxygenase (BPDO-ORHA1; Furusawa et al., 2004 ▶), nitrobenzene dioxygenase (NBDO-O; Friemann et al., 2005 ▶), naphthalene dioxygenase (NDO-O; Kauppi et al., 1998 ▶) and cumene dioxygenase (Dong et al., 2005 ▶) have a long insertion compared with TDO-F close to the Rieske center (residues 118–132 in NDO-O). This loop, which is conserved in all known oxygenase structures, makes subunit interactions with the neighboring catalytic domain of the dioxygenase α-subunit. The involvement of this loop in α-subunit–α-subunit interactions suggests that it is involved in tertiary-structure stabilization. Since the small electron-transport ferredoxins including TDO-F are monomers, such loops are absent in these proteins.
While the overall structures of Rieske ferredoxins from different kingdoms are similar, their redox potentials differ significantly (Colbert et al., 2000 ▶; Brown et al., 2008 ▶). However, there are several noteworthy insertions and deletions in the various structures. The Rieske domain of the water-soluble fragment of mitochondrial cytochrome bc 1 Rieske protein (Iwata et al., 1996 ▶), the soluble respiratory-type Rieske protein (Hunsicker-Wang et al., 2003 ▶), the Rieske protein of the photosynthetic cytochrome b 6 f (Carrell et al., 1997 ▶) and the Rieske subunit of arsenite oxidase (Ellis et al., 2001 ▶) also contain additional elements. There is no equivalent structure in TDO-F to residues 104–128 of the cytochrome bc 1 Rieske protein, which are located on one side of the domain far away from the Rieske center. The two iron-binding loops in the Rieske center of the cytochrome bc 1 fragment are connected to each other by a disulfide bridge that covers one side of the iron center. TDO-F has no disulfide bridge. These differences contribute to the differences in the redox potentials of these proteins. The differences between the individual Rieske dioxygenase ferredoxins themselves contribute to the difference in specificities for their Rieske oxygenases. It has been shown that some Rieske oxygenase ferredoxins can indeed transfer electrons to oxygenases from a different system (Chi-Li Yu, personal communication), albeit with differential efficiency. An extreme example of this is the functioning of dicamba dioxygenase from P. maltophilia engineered into plant chloroplasts without its corresponding ferredoxin components (Behrens et al., 2007 ▶).
3.1.3. Toluene 2,3-dioxygenase
TDO-O contains one αβ heterodimer in the asymmetric unit, corresponding to a solvent content of 82%. In spite of the high solvent content, the crystal packing and the progress of refinement suggest that there are no further molecules in the asymmetric unit. The structure has been refined to a final R value of 18.5% (R free = 20.8%) at 3.2 Å resolution (Table 1 ▶). 14 N-terminal residues, residues 239–240, ten C-terminal residues of the α-subunit and seven N-terminal residues of the β-subunit could not be located in the electron-density map. Two residues (Glu310 of the α-subunit and Asp9 of the β-subunit) are in disallowed regions of the Ramachandran plot (Laskowski et al., 1993 ▶). In addition, four residues (Cys141, Thr225 and Met242 in the α-subunit and Leu140 in the β-subunit) are in the generously allowed region of the Ramachandran plot. TDO-O has previously been suggested to have an α2β2 subunit composition (Jiang et al., 1996 ▶). However, the structure of TDO-O reveals an α3β3 hexamer as found in NDO-O (Kauppi et al., 1998 ▶), NBDO-O (Friemann et al., 2005 ▶) and BPDO-ORHA1 (Furusawa et al., 2004 ▶) (Figs. 2 ▶ c and 2 ▶ d). The overall structure is mushroom-shaped, with the cap and stem composed of the α3 and β3 subunits, respectively.
The α-subunit contains a Rieske domain (residues 55–173) with a Rieske [2Fe–2S] center and a catalytic domain (residues 1–54 and 174–450) that contains mononuclear iron at the active site. The structure of the catalytic domain belongs to the helix-grip fold (Iyer et al., 2001 ▶) and consists of a nine-stranded antiparallel β-sheet surrounded by 12 α-helices. The completely conserved residues His222, His228 and Asp376 coordinate the mononuclear iron (Fig. 3 ▶).
Figure 3.

Stereo representation of the active site of TDO-O with toluene bound. The F obs − F calc map was computed before toluene was modeled. Residues that coordinate (light green) the mononuclear iron (magenta) and those that line the active site (green) are shown in stick representation. This figure was produced using PyMOL (http://www.pymol.org).
