

Abstract
The NS3 helicase from hepatitis C virus is a prototypical DEx(H/D) RNA helicase. NS3 has been shown to unwind RNA in a discontinuous manner, pausing after long apparent steps of unwinding. We systematically examined the effects of duplex stability and ionic conditions on the periodicity of the NS3 unwinding cycle. The kinetic step size for NS3 unwinding was examined on diverse substrate sequences. The kinetic step size (16 bp/step) was found to be independent of RNA duplex stability and composition, but it exhibited strong dependence on monovalent salt concentration, decreasing to ∼11 bp/step at low [NaCl]. We addressed this behavior by analyzing the oligomeric state of NS3 at various salt concentrations. Whereas only NS3 oligomers are capable of processive unwinding, we found that monomeric NS3 is an active helicase that unwinds with low processivity. We demonstrate that low salt conditions enhance unwinding by monomeric NS3, which is likely to account for the reduction in apparent step size under low salt conditions. Based on results reported here, as well as available structural and single molecule data, we present an unwinding mechanism that addresses the apparent periodicity of NS3 unwinding, the magnitude of the step size, and that integrates the various stepwise motions observed for NS3. We propose that the large kinetic step size of NS3 unwinding reflects a delayed, periodic release of the separated RNA product strand from a secondary binding site that is located in the NTPase domain (Domain II) of NS3. These findings suggest that the mechanism of product release represents an important and unexplored feature of helicase mechanism.
The DEx(H/D) proteins represent a large and ubiquitous family of putative RNA helicases (1, 2). The members of this highly conserved protein family are present in the vast majority of organisms, from viruses to humans, and are involved in virtually every known aspect of RNA metabolism (3). Despite the importance of these proteins, only a small fraction of them has been well characterized, and the mechanism of their action is poorly understood. Although they are usually called “RNA helicases” because of the NTP-dependent RNA unwinding activity displayed by many DEx(H/D) proteins in vitro, other activities, such as ribonucleoprotein remodeling and RNA strand annealing, have been described for several proteins of the family, and these activities may be relevant to their functions in vivo (2, 4-6). The physiological functions and targets of most DEx(H/D) proteins remain unknown.
The nonstructural protein 3 (NS3) from hepatitis C virus (HCV)3 possesses robust RNA and DNA unwinding activities and is a prototypical member of the DEx(H/D) family of ATPase proteins (7). NS3 is an essential component of the HCV replication machinery, and it is an important drug target for anti-HCV therapy. NS3 is one of the most exhaustively studied RNA helicases, and a wealth of structural and biochemical information is available for this enzyme (8-10). Ensemble and single molecule studies have established that NS3 is a processive helicase, capable of making multiple unwinding steps of well defined size without dissociating from the substrate (11, 12). This stepping behavior involves alternating pauses and rapid unwinding events and is similar to the behavior displayed by cytoskeletal motor proteins. Despite these important findings, the molecular mechanism of NS3 unwinding remains largely obscure. The pauses are likely to be caused by the necessity to reset the conformation of the helicase-RNA complex after each unwinding step, consistent with an inch-worm model of unwinding. However, the mechanism by which pauses are triggered and the role of ATP in this process are not understood. The number of base pairs unwound by NS3 per step appears to be one of the largest reported for helicases to date, and it significantly exceeds the footprint of a monomeric NS3 bound to an RNA substrate (6-8 nt) (8). The structural basis for these large steps is unknown, although it has been suggested that it may be related to the oligomeric state of NS3 in its active form (12).
Kinetic methods have long been employed to study helicase activity, and they provide essential tools for dissecting the mechanism of helicase function (13-17). One of the fundamental parameters that can be measured is the kinetic step size, which is defined as the number of base pairs unwound per rate-limiting step. The kinetic step size is often reflected in a periodic mode of unwinding that is commonly displayed by processive helicases that make multiple repetitive unwinding steps before dissociating from the nucleic acid substrate (13).
To characterize the mechanism of RNA unwinding by NS3 and to develop a physical explanation for its unusual stepping behavior, we systematically studied the dependence of the NS3 kinetic step size on duplex stability and ionic conditions. We find that the step size of NS3 shows no dependence on duplex stability or sequence, in contrast to its processivity and unwinding rate constant, which have been previously shown to strongly depend on duplex stability (18, 19). However, we observe that the apparent kinetic step size of RNA unwinding by NS3 is sensitive to monovalent salt concentration and is reduced to ∼11 bp under low salt conditions. We address this behavior by analyzing the oligomeric state of NS3 at various salt concentrations. Whereas NS3 oligomers are capable of processive unwinding, we find that monomeric NS3 is an active helicase that possesses low processivity. We demonstrate that low salt conditions enhance unwinding by monomeric NS3 and are likely to account for the changes in kinetic step size of NS3 under low salt conditions. Combining these results with data from previous studies, we propose a physical unwinding mechanism that explains the kinetic step size and the diverse kinetic behaviors that are displayed by NS3.
EXPERIMENTAL PROCEDURES
Materials—DNA oligonucleotides were obtained from Keck Facility, Yale University. Buffers, salts, and ATP were from Sigma. Phosphorothioate NTPs were purchased from TriLink Biotechnologies. Full-length NS3 (genotype 1a) was expressed and purified as described (20). The expressed protein contained a tag of following sequence at its N terminus, MRGSHHHHHHGSDYKDDDDKA. This particular tag, which greatly facilitates NS3 purification, does not influence unwinding step size or functional complex formation characteristics when compared with untagged protein (data not shown).4
Preparation of RNS Duplexes—RNA strands were prepared by T7 transcription of PCR-generated DNA templates using the MEGAshortscript RNA transcription kit (Ambion). For each substrate, the top strand RNA (Fig. 2) was synthesized in four separate transcription reactions. Each of the reactions contained one of the α-phosphorothioate NTPs along with the four regular NTPs. To achieve a suitable level of phosphorothioate incorporation (∼0.7 nicks per top strand upon iodine cleavage), the concentration of each α-phosphorothioate NTP was equal to that of the corresponding NTP (3.5 mm). After transcription, the four RNA products were purified on denaturing PAGE and combined in equimolar proportions to yield a pool of top strand RNAs containing phosphorothioate linkages at random positions. The bottom strands were unmodified RNAs.
