

Abstract
Arginine deprivation as an anti-cancer therapy has historically been met with limited success. The development of pegylated arginine deiminase (ADI-PEG20) has renewed interest in arginine deprivation for the treatment of some cancers. The efficacy of ADI-PEG20 is directly correlated with argininosuccinate synthetase (ASS) deficiency. CWR22Rv1 prostate cancer cells do not express ASS, the rate-limiting enzyme in arginine synthesis, and are susceptible to ADI-PEG20 in vitro. Interestingly, apoptosis by 0.3 μg/mL ADI-PEG20 occurs 96 hours post treatment and is caspase-independent. The effect of ADI-PEG20 in vivo reveals reduced tumor activity by microPET as well as reduced tumor growth as a monotherapy and in combination with docetaxel against CWR22Rv1 mouse xenografts. In addition, we demonstrate autophagy is induced by single amino acid depletion by ADI-PEG20. Here, autophagy is an early event that is detected within 1 to 4 hours of 0.3 μg/mL ADI-PEG20 treatment and is an initial protective response to ADI-PEG20 in CWR22Rv1 cells. Significantly, the inhibition of autophagy by chloroquine and Beclin1 siRNA knockdown enhances and accelerates ADI-PEG20-induced cell death. PC3 cells, which express reduced ASS, also undergo autophagy and are responsive to autophagy inhibition and ADI-PEG20 treatment. In contrast, LNCaP cells highly express ASS and are therefore resistant to both ADI-PEG20 and autophagic inhibition. These data point to an interrelationship among ASS deficiency, autophagy, and cell death by ADI-PEG20. Finally, a tissue microarray of 88 prostate tumor samples lacked expression of ASS, indicating ADI-PEG20 is a potential novel therapy for the treatment of prostate cancer.
Keywords: Autophagy, arginine deiminase, prostate cancer
Introduction
The initial observations that various tumor cells are susceptible to arginine deprivation were made over 40 years ago, though appropriate therapeutic methods have hindered further development of this approach until recently. Arginine deiminase (ADI), an enzyme isolated from Mycoplasma (1, 2), degrades arginine into its citrulline precursor. In its native form, it is strongly antigenic with a half-life of 5 hours (3). Conjugation to 20,000mw polyethylene glycol (ADI-PEG20) decreases antigenicity as well as dramatically increases serum half-life, allowing weekly administration that reduces plasma arginine to undetectable levels (4, 5). Various tumor types (hepatocellular carcinomas, melanomas, mesotheliomas, renal cell carcinomas, pancreatic carcinomas) have been shown to lack expression of argininosuccinate synthetase (ASS) (4, 6–8), a ubiquitous enzyme involved in the two-step synthesis of arginine from citrulline (9). Unable to synthesize their own arginine, ASS deficient cells depend on relatively inefficient amino acid transporters (10). In the setting of ASS deficiency, ADI-PEG20 depletes intracellular arginine by reducing extracellular levels available for transmembrane uptake while unaffecting cells with preserved ASS expression capable of endogenous arginine biosynthesis (11). Previous in vitro studies show the growth of prostate cancer PC3 cells is inhibited when arginine is eliminated from cell culture media (12), indicating ADI-PEG20 may be an effective therapy for prostate cancers.
The anti-tumor effects of ADI-PEG20 elicit a G1 cell cycle arrest with eventual apoptosis in a number of tumor cell lines (13). In addition, ADI-PEG20 is anti-angiogenic, inhibiting migration and tube formation in HUVE cells (14) and neovascularization of neuroblastomas in vivo (15). However, other cellular effects of arginine starvation by ADI-PEG20 are still unknown.
Nutrient depletion triggers a process called macroautophagy (hereafter referred to as autophagy), an evolutionary conserved eukaryotic process in which organelles and bulk proteins are turned over by lysosomal activity. Autophagy serves to provide ATP and other macromolecules as energy sources during metabolic stress (16, 17). The most distinctive feature of autophagy is the formation of the autophagosome, a double membrane vesicle that fuses with lysosomes for hydrolytic cleavage of engulfed proteins and organelles. In mammalian cells, microtubule-associated protein 1 light chain 3 (LC3) is processed by lipid conjugation to phosphatidylethanolamine for insertion into the autophagosome membrane (18). Translocation and processing of an eGFP-LC3 fusion protein are often used as markers for autophagic activity.