The Rieske domain is dominated by three antiparallel β-sheets and the Rieske [2Fe–2S] center, which is coordinated by two histidines (His98 and His119) and two cysteines (Cys96 and Cys116). The Rieske domain shares high similarity to the equivalent domains of NDO-O, NBDO-O and BPDO-ORHA1 as well as the Rieske ferredoxins TDO-F and BPDO-FLB400.
The β-subunit, the main function of which appears to be structural (Beil et al., 1998 ▶; Friemann et al., 2005 ▶; Jiang et al., 1999 ▶; Kauppi et al., 1998 ▶; Parales, Parales et al., 1998 ▶; Parales, Emig et al., 1998 ▶; Tan & Cheong, 1994 ▶), is dominated by a long twisted six-stranded β-sheet and its structure is similar to those of a number of functionally unrelated proteins (Arand et al., 2003 ▶; Bullock et al., 1996 ▶; Hoelz et al., 2003 ▶; Johnson et al., 2002 ▶; Kerfeld et al., 2003 ▶; Kim et al., 1997 ▶; Lundqvist et al., 1994 ▶; Wang et al., 1997 ▶). A heteroatom density was observed between the β-subunits that are related by the threefold crystallographic symmetry and belong to the same hexamer. The side chain of His24 from each of the three β-subunits coordinates this structural metal ion (Supplementary Fig. 41). Of the currently known RO structures, TDO-O is most similar to biphenyl dioxygenase from Rhodococcus sp. strain RHA1 (BPDO-ORHA1; 77% sequence identity), which also belongs to the toluene/biphenyl dioxygenase family (Gibson & Parales, 2000 ▶). The overall structures are almost identical; superposition of the αβ heterodimers (594 Cα atoms) gives an r.m.s. deviation of 0.47 Å. The only structural difference was located in one loop (residues 238–247) of the α-subunit at the entrance of the active site (Supplementary Fig. 51). The loop has high temperature factors and is completely absent in the BPDO-ORHA1 structure.
NDO-O and NBDO-O both belong to the naphthalene dioxygenase family (Gibson & Parales, 2000 ▶) of ROs and share 27% and 29% sequence identity with TDO-O, respectively. The naphthalene dioxygenase family enzymes also show the largest structural differences in the loops at the entrance of the active site. The structural differences in the β-subunit are mainly located in three of the loops connecting the strands in the long twisted six-stranded β-sheet (Supplementary Fig. 51). One of these loops stacks against the Rieske domain of the α-subunit in NBDO-O and NDO-O. This loop contains a short 310-helix in TDO-O and is more directed towards the β-subunit. The N-terminal helix of the β-subunit in NDO-O and NBDO-O is missing in TDO-O.
3.2. The active site of 2,3-toluene dioxygenase
The active site in the α-subunit is remarkably similar to the active site of BPDO-ORHA1. 17 mainly hydrophobic residues line the ellipse-shaped substrate pocket; only one of these residues differs between TDO-O and BPDO-ORHA1 (Val309 in TDO-O, Ala311 in BPDO-ORHA1; Fig. 3 ▶). TDO-O can use biphenyl as a substrate but it is unknown whether it can use polychlorinated biphenyls (PCBs). However, BPDO-ORHA1 and other biphenyl dioxygenases use several substituted PCBs as substrates (Erickson & Mondello, 1992 ▶; Fukuda et al., 1994 ▶; Furukawa & Miyazaki, 1986 ▶; Masai et al., 1995 ▶).
TDO-O was cocrystallized with toluene, which could be identified in the difference electron-density map of the active site (Fig. 3 ▶). It can be assumed that a dioxygen molecule binds to the active-site mononuclear iron similar to that shown in NDO-O (Karlsson et al., 2003 ▶). At 3.2 Å resolution the map quality is not sufficient to determine the direction of the toluene methyl group. Both possible orientations of the toluene in the active site place the carbon atoms in the ortho and meta positions closest to the modelled dioxygen (about 4.3 Å from the mononuclear iron). This would account for the formation of the cis-(1R,2S)-dihydroxy-3-methylcyclohexa-3,5-diene product. Since the active sites of TDO-O and BPDO-ORHA1 are highly conserved, we have chosen to position the toluene molecule in the orientation in which it overlaps with one of the biphenyl rings in the structure of BPDO-ORHA1 in complex with biphenyl (Furusawa et al., 2004 ▶; Supplementary Fig. 61). The toluene in the active site is stabilized by a set of van der Waals and hydrophobic interactions. Phe366, Phe216 and Ile324 surround one side of the substrate. Ala223 and the C∊ atom of the iron-bound His222 also are involved in stabilizing the binding by these interactions. His311 is at a slightly greater distance (4.0 Å) and might also be involved in orienting the substrate in the active site (Fig. 3 ▶).