FIGURE 2.

Sequences of the RNS substrates used in this study. The duplex part is either 80 (RAND1 and RAND2) or 82 bp. All substrates have identical single-stranded 3′-overhangs of 18 nt. The shorter and longer strands are referred to as top and bottom strands, respectively, throughout the text. For substrates FLAT1 and FLAT2, the flanking sequences that were introduced to prevent incorrect annealing of strands are underlined. The boxed sequence in FLAT2 substrate represents the “GC-clamp” (see text for details).
To prepare RNS duplexes, the pool of top strand RNS oligonucleotides was 5′-end-labeled with [γ-32P]ATP (PerkinElmer Life Sciences) using T4 polynucleotide kinase (New England Biolabs) and annealed with 2× molar excess of unlabeled complementary bottom strand in the annealing buffer (20 mm MOPS-NaOH, pH 6.5, 30 mm NaCl and 1 mm EDTA) by heating to 95 °C and then cooling to room temperature over 30 min. The phosphorothioate linkages in the top strands were then cleaved by addition of iodine to 0.1 mm final concentration and incubating for 15 min at room temperature. The cleaved substrates were then purified by electrophoresis on a semi-denaturing polyacrylamide gel (15% acrylamide, 3 m urea, and 0.5× Tris borate electrophoresis buffer, TBE).
NS3 Unwinding Kinetics—Quench-flow unwinding experiments were performed using a model RQF-3 KinTek quench-flow apparatus maintained at 37 °C by a circulating water bath. Prior to initiation of unwinding, the radiolabeled RNS duplexes (2 nm) were preincubated with 200 nm NS3 in the standard reaction buffer (20 mm MOPS-NaOH, pH 6.5, 30 mm NaCl, 3 mm MgCl2, 2 mm dithiothreitol, 1% glycerol, 0.2% Triton X-100) for 1 h at 37 °C. Unwinding reactions were initiated in the quench-flow apparatus by mixing the preincubated reaction mix with an equal volume of 8 mm ATP and 4 μm trap oligonucleotide in the standard reaction buffer. The oligonucleotide used as a trap for NS3 was a 60-mer DNA with the sequence CATATGAGTCGTATCGTG(T)42. The unwinding reactions were quenched at various times (0.005 to 5 s) with another volume of quench buffer containing 30 mm EDTA, 25% sucrose, 1% SDS, 0.015% xylene cyanol, and 0.015% bromphenol blue. The addition of EDTA and SDS causes NS3 to dissociate from the RNA. Because all partially unwound duplexes immediately reassociate upon NS3 dissociation, only those product lengths that were completely unwound before quenching of the reaction were detected. The quenched reactions were analyzed by electrophoresis on a semi-denaturing polyacrylamide gel (15% acrylamide, 3 m urea, 0.5× TBE buffer). No detectable spontaneous duplex dissociation occurred in the gel under these conditions. The electrophoretic bands were visualized and quantified using a Storm 840 PhosphorImager and Image-Quant software (GE Healthcare). The fraction unwound was determined for each time point and for each RNS duplex length using the following formula: Aunw = Iunw/Iheat, where Iunw and Iheat are relative intensities of electrophoretic bands corresponding to the unwinding reaction and heat-denatured substrate, respectively, corrected for total amounts of radioactivity loaded onto the respective gel lanes.
Analysis of Unwinding Time Courses—Individual time courses of NS3 unwinding were analyzed using the analytical solution for Scheme 1 obtained through Laplace transform (37) and continuous in both k and n as shown in Equation 1,
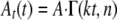 |
(Eq.1) |
where At(t) is the fraction unwound; t
is unwinding time; A is the maximal unwinding amplitude, and
Γ(kt, n) is incomplete γ function, defined as shown in
Equation 2,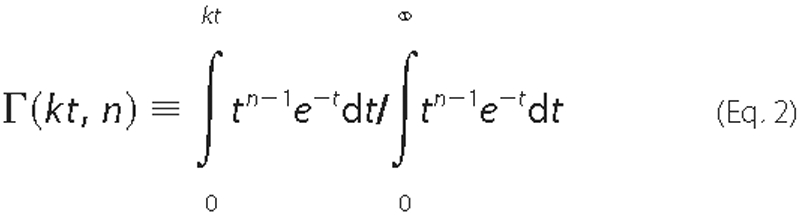
The fitting of time courses to Equation 1 was performed using a custom-written script for MATLAB 7.0. This script will be made available at Pyle laboratory website. Fitting was performed individually for each unwinding time course. All unwinding parameters (unwinding amplitude, kinetic rate constant, and number of steps) were allowed to float.
RESULTS
To determine the kinetic step size of NS3 under diverse reaction conditions, we conducted unwinding experiments with a combinatorial RNA library of randomly nicked substrates, using methodologies that were previously developed in our laboratory (12). This approach takes advantage of the fact that NS3 translocates along the “bottom strand” of the duplex and readily displaces “top strand” sequences that contain lesions or discontinuities (Fig. 1) (20). During the course of unwinding, when the helicase passes a nick, the unwound 5′-portion of the top strand is released, thereby reporting on the progress of helicase translocation and duplex unwinding at a particular location and time. We have employed this approach to examine intermediate states of unwinding using a pool of RNA substrates that contain nicks at random positions in the top strand.
FIGURE 1.

General scheme for the RNS unwinding experiments. The pool of substrates with randomly nicked top strand is created by annealing the 5′-labeled top strand RNA containing randomly incorporated phosphorothioate linkages (less than 1 incorporation per top strand molecule) with the complementary bottom strand, followed by iodine cleavage. NS3 is prebound to the single-stranded overhangs of the substrate molecules in the absence of ATP, and then the synchronized unwinding reaction is initiated by ATP addition. The radioactively labeled 5′-terminal portions of the top strand are released when unwinding of that region is complete and they are subsequently analyzed by gel electrophoresis.
The nicks are generated by “doping” the top strand with phosphorothioate linkages during the T7 transcription reaction. The top strand RNAs containing phosphorothioates are then 5′-end-labeled with 32P and annealed to the bottom strands, and the phosphorothioate linkages are cleaved by iodine. The resulting pool of randomly nicked substrates is then subjected to an unwinding reaction, and the resultant products are resolved by gel electrophoresis. Because the nick positions are completely random, every possible nucleotide position along the top strand is represented by a fraction of molecules in the substrate pool, and the amount of the unwound product can be independently determined for all the RNS lengths by quantitating the corresponding bands on the gel.