Autophagy has recently gained much attention for its paradoxical roles in cell survival and cell death, particularly in the pathogenesis as well as the treatment of cancer (19, 20). Regulation of autophagy is highly complex with inputs from the cellular environment through the PI3K/Akt/mTOR pathway (21), members of the Bcl2 family (22), p53 (23), and death associated protein kinases (DAPk) (24). Not surprisingly, there is an intricate relationship between autophagy and apoptosis. Whether autophagy enables cells to survive or enhances their death is context-driven, depending on the type of stimuli, nutrient availability, organism development, and apoptotic status. We hypothesize prostate cancer cells that are ASS deficient are sensitive to arginine deprivation by ADI-PEG20 and consequently, undergo autophagy as an initial survival response.
In this study, we show susceptibility of several prostate cancer cell lines to ADI-PEG20 correlates with the absence of ASS expression. Due to the lack of ASS, ADI-PEG20 induces a late caspase-independent cell death in CWR22Rv1 in vitro. Metabolic activity by microPET imaging of CWR22Rv1 xenografts in nude mice was reduced by ADI-PEG20. Tumor growth was significantly inhibited by ADI-PEG20 alone as well as in combination with docetaxel. ADI-PEG20 also induces autophagy within hours of treatment. However, inhibition of autophagy prematurely leads to cell death by ADI-PEG20. With the success of ADI-PEG20 therapy for hepatocellular carcinomas and melanomas and our findings that prostate cancer specimens lack ASS expression, ADI-PEG20 can potentially be extended to clinical trials for prostate cancer. Moreover, combination with standard chemotherapies or autophagy-targeting drugs represents multi-pronged approaches to cancer therapy.
Materials and methods
Reagents
Recombinant ADI formulated with multiple linear 20,000mw polyethylene glycol molecules (ADI-PEG20) was generously provided by DesigneRx Pharmaceuticals, Inc (Vacaville, CA). Specific enzyme activity was 7.4IU/mg. Internal calibration of enzyme IC50 was determined with each batch.
Cells and cell culture
All cell lines were cultured in RPMI-1640 (10% fetal bovine serum, 1% penicillin, streptomycin, glutamine). LNCaP cells were cultured in serum free, phenol free media before 10nM 5α-Dihydrotestosterone (DHT) (Sigma) treatment for 4, 24, and 48 hours. PC3 cells were transiently transfected and CWR22Rv1 cells were stably transfected with eGFP-LC3 plasmid (JUJ) using Effectene (Qiagen).
RT-PCR and qRT-PCR
Total RNA was isolated from cultured cells by TRIzol (Invitrogen) homogenization and reverse transcribed using MMLV (Invitrogen). 100ng cDNA was PCR amplified as described previously (25). Primers: ASS (F) 5′-GACGCTATGTCCAGCAAAG-3′ and (R) 5′-TTGCTTTGCGTACTCCATCAG-3′; GAPDH (F) 5′-ACCACAGTCCATGCCATCAC-3′ and (R) 5′-TCCACCACCCTGTTGCTGTA-3′. Total RNA from primary prostate tissues was reverse transcribed using SuperScriptIII (Invitrogen). 100ng cDNA was amplified by iQ5 iCycler thermal cycler (Bio-Rad) and monitored by SYBRGreen (Invitrogen) for real time PCR. Threshold cycle values were normalized against actin and analyzed using QGene software. Primers: ASS (F) 5′-TGAAATTTGCTGAGCTGGTG-3′ and (R) 5′-ATGTACACCTGGCCCTTGAG-3′; Actin (F) 5′-TCCTTAATGTCACGCACGATTT-3′ and (R) 5′-GAGCGCGGCTACAGCTT-3′.