Just outside the inner coordination sphere of the active-site iron, the side chain of Gln215 is positioned about 3.3 Å from the Fe atom. The corresponding residue in NDO-O and NBDO-O is Asn and has been suggested to participate in a hydrogen-bonded water channel that is of importance for proton transfer to the active site (Karlsson et al., 2003 ▶). This Asn in NDO-O was originally suggested to be a ligand in some stages of the reaction, but such a function has not been verified. It appears that even the longer Gln does not coordinate the active-site iron in TDO-O nor in BPDO-ORHA1, where the corresponding Gln is also outside the inner coordination sphere.
3.3. Structure of apo-toluene 2,3-dioxygenase
The mononuclear iron at the active site of TDO-O is not strongly bound and is easily removed during enzyme preparation (Lee et al., 2005 ▶). The structure of apo-TDO-O was refined at 3.2 Å resolution to final R factors of 24.5% (R free = 27.9%; Table 1 ▶). 14 N-terminal residues, residues 221–225, 239–247 and six C-terminal residues of the α-subunit and seven N-terminal residues of the β-subunit could not be located in the electron-density map. 2.5% and 1.9% of the residues are in the generously allowed and disallowed regions of the Ramachandran plot, respectively (Laskowski et al., 1993 ▶). The structure of apo-TDO-O demonstrates that parts of the structure in both the α- and the β-subunits become flexible and change conformation in the absence of the mononuclear iron (Supplementary Fig. 71). Residues 215–227 of the α-subunit have a different conformation to that observed in iron-bound TDO-O, where residues 220–224 comprising one of the iron ligands (His222) could not be fitted in the electron density. The main-chain O atom of Thr226 of the α-subunit makes a hydrogen bond to the side chain of Trp211 of the β-subunit in iron-bound TDO-O. In apo-TDO-O Thr226 is positioned differently, leading to a conformational change in the area around Trp211 of the β-subunit (Supplementary Fig. 71). One of the loops covering the substrate-binding pocket is also absent in apo-TDO-O. This is the same loop that is flexible in BPDO-ORHA1.
3.4. Electron transfer
3.4.1. Electron transfer from NADH of TDO-R to the Rieske [2Fe–2S] center of TDO-F
TDO-R and TDO-F transfer electrons from NADH to the mononuclear iron at the active site of the dioxygenase. Initially, two electrons are transferred from NADH in the hydride transfer to the FAD in TDO-R (Fig. 1 ▶). Based on the structure of BPDO-RKKS102, NADH could be modeled in the TDO-R structure (Figs. 2 ▶ a and 4 ▶); the nicotinamide ring is sandwiched approximately 3.5 Å from the isoalloxazine ring (Fig. 4 ▶). Two conserved residues in close proximity of the nicotinamide and isoalloxazine rings (Lys48 and Glu157 in TDO-R) have been suggested to affect hydride transfer in GR (Karplus & Schulz, 1989 ▶) and BPDO-RKKS102 (Senda et al., 2000 ▶).
Figure 4.

Modeled interactions between TDO-R and TDO-F. The FAD-binding domain, NADH-binding domain and C-terminal domain of the TDO-R are colored orange, blue and green, respectively. FAD (red) and NADH (blue) are shown in stick representation. NADH has been modeled in the NADH-binding pocket based on the structure of BPDO-RKKS102. TDO-F is colored gray and the Rieske iron–sulfur cluster with its coordinating residues is shown in stick representation. (a) Overall representation of a possible binding site for TDO-F between the FAD-binding and C-terminal domains of TDO-R. (b) Close-up of the modeled interactions for electron transfer between TDO-R and TDO-F via FAD and Trp320 of TDO-R. Two residues (Lys48 and Glu157) that are believed to be involved in hydride transfer in GR and BPDO-RKKS102 are represented. This figure was produced using PyMOL (http://www.pymol.org).