To address the influence of sequence on NS3 unwinding parameters, we synthesized four different randomly nicked RNA substrates (Fig. 2). Each of the substrates consists of an RNA duplex that is flanked by a single-stranded 18-nt 3′-overhang that is required for NS3 binding and initiation of unwinding. The overhang sequence was identical for all of the substrates. The length of the duplex part was either 80 (RAND1 and RAND2 substrates) or 82 bp (FLAT1 and FLAT2), to allow for multiple unwinding steps by NS3. The four RNA duplexes used in this study were designed to explore NS3 behavior in a diverse set of environments, with variations in GC content and stability profiles. RAND 1 and RAND2 are substrates of random sequence and average GC contents of 60.0 and 45.0%, respectively. The substrate FLAT1 is composed of the repeating tetranucleotide sequence ACTG, flanked with short irregular sequences (Fig. 2, underlined letters) to ensure proper annealing of strands. The repeat sequence was selected so that the variations of free energy for base pairs within the repeat were minimal, based on the nearest-neighbor rules (21). This results in nearly uniform thermodynamic stability for the repetitive portion of the substrate duplex. The “flat” profile of thermodynamic stability essentially eliminates the idiosyncrasy originating from “rugged” energetic profiles of substrates with random sequences, thereby providing an important control for unwinding behavior that otherwise might be obscured by energetic irregularities of the duplex. Finally, the substrate FLAT2 is identical to FLAT1, with the exception of 7 bp in the middle of the duplex that have been changed to GCCCCCC (Fig. 2, boxed sequence, nucleotides 37-43). This creates a short stretch of RNA duplex that is considerably more stable than the surrounding sequence and has the potential to promote altered unwinding behavior of NS3 (e.g. cause changes in its unwinding periodicity) because of the sudden increase in its “workload.”
We carried out time-resolved NS3 unwinding experiments with the four RNS duplexes under identical conditions (37 °C, 4 mm ATP), using a rapid quench-flow mixer. The unwinding reactions were quenched at various times (0.005 to 5 s), and the unwinding products were resolved on a semi-denaturing polyacrylamide gel (Fig. 3A). Each experiment resulted in a series of 30-40 unwinding time courses for various product lengths, all of which could be visualized with single nucleotide resolution (Fig. 3B). With the exception of the shortest resolved RNS lengths, all kinetic series exhibited delayed unwinding kinetics (manifested as lag phases in the unwinding time courses), indicating the presence of rate-limiting intermediate states of unwinding.
FIGURE 3.

A, representative polyacrylamide gel showing separation of the unwinding products after performing a time-resolved RNS experiment with substrate FLAT1. The unwinding reactions were initiated by addition of ATP and terminated at various times (0.005 to 5 s) using a quench-flow mixer, and analyzed on a semi-native polyacrylamide gel as described under “Experimental Procedures.” The last two lanes on the right are the RNS substrate heated at 99 °C for 5 min (>95% strand dissociation) and a sequencing ladder, respectively. B, kinetic time courses obtained by quantitating the autoradiogram of the gel shown in A. Product lengths up to ∼50 bp can be resolved with single nucleotide resolution.
Unwinding time courses were analyzed using the n-step kinetic
Scheme 1
(13),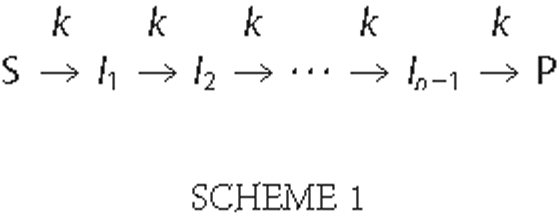
where n represents the number of rate-limiting steps, and n-1 is the number of populated intermediate states of unwinding (I1, I2,···, In-1). S and P correspond to the substrate and final product, respectively, and k is the kinetic rate constant of each step. Fitting of data to Scheme 1 was performed using Equation 1 as described under “Experimental Procedures.” For each RNA substrate, the numbers of rate-limiting kinetic steps determined from the NLLS procedure were plotted versus corresponding RNS lengths (Fig. 4). The average kinetic step sizes (bp/step) and associated standard deviations were then determined by linear regression (Table 1). The unwinding rate constants were found to be independent of RNS duplex length within our experimental uncertainty (Table 1). Despite considerable differences in the thermodynamic stabilities of our substrates, we observed an identical unwinding step size (16 ± 1 bp/step) for all of the substrates under our standard unwinding conditions (20 mm MOPS-NAOH, pH 6.5, 30 mm NaCl). This number provides a more precise estimate than that which was previously reported (18 ± 2 bp/step) (12).
FIGURE 4.

The dependence of the number of rate-limiting steps on RNS duplex length, determined from the analysis of experimental unwinding time courses for each of the RNS substrates. Data for different substrates are shown in different colors as follows: black (RAND1), blue (RAND2), red (FLAT1), and green (FLAT2). The numbers of rate-limiting unwinding steps (symbols) were determined from unwinding time courses by performing the NLLS fitting to Equation 1 as described under “Experimental Procedures.” Solid lines represent linear fits.
TABLE 1.
Kinetic parameters of NS3 unwinding on four RNA substrates
| Substrate | Step size, bp/step | Unwinding rate constant, steps/s |
|---|---|---|
| RAND1 | 16.3 ± 1.1 | 1.1 ± 0.3 |
| RAND2 | 16.5 ± 1.0 | 0.9 ± 0.2 |
| FLAT1 | 16.3 ± 1.1 | 1.3 ± 0.2 |
| FLAT2 | 15.9 ± 1.0 | 1.5 ± 0.3 |
| Average | 16.3 ± 0.3 | 1.2 ± 0.3 |
Kinetic Step Size of NS3 Unwinding Is Sensitive to Monovalent Salt Concentration—It was previously proposed that the large kinetic step size exhibited by NS3 in bulk is the consequence of NS3 dimerization on the RNA substrate (12). However, additional insights were provided by high resolution single molecule studies using optical tweezers (11, 18). These experiments demonstrated that, under conditions of relatively high pulling force, NS3 unwinds RNA duplexes with a somewhat smaller step size of 11 ± 3 bp, which was attributed to unwinding by a monomeric form of NS3. Significantly, NS3 requires relatively high pulling forces to behave as a processive helicase in the optical trap.