Immunoblotting
Cellular lysates were resolved on SDS-PAGE and electrophoretically transferred to PVDF membranes. Membranes were incubated with primary antibody followed by species-specific horseradish peroxidase secondary antibody. Immunoreactive material was detected by chemiluminescence (Pierce Laboratories). Antibodies: actin (Santa Cruz Biotechnology), ASS (BD Biosciences), caspase-3 (Biosource), GAPDH (Chemicon), tubulin (Sigma), Beclin1, phospho-AMP kinase (Thr172), phospho-mTor (Ser2481), phospho-S6 kinase (Thr389), phospho-S6 (Ser235/236), LC3, ERK1/2, and phospho-ERK1/2 (Cell Signaling).
MTT cytotoxicity assay
RWPE-1, LNCaP, PC3, and CWR22Rv1 cells were seeded in 96-well plates and treated with serial dilutions of ADI-PEG20. After 6 days, thiazolyl blue tetrazolium bromide (MTT) (Sigma) was added for a final concentration of 0.5mg/mL. PC3 cells were treated for 3 days in 2% FBS. Formazan crystals were solubilized by 10% SDS. The IC50 is the drug concentration at which 50% of cell growth is inhibited.
FACS Analysis for sub-G1 DNA fragmentation
CWR22Rv1 cells were treated with 0.3μg/mL ADI-PEG20, 100nM paclitaxel (Sigma), or pre-treated with 50μM z-VAD-fmk (MBL International). Cells were analyzed by flow cytometry as described previously (8).
Active Caspase-3 ELISA
CWR22Rv1 cells were seeded in 6-well plates and treated with 100nM paclitaxel or 0.3μg/mL ADI-PEG20 for 24 hours. Treatment groups were compared to cells pretreated with 50μM z-VAD-fmk for 2 hours before assaying for activated caspase-3 by ELISA (R&D Systems).
MicroPET imaging
Nude mice with CWR22Rv1 subcutaneous xenografts were injected via tail vein with 120mCi of 18F-FDG and imaged by positron emission tomography as described previously (26) before and after 5IU ADI-PEG20 treatment of 4 or 24 hours. Standard uptake values (SUV) were computed by dividing the activity concentration in each voxel by the injected dose and multiplying by animal weight. Absolute uptake values of posttreatment images were normalized to pretreatment images prior to analysis.
Xenograft efficacy studies
For tumorigenesis, 1x106 CWR22Rv1 cells were injected subcutaneously into the bilateral flanks of male athymic Balb/c mice (Harlan Sprague Dawley, Inc). Mice received weekly 0.5mL intraperitoneal injections of sterile PBS (n=4), 10mg/kg docetaxel (n=4), 5 international units (IU) (225 μg/mL) ADI-PEG20 (n=4), or both 10mg/kg docetaxel and 5IU ADI-PEG20 (1mL total volume) (n=4). Tumor dimensions were measured twice weekly. Tumor volumes were calculated by V=0.5236(LxW2), L=length, W=width.
Fluorescence Microscopy for LC3
CWR22Rv1 and PC3 cells overexpressing eGFP-LC3 were seeded on poly-lysine coated coverslips. Cells were treated with 0.3μg/mL ADI-PEG20 for 4 or 24 hours or 2μM rapamycin for 4 hours. Cells were fixed, mounted using SlowFade with DAPI (Invitrogen), and examined under a 60x lens on an Olympus BX61 motorized reflected fluorescence microscope with an AMCA filter (excitation, 350nm/emission, 460nm) for DAPI and FITC filter (excitation, 480nm/emission, 535nm) for eGFP-LC3 using Slide Book 4.1 software (Intelligent Imaging Innovations).
For live cell imaging, CWR22Rv1 cells overexpressing eGFP-LC3 were plated on 35mm #1 glass bottom dishes (WillCo Wells), treated with 0.3μg/mL ADI-PEG20, and imaged with an IX-71 inverted microscope with a 100x 1.40 NA oil objective (Olympus) and ASI 400 air stream incubator (NEVTEK) set to 37°C. Images were acquired using a spinning disk system.