In TDO-R, there are three shallow depressions that should in principle be possible docking sites for TDO-F. However, one of these is at the entrance for NADH and is not suitable for the one-electron transfer from reduced FAD. The other two depressions are located on each side of the isoalloxazine ring (Supplementary Fig. 71). At one edge of the ring, TDO-F can be placed such that the histidines of the Rieske center are approximately 11 Å from the C8 methyl group of the isoalloxazine ring. At this site, a histidine (from TDO-F) may form a hydrogen bond to Thr129 or Asp132 in TDO-R. A more favorable electron-transfer path could be formed on the opposite edge of the isoalloxazine ring, where a histidine of the Rieske center in TDO-F could be positioned about 6 Å from N3 of the isoalloxazine ring (Fig. 4 ▶ a). Electron transfer could in this case be mediated by the conserved Trp320 in TDO-R that covers this edge of the isoalloxazine (Fig. 4 ▶ b).
Initial rigid-body docking studies were performed with Z-DOCK. The results did not discriminate between the two possible docking sites at each edge of the isoalloxazine ring of TDO-R described above. Both possible docking sites give decent interactions with favorable electron-transfer possibilities.
The recent structure of biphenyl dioxygenase reductase and ferredoxin is consistent with one of our positions for TDO-F occupying a position close to N3 of the isoalloxazine ring, which suggests that this is the predominant electron-transfer pathway (Senda et al., 2007 ▶). A similar complex structure has also been reported for the adrenodoxin reductase–adrenodoxin complex, although this reductase lacks the C-terminal domain (Müller et al., 2001 ▶; Senda et al., 2007 ▶). Of the 13 residues of the reductase that interact with ferredoxin in the biphenyl dioxygenase complex, five are conserved, including the conserved Trp320 and two of the hydrogen-bonding residues, Gln329 and Arg371. Most of the remaining residues are conservative substitutions.
3.4.2. Electron transfer from the TDO-F [2Fe–2S] cluster to the active-site mononuclear iron of TDO-O
A specific electron-transfer pathway is documented in the structure of the complex between the α3 dioxygenase from carbazole 1,9a-dioxygenase and ferredoxin (Ashikawa et al., 2006 ▶). The ferredoxin in this complex binds on the side of the α3 trimer that would represent the top of the cap of the mushroom-shaped α3β3 hexamer. A similar docking site for TDO-F is possible for TDO-O since there is a depression in the molecule in this area (Figs. 5 ▶ d, 5 ▶ e and 5 ▶ f). However, the residues that constitute these surfaces are different in the two structures in this region so the detailed interactions have to differ. Given the lower resolution of the TDO-O structure reported here and the errors introduced in modeling, it is not possible to detail the residue-level changes with confidence.
Figure 5.

Modeled interactions between TDO-F and TDO-O. Color representations are the same as in Fig. 2 ▶. Cys42, Cys61, His44 and His64 belong to TDO-F. Cys116, Cys96, His98 and His119 belong to the α-subunit of TDO-O, while the rest of the residues belong to a neighboring TDO-O α-subunit. (a) and (b) TDO-F is modeled in a shallow depression between two adjacent TDO-O heterodimers. (c) A line is drawn from the Rieske center of TDO-F to the Rieske center of TDO-O. (d), (e) and (f) TDO-F is modelled in accordance with carbazole 1,9a-dioxygenase and ferredoxin at the top of the cap of the mushroom-shaped α3β3 TDO-O with a possible electron-pathway route represented in (f). This figure was produced using PyMOL (http://www.pymol.org).
The Rieske centers of TDO-O are located close to the surface of the molecule in shallow depressions at the interface between the αβ heterodimers. These depressions are most likely to be docking sites for TDO-F, where it can be positioned such that the histidines of its Rieske center are approximately 12 Å from the Rieske center of TDO-O (Fig. 5 ▶). The electron is then transferred from the Rieske center to the mononuclear iron in the neighboring α subunit via an absolutely conserved aspartate as previously suggested (Kauppi et al., 1998 ▶; Parales et al., 1999 ▶).
Previously, an attempt to model the interaction between NDO-O and NDO-F was made using the docking program FTDOCK (Gabb et al., 1997 ▶; Karlsson, 2002 ▶). Several of the best solutions from the docking calculation suggest that the ferredoxin indeed interacts with NDO-O at the corresponding cavity as that described above for TDO-O. The presence of similar topological features in the two closely related proteins NDO-O and TDO-O and in the less related carbazole dioxygenase system suggests very similar modes of interaction between the oxygenase and ferredoxin components of these systems.