To better understand the relationship between step size, oligomerization state, NS3 affinity, and structural integrity of the duplex, we examined the kinetic step size for NS3 under different monovalent salt conditions. RNS unwinding experiments were conducted in a reaction buffer that contained varying concentrations of NaCl (from 0 to 100 mm, in addition to 20 mm MOPS-NaOH, pH 6.5). The numbers of unwinding steps for different RNS duplex lengths were determined by fitting the resultant time courses to Equation 1 for substrate FLAT1 under various salt concentrations (Fig. 5A). The kinetic step sizes at different salt concentrations were determined from the slope of the linear fit to the data, revealing a substantial dependence of step size on monovalent salt concentration (Fig. 5B). Indeed, under conditions of lowest ionic strength, the NS3 step size reduces to 11 bp, which is exactly the value observed in the optical tweezer experiments.
FIGURE 5.

The apparent kinetic step size of RNA unwinding by NS3 is sensitive to monovalent salt concentration. A, numbers of kinetic steps determined for the substrate FLAT1 at 0, 30, and 60 mm NaCl (in addition to 20 mm MOPS-NaOH, pH 6.5) plotted versus RNS duplex length. B, apparent step size of NS3 unwinding at varying NaCl concentrations determined from the slopes of linear fits from a. Except for 100 mm NaCl, duplicate experiments are shown.
Determination of the Functional Oligomeric State of NS3 on RNA Substrates—We set out to determine whether the oligomeric state of NS3 in the functional RNA complex can be modulated by salt concentration and thereby account for the observed changes of the unwinding step size. To study the kinetics of functional complex formation, we performed “double mixing” experiments (Fig. 6), in which the single cycle reaction amplitude of NS3 unwinding (Aunw) is measured as a function of time of preincubation of NS3 with RNA substrate. It has been established that, under single cycle conditions, Aunw is dependent on both functional complex concentration and RNA duplex length (13) as shown in Equation 3,
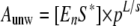 |
(Eq.3) |
where [EnS*] is concentration of NS3-RNA functional complex; p is processivity per unwinding step; L is the duplex length, and s is unwinding step size.
FIGURE 6.

Top, the general scheme of formation of functional NS3-RNA complex, where S is an RNA substrate; E is NS3 protein; P is unwound product; EnS* is the functional complex, and n is number of NS3 molecules bound to the RNA substrate in the functional complex. Bottom, the experimental design of functional complex formation experiments. The RNA substrate is preincubated with NS3 for variable time Δt, after which unwinding is initiated by the addition of ATP and trap oligonucleotide. The trap oligonucleotide is included to sequester any unbound protein and to prevent re-binding of NS3 to the RNA substrate during the unwinding reaction (single cycle unwinding conditions). The unwinding reactions are allowed to proceed to completion (90 s), after which unwinding amplitudes (Aunw) are determined for each Δt by resolving the unwound products by native PAGE.
Equation 3 describes the relationship between the measured amount of unwound product and the concentration of the functional complex. One important implication of Equation 3 is that if observed unwinding is produced by a homogeneous population of active NS3 species, then the kinetics of functional complex formation measured with different RNA duplex lengths should be identical and differ only in their amplitudes. However, if unwinding results from a heterogeneous mixture of functionally active species (e.g. different oligomeric states of NS3), and at least some of these species have substantially different levels of processivity, then different kinetics of functional complex formation can be observed for different RNA duplex lengths. For instance, if nonprocessive species are present, they will only contribute to kinetics of functional complex formation measured with very short RNA duplexes. Measuring functional complex formation kinetics with duplexes of variable length can therefore enable one to resolve different active species of NS3.
To establish if NS3 unwinding can be attributed to a mixed population of functional species, we measured the kinetics of functional complex formation using RNA substrates with an identical 3-overhang (18 nt) and variable duplex lengths (12, 24, and 40 bp). The accumulation of functional complex for the shortest of the substrates (12 bp) displayed a small but readily detectable burst, which was followed by a much slower exponential phase. Kinetics measured with either 24- or 40-bp substrates did not reveal a burst, and displayed only the slow phase of functional complex formation (Fig. 7). Given that the burst is only observed with the short duplex substrate, the data suggest that the burst represents a population of functional complexes that form rapidly but possess very low processivity. Accordingly, the gradual, exponential phase of functional complex formation is likely to represent a population of complexes that assembles more slowly, but which is processive and capable of unwinding 24- and 40-bp RNA duplexes.
FIGURE 7.

A, kinetics of formation of the functional NS3-RNA complex measured with 0.5 nm of 40-bp (circles), 24-bp (squares), and 12-bp (triangles) RNA substrates and 50 nm NS3 (all concentrations are before ATP addition). All RNA substrates had identical 18-nt 3′-single strand overhang. After preincubating NS3 with RNA substrates for the indicated time, unwinding reactions were initiated by the addition of ATP and a trap DNA nucleotide to concentrations of 4 mm and 2 μm, respectively. After 90 s, the unwinding reactions were stopped by the addition of EDTA and SDS to 12 mm and 0.5%, respectively, and unwound products were analyzed by a native PAGE. B, amplitudes of the burst and slow kinetic phase from a, plotted versus RNA duplex length. The smooth curves are included solely to show the trends in the data.
To characterize the nonprocessive NS3 species, we measured the kinetics of functional complex formation on a 12-bp RNA substrate under conditions identical to those employed previously (Fig. 7A), but under conditions where unwinding reactions were initiated by the addition of ATP and a large excess of unlabeled top strand oligonucleotide. In addition to trapping unbound NS3 molecules, this complementary trap increases apparent unwinding of the helicase by annealing to the bottom strand and promoting the dissociation of the radiolabeled top strand from partially unwound duplexes (20). Significantly, the inclusion of complementary trap greatly increases the amplitude of the burst (Fig. 8). Re-annealing of completely separated RNA strands is extremely slow at the RNA concentration used in this experiment (0.5 nm); therefore, the observed increase in the burst amplitude must originate from annealing of the trap oligonucleotide to the bottom strand before the strands have been completely separated, e.g. to a partially unwound RNA substrate during the unwinding reaction.