Inhibition of autophagy
CWR22Rv1 cells were treated with 25μM chloroquine (Sigma), 0.1μg/mL ADI-PEG20, or both for 24, 48, 72, and 96 hours. LNCaP cells were treated as above except with 0.3μg/mL ADI-PEG20. Cells were analyzed by FACS analysis as described previously. CWR22Rv1 cells were seeded in 6-well plates then transiently transfected with 100pmol eGFP siRNA (Ambion) or Beclin1 siRNA ON-TARGETplus SMARTpool (Dharmacon) using DharmaFECT reagent (Dharmacon). Cells were treated with 0.3μg/mL ADI-PEG20 for 24 or 48 hours the following day and analyzed by FACS analysis as described previously.
PC3 cells were treated with 0.3μg/mL ADI-PEG20, 5μg/mL ADI-PEG20, 1mM 3-methyladenine (Sigma), or both for 24, 48, and 72 hours and analyzed by MTT as described previously.
ASS immunohistochemistry
Formalin-fixed, paraffin-embedded archival material from 88 prostate tumors and 59 normal prostate samples were obtained. Tumors represent a range of Gleason grades (3+3=6 to 4+5=9). Hematoxylin and eosin stained sections were made from each block to define representative tumor regions, and a tumor microarray (TMA) was constructed. TMA paraffin blocks were sectioned at 4μm and transferred to glass slides. Immunohistochemistry was performed using α-ASS monoclonal mouse antibody (DesigneRx Pharmacologies) at 2.2μg/mL. Normal liver was used as a positive control. Omission of primary antibody was used as negative control. Sections were counterstained with Gill’s hematoxylin and fixed. Slides were independently examined by a board certified anatomic pathologist (RGE) three times and scored by percentage of cells stained.
Results
Sensitivity to ADI-PEG20 correlates with ASS expression
ASS expression in three commonly cultured prostate carcinoma cell lines (LNCaP, PC3, CWR22Rv1) was evaluated for mRNA and protein levels. LNCaP is androgen dependent whereas PC3 and CWR22Rv1 are androgen independent. The normal immortalized cell line RWPE-1 was used to evaluate ASS expression in non-cancerous prostate cells. All cell lines expressed ASS mRNA determined by RT-PCR except CWR22Rv1 (Fig 1a). Quantitative real time PCR of ASS mRNA in the prostate cancer cell lines revealed that, relative to CWR22Rv1, LNCaP and PC3 expressed ASS transcript 6.7 and 1.4 times greater, respectively. Western blot analysis showed CWR22Rv1 did not express ASS protein; in contrast, PC3 expressed moderate levels while LNCaP and RWPE-1 expressed high levels of ASS (Fig 1b). Disparity between ASS mRNA and protein levels is potentially attributed to nonproductive, alternatively spliced transcripts or pseudeogenes1. The relationship between androgen status and ASS expression was further examined by treating LNCaP cells with 10nM DHT (Fig 1c), revealing androgens do not regulate ASS expression.
Figure 1. Prostate cancer cell lines profiled for ASS expression and ADI-PEG20 sensitivity.