4. Conclusions
The toluene 2,3-dioxygenase system was one of the first systems among the bacterial non-heme iron Rieske oxygenases for which all three components were purified. It is interesting that this is also the first system for which the structures of all three components have been reported. We also report a structure of the oxygenase that lacks the mononuclear iron and describe the corresponding structural changes in the active site. The labile nature of the iron and the commonly observed inability to recover activity after loss of iron may be related to the observed structural changes. The structures described have allowed us to propose a model for electron transfer from NADH bound to TDO-R to mononuclear iron at the active site of TDO-O. There are some similarities in the interactions between TDO-R and TDO-F and those reported for the analogous proteins in the biphenyl dioxygenase system. There are also similarities in the interactions between TDO-F and TDO-O and their counterparts in the carbazole dioxygenase system. While the overall surface shapes are conserved, the residues that line the surfaces are different. The conserved features suggest a common model for electron transport. We propose that the differences in the observed interfaces provide the differences in the specificities of the ferredoxin for a given reductase and oxygenase. Further studies will be required to understand the co-evolution of the individual proteins in the system, possibly from a common set of ancestors.
Supplementary Material
PDB reference: TDO-R, 3ef6, r3ef6sf
PDB reference: TDO-F, 3dqy, r3dqysf
PDB reference: TDO-O, 3en1, r3en1sf
PDB reference: apo-TDO-O, 3eqq, r3eqqsf
Supplementary material file. DOI: 10.1107/S0907444908036524/gx5137sup1.pdf
Acknowledgments
This work was supported by the Swedish Research Council (to HE) and NIH grant GM062904-03 (to SR). We would like to acknowledge the staff of beamlines ID14-4 at ESRF and IMCA-CAT at APS. Use of the IMCA-CAT beamline 17-ID at the APS was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Illinois Institute of Technology. Research was carried out in part at the X6A beamline, National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the US Department of Energy under contract No. DE-AC02-98CH10886. X6A is funded by NIH/NIGMS under agreement Y1 GM-0080-03.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: GX5137). Services for accessing this material are described at the back of the journal.
References
- Arand, M., Hällberg, B. M., Zou, J., Bergfors, T., Oesch, F., van der Werf, M. J., de Bont, J. A., Jones, T. A. & Mowbray, S. L. (2003). EMBO J.22, 2583–2592. [DOI] [PMC free article] [PubMed]
- Ashikawa, Y., Fujimoto, Z., Noguchi, H., Habe, H., Omori, T., Yamane, H. & Nojiri, H. (2006). Structure, 14, 1779–1789. [DOI] [PubMed]
- Behrens, M. R., Mutlu, N., Chakraborty, S., Dumitru, R., Jiang, W. Z., Lavallee, B. J., Herman, P. L., Clemente, T. E. & Weeks, D. P. (2007). Science, 316, 1185–1188. [DOI] [PubMed]
- Beil, S., Mason, J. R., Timmis, K. N. & Pieper, D. H. (1998). J. Bacteriol.180, 5520–5528. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F. & Hermans, J. (1981). Intermolecular Forces, edited by B. Pullman, pp. 331–342. Dordrecht: Reidel.
- Berendsen, H. J. C., van der Spoel, D. & van Drunen, R. (1995). Comput. Phys. Commun.91, 43–56.
