

Abstract
Respiratory Distress Syndrome (RDS) is due to deficiency of surfactant and commonly occurs in preterm babies. We report the first confirmed case in Northern Ireland of ABCA3 transporter deficiency which is a rare but important cause of RDS in term babies.
A 38 week gestation female infant developed respiratory distress at four hours of age. Chest radiography was consistent with RDS. The baby required repeated doses of surfactant, each resulting in transient periods of decreased ventilatory requirement and improvement in blood gases, but unfortunately she did not survive.
DNA sequencing demonstrated two different mutations in the ABCA3 gene, one inherited from each parent. The baby was therefore a compound heterozygote, and both mutations were thought to be functionally significant.
ABCA3 transporter deficiency is a genetic disorder that is increasingly recognized as a cause of RDS in term babies in whom congenital deficiency of surfactant B and abnormalities of surfactant protein C have been excluded. It should be considered in mature babies who develop severe RDS.
INTRODUCTION
Respiratory Distress Syndrome (RDS) is due to deficiency of surfactant and commonly occurs in preterm babies1. However, the condition can also occur in term babies and may be due to abnormalities of surfactant production. Surfactant protein B deficiency and abnormalities of protein C are recognized causes of abnormal surfactant2. Recent studies indicate that mutations in the ABCA3 transporter gene are a significant cause of RDS in term babies3. We report the first confirmed case of ABCA3 transporter deficiency in Northern Ireland.
CASE REPORT
A baby girl was born at 38 weeks gestation to non-consanguineous parents. Her mother was a primigravida whose membranes had ruptured three weeks prior to delivery. She had been treated with antibiotics for five days but was systemically well. The baby required no active resuscitation at birth but developed severe respiratory distress by four hours of age.
Initial investigations showed a high white cell count, a normal C-reactive protein and a white-out of both lung fields on chest radiograph, consistent with RDS (Figure 1). Blood cultures, respiratory viral screen, immunology profile, karyotype, ultrasound of abdomen and echocardiograph were all normal.
Fig 1.

Chest radiograph showing severe RDS
She was treated with antibiotics and required mechanical ventilation and surfactant replacement. Her condition did not improve as expected, and she subsequently required high frequency oscillatory ventilation and multiple additional doses of surfactant. There were brief periods of decreased ventilatory requirements and improvement in blood gases following each dose of surfactant, but the effects wore off 18 to 24 hours after each treatment.
A provisional diagnosis of congenital surfactant deficiency was made. Broncho-alveolar lavage (BAL) was performed to test for surfactant proteins B and C, and blood was obtained for DNA analysis. The baby's condition continued to deteriorate and on day 25 of life, after discussions with parents and family, intensive care was withdrawn and she died peacefully.
Subsequently, surfactant protein B was detected in adequate concentrations in BAL fluid. DNA sequencing demonstrated a mutation in coding exon 8 on one allele of the baby's ABCA3 gene resulting in substitution of arginine for glycine (G378R, where G refers to Glycine and R refers to Arginine). There was a second mutation in the last base coding exon 18 that led to substitution of arginine for glycine in codon 1002 (G1002R). Both were non-conservative substitutions. One of these mutations was detected in analysis of the mother's DNA and the other mutation in the father's. Hence the baby was a compound heterozygote, with critically significant effects on her lung function (Fig 2).
Fig 2.
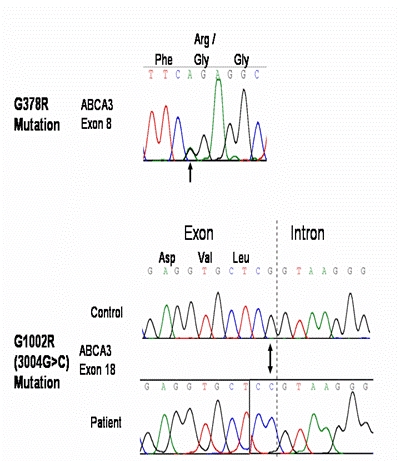
DNA sequencing showing two ABCA 3 mutations (arrowed)
DISCUSSION
Surfactant is a complex substance containing phospholipids and four different types of surfactant proteins: hydrophilic proteins SP-A and SP-D and the hydrophobic proteins SP-B and SP-C. Surfactant is produced in the endoplasmic reticulum of the pulmonary type II alveolar cells. It is stored in the lamellar bodies and released from cells by exocytosis. After exocytosis surfactant undergoes a series of morphological changes to form a multilayered lipid rich coating at the air/ liquid interface in the alveoli.
Surfactant lowers alveolar surface tension, thereby stabilizing the lungs at the end of expiration, and preventing endexpiratory collapse. It also protects lungs against epithelial injury and provides a barrier against infection. Deficiency is common in preterm babies and results in respiratory distress syndrome, which is effectively treated with surfactant replacement. Recently, deficiency of surfactant proteins B and C, and of ABCA3 have been shown to be causes of RDS in more mature, term babies.
ABCA3 is a 1704 amino acid protein of the ATP binding cassette transporter family, whose exact function is as yet unknown. It is widely expressed in type II epithelial cells of the lung and is detected in the limiting membranes of the lamellar bodies which suggests that it is involved in the inward transport of lipids for the production of surfactant. Mutations of the ABCA3 gene may lead to the transport of abnormal lipids into these bodies leading to the production of abnormal or absent surfactant4. Evidence is accumulating of a range of different mutations in the ABCA3 gene5.
The carrier rate of this condition is unknown but was thought to be extremely rare. ABCA3 transporter deficiency may be more common than originally believed6. The baby described here was unfortunate to have inherited two different mutations from her parents, leading to abnormal surfactant function and clinical features of severe RDS. Currently the only curative treatment available is lung transplantation without which most babies will not survive beyond 3 to 6 months of life. This treatment is however not readily available and highly problematic in very young infants.
Acknowledgments
We gratefully acknowledge the assistance of Dr Larry Nogee, Associate Professor of Paediatrics, John Hopkins Hospital, Baltimore and his staff for DNA analysis and helpful comments on the laboratory results. The authors have no conflict of interest.
REFERENCES
- 1.Rennie J. Roberton's Textbook of Neonatology. 4th edition. London: Churchill Livingstone; 2005. [Google Scholar]
- 2.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350(13):1296–303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 3.Whitsett JA, Wert SE, Trapnell BC. Genetic disorders influencing lung formation and function at birth. Hum Mol Genet. 2004;13(Spec No 2):R207–15. doi: 10.1093/hmg/ddh252. [DOI] [PubMed] [Google Scholar]
- 4.Garmany TH, Moxley MA, White FV, Dean M, Hull WM, Whitsett JA, et al. Surfactant composition and function in patients with ABCA3 mutations. Pediatr Res. 2006;59(6):801–5. doi: 10.1203/01.pdr.0000219311.14291.df. [DOI] [PubMed] [Google Scholar]
- 5.Saugstad OD, Hansen TW, Rønnestad A, Nakstad B, Tølløfsrud PA, et al. Novel mutations in the gene encoding ATP binding cassette protein member A3 (ABCA3) resulting in fatal neonatal lung disease. Acta Paediatr. 2007;96(2):185–90. doi: 10.1111/j.1651-2227.2007.00016.x. [DOI] [PubMed] [Google Scholar]
- 6.Garmany TH, Wambach JA, Heins HB, Watkins-Torry JM, Wegner DJ, Bennet K, et al. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008;63(6):645–9. doi: 10.1203/PDR.0b013e31816fdbeb. [DOI] [PMC free article] [PubMed] [Google Scholar]