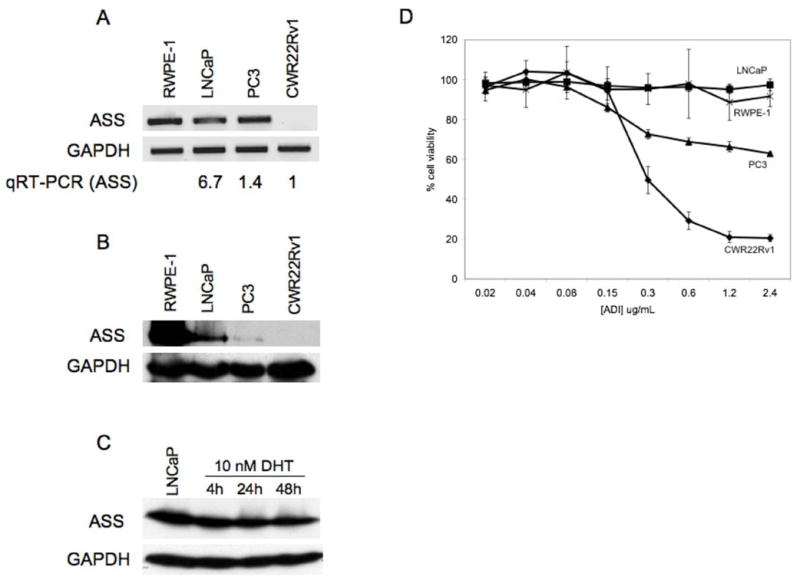
A, RWPE-1, LNCaP, PC3, and CWR22Rv1 were examined for ASS mRNA by RT-PCR and B, ASS protein by immunoblotting. C, Immunoblot for 10nM DHT timecourse of LNCaP against α-ASS. D, Cell lines were treated by ADI-PEG20 at 0.02, 0.04, 0.08, 0.15, 0.3, 0.6, 1.2, and 2.4μg/mL for 3 (PC3) or 6 days before MTT assay. Values are reported as mean±SD.
To evaluate the effect of ADI-PEG20 on prostate carcinoma, the previously described cell lines were treated with ADI-PEG20 over a broad dose range and assayed for cytotoxicity with MTT. CWR22Rv1 was the most sensitive to ADI-PEG20 with an IC50 of 0.3μg/mL. PC3 was moderately sensitive to ADI-PEG20, while LNCaP and RWPE-1 were not responsive to ADI-PEG20 (Fig 1d). Taken together, these data confirm that ASS protein levels inversely correlate with sensitivity to ADI-PEG20. CWR22Rv1 was subsequently chosen as the model cell line for future experiments.
ADI-PEG20 induces caspase-independent apoptosis in CWR22Rv1 in vitro
To study whether the reduced viability of CWR22Rv1 upon ADI-PEG20 treatment is due to cell growth arrest, apoptosis, or both, we subjected treated and untreated cells to FACS analysis. The sub G1 DNA content was used as an indicator of apoptosis induced by ADI-PEG20. CWR22Rv1 cells were treated with 0.3μg/mL ADI-PEG20 for 4, 24, 48, 72, and 96 hours. Apoptosis was not induced until 4 days post-treatment, when about 30% of cells had undergone apoptosis (Fig 2a). While DNA fragmentation is considered the defining endpoint in apoptosis, caspase cleavage is an early marker for classical apoptosis. Interestingly, cleavage of caspase-3 into its activated 17kDa fragment was undetected after ADI-PEG20 (Fig 2b).
Figure 2. ADI-PEG20 induces caspase-independent apoptosis in CWR22Rv1.

A, 0.3μg/mL ADI-PEG20 timecourse of CWR22Rv1 before FACS analysis for sub-G1 DNA content. Values are reported as mean±SE. B, Immunoblot for 0.3μg/mL ADI-PEG20 timecourse of CWR22Rv1. α-Caspase-3 detects the 32kDa pro-form and the activated 17kDa cleavage product. C, CWR22Rv1 cells were treated with vehicle (untreated), 100nM paclitaxel, or 0.3μg/mL ADI-PEG20 for 24 hours and compared to 2 hour pre-treatment with 50μM z-VAD-fmk before caspase-3 ELISA. Values were normalized to vehicle. D, CWR22Rv1 cells were treated with 50μM z-VAD-fmk, 0.3μg/mL ADI-PEG20, ADI-PEG20+z-VAD-fmk for 96 hours, and 100nM paclitaxel, paclitaxel+z-VAD-fmk for 24 hours before FACS analysis for sub-G1 DNA content.
Caspase-independent cell death was further investigated with z-VAD-fmk, a pan-caspase inhibitor. Caspase inhibition was confirmed with caspase-3 ELISA (Fig 2c). z-VAD-fmk led to a 50% reduction of activated caspase-3 levels in cells treated with paclitaxel, a standard chemotherapy for advanced and metastatic prostate cancer. However, ADI-PEG20 did not significantly alter active caspase-3 levels. While z-VAD-fmk attenuated apoptosis in cells treated with paclitaxel by approximately 50%, it did not affect the fraction of apoptotic cells after ADI-PEG20 (Fig 2d). These data suggest that cell death mediated by ADI-PEG20 is independent of caspase-mediated pathways.