- Brown, E. N., Friemann, R., Karlsson, A., Parales, J. V., Couture, M. M., Eltis, L. D. & Ramaswamy, S. (2008). J. Biol. Inorg. Chem.13, 1301–1313. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Bullock, T. L., Clarkson, W. D., Kent, H. M. & Stewart, M. (1996). J. Mol. Biol.260, 422–431. [DOI] [PubMed]
- Carrell, C. J., Zhang, H., Cramer, W. A. & Smith, J. L. (1997). Structure, 5, 1613–1625. [DOI] [PubMed]
- Chen, R., Li, L. & Weng, Z. (2003). Proteins, 52, 80–87. [DOI] [PubMed]
- Chen, Z. W., Koh, M., Van Driessche, G., Van Beeumen, J. J., Bartsch, R. G., Meyer, T. E., Cusanovich, M. A. & Mathews, F. S. (1994). Science, 266, 430–432. [DOI] [PubMed]
- Colbert, C. L., Couture, M. M., Eltis, L. D. & Bolin, J. T. (2000). Structure Fold. Des.8, 1267–1278. [DOI] [PubMed]
- Correll, C. C., Batie, C. J., Ballou, D. P. & Ludwig, M. L. (1992). Science, 258, 1604–1610. [DOI] [PubMed]
- Dong, X., Fushinobu, S., Fukuda, E., Terada, T., Nakamura, S., Shimizu, K., Nojiri, H., Omori, T., Shoun, H. & Wakagi, T. (2005). J. Bacteriol.187, 2483–2490. [DOI] [PMC free article] [PubMed]
- Ellis, P. J., Conrads, T., Hille, R. & Kuhn, P. (2001). Structure, 9, 125–132. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Erickson, B. D. & Mondello, F. J. (1992). J. Bacteriol.174, 2903–2912. [DOI] [PMC free article] [PubMed]
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995). J. Chem. Phys.103, 8577–8593.
- Ferraro, D. J., Brown, E. N., Yu, C. L., Parales, R. E., Gibson, D. T. & Ramaswamy, S. (2007). BMC Struct. Biol.7, 10. [DOI] [PMC free article] [PubMed]
- Ferraro, D. J., Gakhar, L. & Ramaswamy, S. (2005). Biochem. Biophys. Res. Commun.338, 175–190. [DOI] [PubMed]
- Friemann, R., Ivkovic Jensen, M. M., Lessner, D. J., Yu, C.-L., Gibson, D. T., Parales, R. E., Eklund, H. & Ramaswamy, S. (2005). J. Mol. Biol.348, 1139–1151. [DOI] [PubMed]
- Fukuda, M., Yasukochi, Y., Kikuchi, Y., Nagata, Y., Kimbara, K., Horiuchi, H., Takagi, M. & Yano, K. (1994). Biochem. Biophys. Res. Commun.202, 850–856. [DOI] [PubMed]
- Furukawa, K. & Miyazaki, T. (1986). J. Bacteriol.166, 392–398. [DOI] [PMC free article] [PubMed]
- Furusawa, Y., Nagarajan, V., Tanokura, M., Masai, E., Fukuda, M. & Senda, T. (2004). J. Mol. Biol.342, 1041–1052. [DOI] [PubMed]
- Gabb, H. A., Jackson, R. M. & Sternberg, M. J. (1997). J. Mol. Biol.272, 106–120. [DOI] [PubMed]
- Gakhar, L., Malik, Z. A., Allen, C. C., Lipscomb, D. A., Larkin, M. J. & Ramaswamy, S. (2005). J. Bacteriol.187, 7222–7231. [DOI] [PMC free article] [PubMed]
- Gibson, D. T., Hensley, M., Yoshioka, H. & Mabry, T. J. (1970). Biochemistry, 9, 1626–1630. [DOI] [PubMed]
- Gibson, D. T., Koch, J. R. & Kallio, R. E. (1968). Biochemistry, 7, 2653–2662. [DOI] [PubMed]
- Gibson, D. T. & Parales, R. E. (2000). Curr. Opin. Biotechnol.11, 236–243. [DOI] [PubMed]
- Greenberg, M. M. (1997). Environ. Res.72, 1–7. [DOI] [PubMed]
- Hoelz, A., Nairn, A. C. & Kuriyan, J. (2003). Mol. Cell, 11, 1241–1251. [DOI] [PubMed]