FIGURE 8.

Kinetics of formation of the functional NS3-RNA complex measured with a 12-bp RNA substrate using either noncomplementary trap (a DNA oligonucleotide of unrelated sequence, open circles) or complementary trap (unlabeled 12-nt top strand RNA oligonucleotide, filled circles). RNA and NS3 concentrations were 0.5 and 20 nm, respectively. Unwinding reactions were initiated by the addition of ATP and trap oligonucleotide to final concentrations of 4 mm and 2 μm, respectively.
Longer substrates (24, 34, and 40 bp) also displayed a readily detectable burst in the kinetics of functional complex formation when reactions were initiated with complementary trap (data not shown). These same substrates had not displayed a burst when a noncomplementary trap was employed, which indicates that the inclusion of complementary trap allows the detection of activity by nonprocessive species. To monitor unwinding by all of the various functional complexes (both nonprocessive and processive), we therefore used complementary top strand trap in all subsequent experiments. Importantly, the inclusion of complementary trap was found to specifically increase the unwinding amplitude without altering the unwinding kinetics (data not shown).
Using this experimental setup, we wished to determine whether the various types of functional NS3 species can be attributed to different oligomeric states of NS3. Functional complex formation was monitored using the 34-bp duplex at constant NS3 concentration (50 nm) and varying RNA substrate concentration (Fig. 9). Unwinding reactions were initiated by the addition of ATP and unlabeled 34-nt top strand oligonucleotide. Under conditions that promote oligomerization of NS3 (10 nm RNA, 50 nm NS3), only a small burst is observed, and the majority of the unwinding amplitude results from the slow exponential phase. By contrast, under conditions that facilitate formation of monomeric species (100 nm RNA, 50 nm NS3), the burst amplitude is dramatically increased, although the amplitude of the slow phase decreased. On the basis of these results, we conclude that the burst accounts for unwinding by monomeric NS3, which can begin unwinding as soon as it binds the substrate. The slow phase is likely to reflect unwinding by oligomeric NS3 species, which require more time as multiple molecules assemble appropriately. Because no burst is observed for the 34-bp substrate when noncomplementary trap is used, the data are consistent with low processivity by the monomeric NS3 species and processive behavior by oligomeric NS3 species.
FIGURE 9.

Kinetics of formation of the functional NS3-RNA complex measured with a 34-bp RNA substrate with 20-nt 3′-overhang. Unlabeled 34-nt top strand RNA was used as trap oligonucleotide (2 μm final concentration). NS3 concentration was 50 nm in all experiments. RNA substrate concentrations were 100 nm (circles), 50 nm (squares), and 10 nm (triangles).
Having developed an approach for differentiating the monomeric NS3-RNA complex from other functional oligomeric species, we wanted to determine how each of these states is influenced by the ionic strength of the system. To this end, the kinetics of functional complex formation was measured with a 34-bp duplex and complementary top strand trap in varying concentrations of sodium chloride (0 versus 30 mm NaCl; Fig 10). At all [RNA]/[NS3] ratios tested, low salt conditions led to a significant increase in the burst amplitude, suggesting that unwinding by monomeric NS3 is promoted by low NaCl concentrations. Significantly, these same salt conditions were observed to reduce the kinetic step size of NS3 (Fig. 5A), suggesting that the smaller kinetic step size in low [NaCl] is attributable to unwinding by a monomeric NS3 species.
FIGURE 10.

Kinetics of formation of the functional NS3-RNA complex measured with a 34-bp RNA substrate with 20-nt 3′-overhang at 0 mm NaCl (filled symbols) and 30 mm NaCl (open symbols). The indicated NaCl concentrations are in addition to 20 mm MOPS-NaOH, pH 6.5. Other conditions are as in Fig. 9.
The oligomeric state of NS3 can be modulated by length of the single-stranded overhang (15). Indeed, formation of a monomeric species is likely to be promoted by shorter overhangs, which should favor loading of single helicase molecules (11). To provide an independent test of NS3 oligomerization state and its correlation with burst kinetics, we monitored the kinetics of functional complex formation as a function of single-stranded 3′-tail length (6, 10, and 20 nts), at varying ratios of [NS3] to [RNA]. RNA substrates with shorter overhangs (6 or 10 nt) exhibited severe reductions in the amplitude of the slow exponential phase for functional complex formation under conditions that facilitate NS3 oligomerization ([NS3] > [RNA]), whereas the fast burst phase remains intact at all overhang lengths (Fig. 11). The slow phase is completely absent under conditions that shift the oligomerization equilibrium toward an exclusively monomeric form of NS3 ([NS3] = [RNA]) (Fig. 11). These results indicate that shorter overhangs can only accommodate monomeric NS3, and a longer overhang (20 nt) is required for efficient assembly of NS3 oligomers. In addition, these data provide independent evidence that the burst phase of reaction represents activity by the NS3 monomer and that the burst is an appropriate metric for monitoring behavior of the monomeric species. These results are consistent with the size of the NS3-binding site on single-stranded RNA, which is 8-11 nt based on binding density of NS3 in solution (22).
FIGURE 11.

Kinetics of formation of the functional NS3-RNA complex measured with a 34-bp RNA substrate with varying lengths of the single-stranded 3′-tail: 6 nt (triangles), 10 nt (squares), and 20 nt (circles). RNA concentration was 25 nm (green symbols and lines), 50 nm (red symbols and lines), and 100 nm (black symbols and lines). NS3 concentration was 50 nm in all experiments. Low salt conditions are shown (0 mm NaCl); experiments in the presence of 30 mm NaCl produced similar results (data not shown).