ADI-PEG20 decreases global tumor metabolic activity
The immediate effect of ADI-PEG20 in vivo was examined using positron-emission tomography. Global tumor metabolism of glucose consumption was monitored by 18F-fluorodeoxyglucose (18F-FDG) in CWR22Rv1 mouse xenografts. MicroPET scans were performed before and after ADI-PEG20 treatment of 4 or 24 hours. 18F-FDG uptake in CWR22Rv1 tumors (arrows) did not change after 4 hours of treatment. In contrast, 18F-FDG uptake was decreased after 24 hours of ADI-PEG20. Tumor SUV decreased 30% after treatment (0.00086 vs. 0.0006), indicating reduced metabolic activity (Fig 3a).
Figure 3. ADI-PEG20 is an effective agent in vivo.

A, Mice with CWR22Rv1 xenografts were imaged by PET using 18F-FDG before and after treatment with 5IU ADI-PEG20 for 4 or 24 hours. B, Mice with CWR22Rv1 xenografts were treated with PBS vehicle, 10mg/kg docetaxel, 5IU ADI-PEG20, or 5IU ADI-PEG20+10mg/kg docetaxel weekly. Tumor volumes are reported as mean±SE.
ADI-PEG20 retards CWR22Rv1 tumor growth in vivo and synergizes with taxane
To determine the long term effects of ADI-PEG20 in vivo, nude athymic mice with subcutaneous CWR22Rv1 xenografts were injected intraperitoneally with control PBS or 5IU ADI-PEG20 weekly. Tumors from ADI-PEG20 mice were significantly smaller than tumors from control mice (157.6mm3 vs. 1108.99mm3) at 13 days after initiation of treatment when control mice were euthanized. The effects of ADI-PEG20 were compared to the current standard of care for hormone refractory prostate cancer patients, docetaxel alone (27), and docetaxel in combination. Docetaxel mice (10 mg/kg) had tumors that were smaller but not statistically significant from control mice. However, the combination of ADI-PEG20 and docetaxel had a synergistic effect on tumor growth inhibition. Tumors from ADI-PEG20 mice reached an average of 910 mm3 at the end of the study while tumors from ADI-PEG20/docetaxel treated mice were about 75% smaller (Fig 3b).
ADI-PEG20 induces autophagy in prostate cancer cells
Arginine degradation by ADI-PEG20 causes metabolic stress to auxotrophic cells. Nutrient starvation such as complete amino acid deprivation is a known inducer of autophagy (28). To determine whether single amino acid deprivation is sufficient to trigger autophagy, CWR22Rv1 cells stably expressing eGFP-LC3 were examined under fluorescence microscopy. Under normal conditions, LC3-I is uniformly distributed throughout the nucleus and cytoplasm. During autophagy, LC3-I is processed into LC-II and translocates into autophagosome membranes, appearing as bright punctae (29). LC3-II localization was seen in fixed CWR22Rv1 cells after 4 and 24 hours of 0.3μg/mL ADI-PEG20 treatment. Rapamycin, an inhibitor of mTOR, was used as a positive control (Fig 4a, top). Live cell imaging of CWR22Rv1 cells revealed rapid and intense autophagosome formation after only 90 minutes of ADI-PEG20 (Fig 4a, bottom). Rapamycin or ADI-PEG20 significantly increased the number of cells undergoing autophagy to 15% (Fig 4a). The LC3-II fragment appeared as early as 30 minutes of ADI-PEG20 and persisted after 24 hours of arginine deprivation. Increase in total autophagic flux was confirmed with chloroquine (30), an autophagy inhibitor that disrupts lysosomal function (Fig 4b) and prevents completion of autophagy, resulting in an accumulation of LC3-II. In addition, potential off-target effects of chloroquine did not lead to caspase-3 cleavage.