- Hunsicker-Wang, L. M., Heine, A., Chen, Y., Luna, E. P., Todaro, T., Zhang, Y. M., Williams, P. A., McRee, D. E., Hirst, J., Stout, C. D. & Fee, J. A. (2003). Biochemistry, 42, 7303–7317. [DOI] [PubMed]
- Iwata, S., Saynovits, M., Link, T. A. & Michel, H. (1996). Structure, 4, 567–579. [DOI] [PubMed]
- Iyer, L. M., Koonin, E. V. & Aravind, L. (2001). Proteins, 43, 134–144. [DOI] [PubMed]
- Jakoncic, J., Jouanneau, Y., Meyer, C. & Stojanoff, V. (2007). FEBS J.274, 2470–2481. [DOI] [PubMed]
- Jiang, H., Parales, R. E. & Gibson, D. T. (1999). Appl. Environ. Microbiol.65, 315–318. [DOI] [PMC free article] [PubMed]
- Jiang, H., Parales, R. E., Lynch, N. A. & Gibson, D. T. (1996). J. Bacteriol.178, 3133–3139. [DOI] [PMC free article] [PubMed]
- Johnson, G. R., Jain, R. K. & Spain, J. C. (2002). J. Bacteriol.184, 4219–4232. [DOI] [PMC free article] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Karlsson, A. (2002). PhD thesis, Swedish University of Agricultural Sciences, Uppsala, Sweden. [DOI] [PubMed]
- Karlsson, A., Beharry, Z. M., Eby, D. M., Coulter, E. D., Neidle, E. L., Kurtz, D. M. Jr, Eklund, H. & Ramaswamy, S. (2002). J. Mol. Biol.318, 261–272. [DOI] [PubMed]
- Karlsson, A., Parales, J. V., Parales, R. E., Gibson, D. T., Eklund, H. & Ramaswamy, S. (2003). Science299, 1039–1042. [DOI] [PubMed]
- Karplus, P. A. & Schulz, G. E. (1989). J. Mol. Biol.210, 163–180. [DOI] [PubMed]
- Kauppi, B., Lee, K., Carredano, E., Parales, R. E., Gibson, D. T., Eklund, H. & Ramaswamy, S. (1998). Structure, 6, 571–586. [DOI] [PubMed]
- Kerfeld, C. A., Sawaya, M. R., Brahmandam, V., Cascio, D., Ho, K. K., Trevithick-Sutton, C. C., Krogmann, D. W. & Yeates, T. O. (2003). Structure, 11, 55–65. [DOI] [PubMed]
- Kim, S. W., Cha, S. S., Cho, H. S., Kim, J. S., Ha, N. C., Cho, M. J., Joo, S., Kim, K. K., Choi, K. Y. & Oh, B.-H. (1997). Biochemistry, 36, 14030–14036. [DOI] [PubMed]
- Lantwin, C. B., Schlichting, I., Kabsch, W., Pai, E. F. & Krauth-Siegel, R. L. (1994). Proteins, 18, 161–173. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lee, K., Friemann, R., Parales, J. V., Gibson, D. T. & Ramaswamy, S. (2005). Acta Cryst. F61, 669–672. [DOI] [PMC free article] [PubMed]
- Lindahl, E., Hess, B. & van der Spoel, D. (2001). J. Mol. Model.7, 306–317.
- Lundqvist, T., Rice, J., Hodge, C. N., Basarab, G. S., Pierce, J. & Lindqvist, Y. (1994). Structure, 2, 937–944. [DOI] [PubMed]
- Martins, B. M., Svetlitchnaia, T. & Dobbek, H. (2005). Structure, 13, 817–824. [DOI] [PubMed]
- Masai, E., Yamada, A., Healy, J. M., Hatta, T., Kimbara, K., Fukuda, M. & Yano, K. (1995). Appl. Environ. Microbiol.61, 2079–2085. [DOI] [PMC free article] [PubMed]
- Mate, M. J., Ortiz-Lombardia, M., Boitel, B., Haouz, A., Tello, D., Susin, S. A., Penninger, J., Kroemer, G. & Alzari, P. M. (2002). Nature Struct. Biol.9, 442–446. [DOI] [PubMed]
- Mattevi, A., Obmolova, G., Sokatch, J. R., Betzel, C. & Hol, W. G. (1992). Proteins, 13, 336–351. [DOI] [PubMed]
- Miyamoto, S. & Kollman, P. A. (1992). J. Comput. Chem.13, 952–962.