DISCUSSION
It is becoming increasingly clear that NS3 and related helicases translocate along the backbone by inching forward one nucleotide at a time, through a process that is likely to involve the ATP-dependent flexing of Domains I and II (9, 10, 23-26). However, these single nucleotide steps are not the only form of periodic, dynamic behavior displayed by helicases, and they are not rate-limiting when helicase unwinding is monitored in bulk or in single molecule experiments. In the case of NS3, both single molecule force and FRET experiments have shown that the operative step size for duplex unwinding is approximately 3 bp, although translocation may occur in smaller steps. Thus, helicases appear to melt or rip the duplex in larger periodic increments, which is consistent with the large “kinetic step sizes” that have historically been reported for SF1 helicases such as UvrD and Rep (13, 26). And yet the stepping story does not stop there; helicases have additional mechanical behaviors that can be coupled with unwinding, and which can become rate-limiting. This is particularly true for NS3 that displays a very large, periodic, rate-limiting step (kinetic step size) of 18 bp (12) when operating at high speeds on long duplexes. In this work, we set out to explore the nature of this large kinetic step and to characterize its behavior under diverse reaction conditions to develop a physical model for this remarkable phenomenon.
To determine whether the 18-bp step reported previously was because of sequence idiosyncrasies, we synthesized four different duplex substrates with varying types of thermodynamic and sequence landscapes. Two of the substrates were random sequence RNAs, with moderate differences in G-C content. The third substrate contained a continuous repeat of the 4-bp types and is therefore considered energetically flat. The final substrate contained a large G-C clamp in its center, to test whether NS3 would stop or change its apparent step size upon encounter with a thermodynamic barrier. Despite all of this variation, each of the four substrates were readily unwound by NS3, and the step size was strikingly constant (16 ± 1 bp), which is the same, within error as that reported previously (18 ± 2) (12). This finding indicates that the large steps are not related to the thermodynamic attributes of the duplex and are therefore unlikely to be coupled with the actual strand-opening event (which should be dependent on sequence and force, see below). Rather, this finding suggests that the large steps are related to physical attributes of the protein and its association with the nucleic acid.
The energetics of macromolecular interactions are strongly affected by ionic strength. As ionic strength is reduced, protein-RNA interactions become tighter, and the stability of an RNA duplex is decreased. We therefore analyzed the influence of ionic strength on the kinetic step size of NS3. Remarkably, the step size decreases from 16 to 11 bp when salt concentration is reduced from the physiological range to zero additional monovalent salt. A step size value of 11 bp is highly significant because we have observed it before. The same step size was directly visualized in single molecule optical tweezer experiments on NS3, which display repetitive cycles of RNA unwinding and pausing every 11 bp (11). There is a similarity between the optical tweezer experiments and the RNS experiments conducted at low salt, and in both cases the NS3-RNA interactions are enhanced, either through applied tension at the junction or through electrostatic effects, respectively. These conditions may promote activity by a form of NS3 that is not typically observed.
The optical tweezer experiments on RNA unwinding by NS3 provided kinetic parameters that agreed well with those we had obtained using RNS experiments in bulk. In addition to a periodic, stepwise unwinding mechanism, the pause durations, unwinding velocities, the Km values for ATP binding, and other parameters were almost the same within error (11, 12). However, there were important differences in the behavior of NS3 in the optical tweezers. In addition to the smaller step size (11 bp), the tweezer experiments reflected activity by NS3-RNA complexes that formed very rapidly (functional NS3-RNA complexes form slowly in bulk), and processive unwinding by NS3 was only observed when RNA hairpins were subjected to high pulling forces (NS3 is processive in bulk). Based on these and other observations, it was suggested that the experimental setup in the optical tweezers enables NS3 to function as a monomer, whereas in the absence of force, NS3 functions as an oligomer (11).
Given the apparent correlation between step size and oligomeric state of NS3, we set out to create a monomeric form of NS3 to examine its properties and determine whether it has an altered step size. The monomeric form of NS3 was examined using a combination of approaches that included the use of high [RNA] to [NS3] ratios, analysis of rapidly forming RNA-NS3 complexes, short substrates for detection of activity by nonprocessive species, and short overhangs that only permit assembly of monomeric states. These experiments revealed distinct active states of NS3, one of which is a nonprocessive but highly reactive monomer. This population of NS3 dominates the reaction under conditions in which the measured kinetic step size is 11, whereas more processive states of NS3 dominate under conditions in which the step size is 16, thereby demonstrating a link between oligomeric state and kinetic step size of the helicase. These experiments reconcile the apparent discrepancy in reported step sizes, and they establish a model for unwinding that is consistent with both ensemble and single molecule studies.
These findings implicate an unwinding mechanism in which NS3 monomer is the minimal functional unit capable of RNA remodeling. Indeed, increased processivity appears to be the only characteristic of NS3 unwinding that is strongly affected by oligomerization, as kinetic parameters for unwinding remain essentially the same. This indicates that oligomerization only improves the ability of NS3 to remain associated with the RNA substrate, and it does not result in an altered mechanism of unwinding or translocation. Although processivity of NS3 is enhanced by interactions between multiple NS3 molecules in vitro, it remains to be determined whether this represents the mechanism by which NS3 gains high processivity in vivo. Based on findings reported here and elsewhere (15, 27-29), efficient assembly of NS3 oligomers requires higher than physiological NS3 concentrations and availability of long stretches of single-stranded RNA. In addition, all HCV-encoded proteins are translated as a single polyprotein and therefore produced in equimolar amounts. It is therefore unlikely that NS3 exists in large excess relative to other viral proteins involved in RNA replication, e.g. the RNA polymerase NS5B and the NS5A protein. At the same time, NS3 has been shown to specifically interact with other HCV proteins, which may serve as its processivity cofactors during RNA replication (30-32). Given the multifunctional nature of NS3, it remains possible that its oligomerization plays a functional role in some aspects of its action in vivo. For instance, NS3 has been recently implicated in the assembly of infectious HCV particles (33), where its oligomeric forms could potentially assist in packaging of the full size HCV RNA genome. However, because monomeric NS3 appears to be the minimal active form of the helicase, the following discussion will address possible models of RNA unwinding in the context of the active form of NS3 being a monomer.