Figure 4. ADI-PEG20 induces autophagy in CWR22Rv1.

A, CWR22Rv1 cells overexpressing eGFP-LC3 were treated with 0.3μg/mL ADI-PEG20 for 1.5, 4, and 24 hours, or 2 μM rapamycin for 4 hours. Punctae represent autophagosome formation. Autophagic cells were quantified from random image fields totaling 200 cells and reported as mean±SE. B, Immunoblot for 0.3μg/mL ADI-PEG20 timecourse of CWR22Rv1. α-LC3 detects LC3-I and LC3-II. Autophagic flux was confirmed by co-administering 25μM chloroquine with 0.3μg/mL ADI-PEG20. α-Caspase-3 detects the 32kDa pro-form and the activated 17kDa cleavage product. C, Immunoblot for 0.3μg/mL ADI-PEG20 timecourse of CWR22Rv1 using α-phopsho-AMP kinase, α-phospho-mTor, α-phospho-S6 kinase, α-phospho-S6 protein, α-phospho-ERK1/2, and α-ERK1/2. Loading control for phospho-mTor was verified using α-tubulin (not shown).
Molecular pathways accompanying the induction of autophagy were also investigated. A major nutrient-sensing pathway involves AMPK/TSC/mTOR/S6K. During nutrient starvation, ATP level decreases and AMP level increases, resulting in activation and phosphorylation of AMPK. ADI-PEG20 immediately increased phospho-AMPK levels (Fig 4c). This should lead to inactivation and decreased phosphorylation of mTOR kinase through the inhibition of TSC complex by AMPK induced phosphorylation. Decreased phosphorylation of mTOR was evident soon after ADI-PEG20 treatment (Fig 4c). A downstream mTOR effector, S6K, was inactivated at a later stage (>24 hours) as shown by its own decreased phosphorylation and the decreased phosphorylation of its substrate S6. Transient increase of S6K activity was observed at early ADI-PEG20 timepoints. The exact mechanism of this phenomenon is unclear, but is likely due to feedback of this kinase as reported by others (31). AMPK activation and mTOR down-modulation are compatible with their roles of major autophagy regulators. We also surveyed other kinase pathways relevant to autophagy. ERK1/2 phosphorylation was evident within 30 minutes of ADI-PEG20 treatment, which increased in a time-dependent manner (Fig 4c). ERK1/2 activation has been shown previously to contribute to autophagy induced pro-survival function (32).
Autophagy delays and protects against ADI-PEG20-induced cell death
The paradoxical relationship between autophagy and apoptosis necessitates determination of the causal nature between these two fundamental biological processes after arginine deprivation. Temporally, autophagy precedes apoptosis; thus, inhibition of autophagy may modulate the onset of apoptosis.
Chemical inhibition of autophagy with chloroquine accelerated and enhanced ADI-PEG20-induced cell death in CWR22Rv1 (Fig 5a). By 48 hours, 27% of ADI-PEG20 + chloroquine cells were apoptotic compared to 11% and 6% of cells undergoing apoptosis by chloroquine alone and ADI-PEG20 alone, respectively. Chloroquine further increased ADI-PEG20-induced cell death to 60% after 72 hours. By 96 hours, the effect of chloroquine was abrogated, possibly due to its metabolism. Similarly, siRNA knockdown of Beclin1, essential for autophagosome nucleation (21), also increased the rate of cell death after ADI-PEG20 treatment (Fig 5b). Almost 60% of cells had undergone apoptosis if Beclin1 was knocked down before 48 hours of ADI-PEG20, while ADI-PEG20 alone only led to apoptosis in 30% of cells. In contrast, ADI-PEG20, chloroquine, and the combination of ADI-PEG20 and chloroquine had no effect on apoptosis at all time points in the ASS expressing LNCaP cells (Fig 5c). To complete the characterization of the relationship of ASS expression and sensitivity to ADI-PEG20, we examined cellular response in PC3, a cell line with low ASS levels. Higher doses of ADI-PEG20 (5μg/mL) were required to arrest cell growth completely compared to CWR22Rv1, though lower doses (0.3μg/mL) induced autophagy (Fig 5d). Inhibiting autophagy with 3-methyladenine (3-MA) greatly reduced cell viability following treatment with low dose ADI-PEG20 (Fig 5d). Therefore, ASS protein level correlates with cellular response to ADI-PEG20, including the early induction of autophagy before the late onset of apoptosis.