- Moe, L. A., Bingman, C. A., Wesenberg, G. E., Phillips, G. N. & Fox, B. G. (2006). Acta Cryst. D62, 476–482. [DOI] [PubMed]
- Müller, J. J., Lapko, A., Bourenkov, G., Ruckpaul, K. & Heinemann, U. (2001). J. Biol. Chem.276, 2786–2789. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nam, J. W., Noguchi, H., Fujimoto, Z., Mizuno, H., Ashikawa, Y., Abo, M., Fushinobu, S., Kobashi, N., Wakagi, T., Iwata, K., Yoshida, T., Habe, H., Yamane, H., Omori, T. & Nojiri, H. (2005). Proteins, 58, 779–789. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Nojiri, H., Ashikawa, Y., Noguchi, H., Nam, J. W., Urata, M., Fujimoto, Z., Uchimura, H., Terada, T., Nakamura, S., Shimizu, K., Yoshida, T., Habe, H. & Omori, T. (2005). J. Mol. Biol.351, 355–370. [DOI] [PubMed]
- Parales, J. V., Parales, R. E., Resnick, S. M. & Gibson, D. T. (1998). J. Bacteriol.180, 1194–1199. [DOI] [PMC free article] [PubMed]
- Parales, R. E., Emig, M. D., Lynch, N. A. & Gibson, D. T. (1998). J. Bacteriol.180, 2337–2344. [DOI] [PMC free article] [PubMed]
- Parales, R. E., Parales, J. V. & Gibson, D. T. (1999). J. Bacteriol.181, 1831–1837. [DOI] [PMC free article] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol.6, 458–463. [DOI] [PubMed]
- Rice, D. W., Schulz, G. E. & Guest, J. R. (1984). J. Mol. Biol.174, 483–496. [DOI] [PubMed]
- Schierbeek, A. J., Swarte, M. B., Dijkstra, B. W., Vriend, G., Read, R. J., Hol, W. G., Drenth, J. & Betzel, C. (1989). J. Mol. Biol.206, 365–379. [DOI] [PubMed]
- Schiering, N., Kabsch, W., Moore, M. J., Distefano, M. D., Walsh, C. T. & Pai, E. F. (1991). Nature (London), 352, 168–172. [DOI] [PubMed]
- Schulz, G. E., Schirmer, R. H., Sachsenheimer, W. & Pai, E. F. (1978). Nature (London), 273, 120–124. [DOI] [PubMed]
- Scrutton, N. S., Berry, A., Deonarain, M. P. & Perham, R. N. (1990). Proc. R. Soc. Lond. B Biol. Sci.242, 217–224. [DOI] [PubMed]
- Senda, M., Kishigami, S., Kimura, S., Fukuda, M., Ishida, T. & Senda, T. (2007). J. Mol. Biol.373, 382–400. [DOI] [PubMed]
- Senda, T., Yamada, T., Sakurai, N., Kubota, M., Nishizaki, T., Masai, E., Fukuda, M. & Mitsuidagger, Y. (2000). J. Mol. Biol.304, 397–410. [DOI] [PubMed]
- Subramanian, V., Liu, T. N., Yeh, W. K., Narro, M. & Gibson, D. T. (1981). J. Biol. Chem.256, 2723–2730. [PubMed]
- Subramanian, V., Liu, T. N., Yeh, W. K., Serdar, C. M., Wackett, L. P. & Gibson, D. T. (1985). J. Biol. Chem.260, 2355–2363. [PubMed]
- Tan, H. M. & Cheong, C. M. (1994). Biochem. Biophys. Res. Commun.204, 912–917. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Wang, M., Roberts, D. L., Paschke, R., Shea, T. M., Masters, B. S. & Kim, J. J. (1997). Proc. Natl Acad. Sci. USA, 94, 8411–8416. [DOI] [PMC free article] [PubMed]
- Wierenga, R. K., Drenth, J. & Schulz, G. E. (1983). J. Mol. Biol.167, 725–739. [DOI] [PubMed]
- Ye, H., Cande, C., Stephanou, N. C., Jiang, S., Gurbuxani, S., Larochette, N., Daugas, E., Garrido, C., Kroemer, G. & Wu, H. (2002). Nature Struct. Biol.9, 680–684. [DOI] [PubMed]
- Yeh, W. K., Gibson, D. T. & Liu, T. N. (1977). Biochem. Biophys. Res. Commun.78, 401–410. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TDO-R, 3ef6, r3ef6sf
PDB reference: TDO-F, 3dqy, r3dqysf
PDB reference: TDO-O, 3en1, r3en1sf
PDB reference: apo-TDO-O, 3eqq, r3eqqsf
Supplementary material file. DOI: 10.1107/S0907444908036524/gx5137sup1.pdf
PDB reference: TDO-R, 3ef6, r3ef6sf
PDB reference: TDO-F, 3dqy, r3dqysf
PDB reference: TDO-O, 3en1, r3en1sf
PDB reference: apo-TDO-O, 3eqq, r3eqqsf
Supplementary material file. DOI: 10.1107/S0907444908036524/gx5137sup1.pdf