Single molecule studies on the force dependence of NS3 and the T7 helicases have provided important insights into the physical mechanism of unwinding. For T7 helicase, unwinding rate was shown to increase in response to mechanical force that is applied to destabilize the duplex. Significantly, the gain of unwinding rate correlated with the degree to which mechanical force destabilized the DNA duplex (34). By contrast, the unwinding rate of NS3 was found to be insensitive to any tested levels of pulling force (11). These findings indicate differences in how the helicases interact with nucleic acid substrates, particularly with their single-stranded regions. The T7 ring helicase unwinds DNA by a “strand exclusion” mechanism (35), which implies that one of the unwound strands is threaded into the ring and is involved in extensive interactions with protein subunits of the hexamer, whereas the other is left outside of the ring and is not involved in specific interactions with the helicase. In this model, the destabilizing pulling force is freely transmitted to the single-/double-stranded junction and acts to destabilize the duplex. The fact that unwinding by NS3 helicase cannot be accelerated by pulling on single strands strongly suggests that NS3 interacts with both RNA strands, thus preventing mechanical tension from reaching the junction. This mode of interaction has been directly visualized for NS3 in a FRET-based single molecule study, where both RNA strands were shown to remain associated with NS3 after the substrate duplex was completely unwound (25). To summarize, although T7 helicase appears to act as a simple “wire stripper,” NS3 may use a mechanism that involves specific interactions with both RNA strands after they have been separated. The following model of RNA unwinding by monomeric NS3 aims to consolidate these results with accumulated data on RNA binding, unwinding, and translocation behavior by NS3.
We propose an unwinding mechanism that can be visualized as a cycle of events that includes an 11-bp “super step” (Fig. 12A, counterclockwise starting from the top). According to this model, NS3 translocates directionally along the RNA tracking RNA in 1-nt steps, hydrolyzing one ATP per translocation step (25). Consistent with single molecule FRET data, and single molecule force experiments at low [ATP], unwinding does not take place in single nucleotide steps but in larger steps of ∼3-4 bp, which may represent elementary “rips” of the RNA duplex (11, 18, 25). Thus, each 11-bp super step is composed of three elementary unwinding steps that occur in sequence. Given their dependence on NS3 oligomerization state, observed force dependence of unwinding and FRET observations of RNA sticking, it is likely that the super steps result from interactions between NS3 and the unwound product strands (25). The data are consistent with a model in which the unwound 5′-strand is not immediately released and stays bound to a secondary RNA-binding site through the three unwinding steps shown. After the last unwinding step, the top strand is released from the secondary binding site, thus concluding the 11-bp step. As in many enzymes, product release is rate-limiting (i.e. release of the top strand is the rate-limiting event), and this is why the 11-bp step is the apparent kinetic step size of NS3. Periodic release of the strand is likely to result from conformational or electrostatic strain associated with the accumulation of single-stranded RNA. Although the top strand can be depicted as a loop (Fig. 12A), it may maintain multiple contacts with the protein along its length.
FIGURE 12.

A, model for delayed, periodic strand release by NS3. The cycle depicted here represents a single super step of NS3 unwinding that consists of at least three smaller unwinding steps, as shown (see text for details). Translocation steps of 1 nt along the tracking RNA strand are not shown. B, structure of the NS3 protein showing distribution of positive (blue) and negative (red) electrostatic potential and model for possible exit paths of unwound RNA strands. The model was produced by rigid body docking of the partially unwound DNA substrate from UvrD-DNA complex structure (26) (Protein Data Bank accession number 2IS1) into the NS3 structure (24) (Protein Data Bank accession number 1CU1). The figure was produced in PyMol (DeLano Scientific) with APBS tools.
The presence of a secondary binding site, or “receptacle” for the unwound top strand, is a critical aspect of the model (Fig. 12A), and it is consistent with structural features of the NS3 molecule. Fig. 12B shows the structural model of NS3 in complex with partially unwound duplex that is based on available structures of DNA helicases UvrD (26) and PcrA (36). Interestingly, this model places the unwound portion of the 5′-strand in close proximity to a cluster of arginines and lysines that are exposed on Domain II of NS3 and form an extended region of uninterrupted positive charge. In fact, in an early crystal structure of the NS3 helicase domain (9), this region was assigned as the primary RNA-binding site by a computational analysis of the surface potential, because of its abundant positive charge and elongated shape. This model suggests that the secondary ssRNA-binding site for retaining the top strand may be located on the surface of Domain II and that it is likely to bind ssRNA through electrostatic interactions. The latter is also consistent with the fact that the apparent periodicity of NS3 unwinding can be modulated by changing ionic conditions, presumably via changes in the energetics of electrostatic interactions.
As discussed above, the 11-bp super step appears to be characteristic for monomeric NS3. The mode of NS3-ssRNA interactions illustrated in Fig. 12, A and B, provides an explanation for a larger 16-bp step observed when NS3 oligomers dominate the unwinding reaction. Because super steps represent periodic events of ssRNA release rather than true unwinding steps, oligomeric NS3 may display a larger size of the super step simply because an oligomeric species provides an extended binding site for the separated 5′-strand.
The proposed model points to the step of product release as an important and unexplored part of the NS3 unwinding mechanism. Staying associated with the 5′-RNA strand can allow the helicase to “snap back” while remaining bound to the RNA substrate and thus carry out repetitive cycles of unwinding, as has been directly observed for NS3 in single molecule FRET experiments (25). Such “shuttling” could be important for resolving the highly structured untranslated regions within the HCV RNA, making these regions accessible for the RNA polymerase. Alternatively, “sheltering” a portion of unwound strand on the protein could represent a mechanism by which NS3 counteracts spontaneous re-annealing of separated strands. Future studies will determine the precise role and significance of the product release step for NS3 as well as related SF1 and SF2 helicases.
Acknowledgments
We thank the members of the Pyle laboratory for helpful discussions and comments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM60620 (to A. M. P.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HCV, hepatitis C virus; MOPS, 4-morpholinepropanesulfonic acid; nt, nucleotide; RNS, randomly nicked substrate; ssRNA, single-stranded RNA.
V. Serebrov and R. K. F. Beran, unpublished information.
References
- 1.Gorbalenya, A., and Koonin, E. V. (1993) Curr. Opin. Struct. Biol. 3 419-429 [Google Scholar]
- 2.Pyle, A. M. (2008) Annu. Rev. Biophys. 37 317-336 [DOI] [PubMed] [Google Scholar]
- 3.Jankowsky, E., and Jankowsky, A. (2000) Nucleic Acids Res. 28 333-334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang, Q., and Jankowsky, E. (2005) Biochemistry 44 13591-13601 [DOI] [PubMed] [Google Scholar]
- 5.Jankowsky, E., Gross, C. H., Shuman, S., and Pyle, A. M. (2001) Science 291 121-125 [DOI] [PubMed] [Google Scholar]
- 6.Fairman, M. E., Maroney, P. A., Wang, W., Bowers, H. A., Gollnick, P., Nilsen, T. W., and Jankowsky, E. (2004) Science 304 730-734 [DOI] [PubMed] [Google Scholar]
- 7.Pang, P. S., Jankowsky, E., Planet, P. J., and Pyle, A. M. (2002) EMBO J. 21 1168-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim, J. L., Morgenstern, K. A., Griffith, J. P., Dwyer, M. D., Thomson, J. A., Murcko, M. A., Lin, C., and Caron, P. R. (1998) Structure (Lond.) 6 89-100 [DOI] [PubMed] [Google Scholar]
- 9.Yao, N., Hesson, T., Cable, M., Hong, Z., Kwong, A. D., Le, H. V., and Weber, P. C. (1997) Nat. Struct. Biol. 4 463-467 [DOI] [PubMed] [Google Scholar]
- 10.Cho, H. S., Ha, N. C., Kang, L. W., Chung, K. M., Back, S. H., Jang, S. K., and Oh, B. H. (1998) J. Biol. Chem. 273 15045-15052 [DOI] [PubMed] [Google Scholar]
- 11.Dumont, S., Cheng, W., Serebrov, V., Beran, R. K., Tinoco, I., Jr., Pyle, A. M., and Bustamante, C. (2006) Nature 439 105-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serebrov, V., and Pyle, A. M. (2004) Nature 430 476-480 [DOI] [PubMed] [Google Scholar]
- 13.Ali, J. A., and Lohman, T. M. (1997) Science 275 377-380 [DOI] [PubMed] [Google Scholar]
- 14.Jankowsky, E., Gross, C. H., Shuman, S., and Pyle, A. M. (2000) Nature 403 447-451 [DOI] [PubMed] [Google Scholar]
- 15.Levin, M. K., Wang, Y. H., and Patel, S. S. (2004) J. Biol. Chem. 279 26005-26012 [DOI] [PubMed] [Google Scholar]
- 16.Lucius, A. L., Vindigni, A., Gregorian, R., Ali, J. A., Taylor, A. F., Smith, G. R., and Lohman, T. M. (2002) J. Mol. Biol. 324 409-428 [DOI] [PubMed] [Google Scholar]
- 17.Lohman, T. M., and Bjornson, K. P. (1996) Annu. Rev. Biochem. 65 169-214 [DOI] [PubMed] [Google Scholar]
- 18.Cheng, W., Dumont, S., Tinoco, I., Jr., and Bustamante, C. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 13954-13959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donmez, I., Rajagopal, V., Jeong, Y. J., and Patel, S. S. (2007) J. Biol. Chem. 282 21116-21123 [DOI] [PubMed] [Google Scholar]
- 20.Beran, R. K., Bruno, M. M., Bowers, H. A., Jankowsky, E., and Pyle, A. M. (2006) J. Mol. Biol. 358 974-982 [DOI] [PubMed] [Google Scholar]
- 21.Freier, S. M., Kierzek, R., Jaeger, J. A., Sugimoto, N., Caruthers, M. H., Neilson, T., and Turner, D. H. (1986) Proc. Natl. Acad. Sci. U. S. A 83 9373-9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin, M. K., and Patel, S. S. (2002) J. Biol. Chem. 277 29377-29385 [DOI] [PubMed] [Google Scholar]
- 23.Kim, J. L., Morgenstern, K. A., Lin, C., Fox, T., Dwyer, M. D., Landro, J. A., Chambers, S. P., Markland, W., Lepre, C. A., O'Malley, E. T., Harbeson, S. L., Rice, C. M., Murcko, M. A., Caron, P. R., and Thompson, J. A. (1996) Cell 87 343-355 [DOI] [PubMed] [Google Scholar]
- 24.Yao, N., Reichert, P., Taremi, S. S., Prosise, W. W., and Weber, P. C. (1999) Structure (Lond.) 7 1353-1363 [DOI] [PubMed] [Google Scholar]
- 25.Myong, S., Bruno, M. M., Pyle, A. M., and Ha, T. (2007) Science 317 513-516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, J. Y., and Yang, W. (2006) Cell 127 1349-1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tackett, A. J., Chen, Y., Cameron, C. E., and Raney, K. D. (2005) J. Biol. Chem. 280 10797-10806 [DOI] [PubMed] [Google Scholar]
- 28.Sikora, B., Chen, Y., Lichti, C. F., Harrison, M. K., Jennings, T. A., Tang, Y., Tackett, A. J., Jordan, J. B., Sakon, J., Cameron, C. E., and Raney, K. D. (2008) J. Biol. Chem. 283 11516-11525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levin, M. K., and Patel, S. S. (1999) J. Biol. Chem. 274 31839-31846 [DOI] [PubMed] [Google Scholar]
- 30.Dimitrova, M., Imbert, I., Kieny, M. P., and Schuster, C. (2003) J. Virol. 77 5401-5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindenbach, B. D., and Rice, C. M. (2005) Nature 436 933-938 [DOI] [PubMed] [Google Scholar]
- 32.Jennings, T. A., Chen, Y., Sikora, D., Harrison, M. K., Sikora, B., Huang, L., Jankowsky, E., Fairman, M. E., Cameron, C. E., and Raney, K. D. (2008) Biochemistry 47 1126-1135 [DOI] [PubMed] [Google Scholar]
- 33.Ma, Y., Yates, J., Liang, Y., Lemon, S. M., and Yi, M. (2008) J. Virol. 82 7624-7639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson, D. S., Bai, L., Smith, B. Y., Patel, S. S., and Wang, M. D. (2007) Cell 129 1299-1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donmez, I., and Patel, S. S. (2006) Nucleic Acids Res. 34 4216-4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velankar, S. S., Soultanas, P., Dillingham, M. S., Subramanya, H. S., and Wigley, D. B. (1999) Cell 97 75-84 [DOI] [PubMed] [Google Scholar]
- 37.Lucius, A. L., Maluf, N. K., Fischer, C. J., and Lohman, T. M. (2003) Biophys. J. 85 2224-2239 [DOI] [PMC free article] [PubMed] [Google Scholar]