Figure 5. Inhibition of autophagy accelerates and enhances ADI-PEG20-induced cell death.

A, Timecourse of CWR22Rv1 cells treated with vehicle (untreated), 25μM CQ, 0.1μg/mL ADI-PEG20, or ADI-PEG20+CQ before FACS analysis for sub-G1 content. Values are reported as mean±SE. B, Immunoblot for CWR22Rv1 cells transfected with mock or 100pmol Beclin1 siRNA to assess knockdown. CWR22Rv1 cells were treated with vehicle (untreated), 0.3μg/mL ADI-PEG20, 100pmol eGFP siRNA, 100pmol Beclin1 siRNA, or Beclin1 siRNA+ADI-PEG20 for 24 and 48 hours before FACS analysis for sub-G1 content. C, LNCaP cells were treated and analyzed as described in A with 0.3μg/mL ADI-PEG20. D, Growth of PC3 cells were treated with vehicle (untreated), 0.3μg/mL ADI-PEG20, 1mM 3-MA, 0.3μg/mL ADI-PEG20+1mM 3-MA, 5μg/mL ADI-PEG20, or 5μg/mL ADI-PEG20+1mM 3-MA by MTT assay. Values are reported as mean±SD. PC3 cells overexpressing eGFP-LC3 were treated with 0.3μg/mL ADI-PEG20 for 4 or 24 hours. Punctae represent autophagosome formation. U=untreated, CQ=chloroquine, 3-MA=3-methyladenine, A=ADI-PEG20
ASS expression in prostate cancer tissue
The above results suggest arginine deprivation by ADI-PEG20 may offer a new treatment strategy for prostate cancers in which ASS expression is low. A key question that follows is whether the absence of ASS expression is generalizable among diverse human prostate cancer specimens. We therefore examined ASS expression by immunohistochemistry in our prostate tissue microarray. Of the 88 human prostate tumors, none demonstrated any detectable ASS staining. Strong cytoplasmic ASS staining was observed, indicated by closed arrows, in the luminal cells of benign prostate glands (Fig 6a) and normal prostate tissue (Fig 6b, left). However, no ASS reactivity was detected in prostate cancer glands (open arrows) (Fig. 6a) or tissue (Fig 6b, right). Among 59 samples of normal prostate tissue, 27% expressed ASS to some degree. Of the 16 samples showing ASS expression, 2 were found to have ASS in >75% of the cells while the remaining 14 showed expression in <25% of the cells. In addition, ASS mRNA expression was evaluated in 6 primary prostate tumor tissues and 2 primary benign prostatic hyperplasia tissues. ASS mRNA was almost absent in specimen 108 and reduced in all other samples (Fig 6c). The differential expression of ASS is in contrast to hepatocytes, which heavily depend on ASS function for the urea cycle, and uniformly stained for cytoplasmic ASS protein (Fig 6d).
Figure 6. ASS Expression in Prostate Tissue.

A, Prostate cancer tissue with ASS(+) benign glands (closed arrows) and ASS(−) cancerous glands (open arrows) by immunohistochemistry. B, Left, Normal prostate tissue. Closed arrows point to luminal ASS staining. Right, Prostate cancer tissue with no ASS reactivity. C, mRNA from primary prostate tissue was examined for ASS expression by RT-PCR. D, Normal liver as a positive control for cytoplasmic ASS staining. BPH=Benign prostatic hyperplasia.
Discussion
In this report, we showed ADI-PEG20 can effectively induce cell death in prostate cancer cells with low or absent ASS expression. It also sensitizes cells to treatment with docetaxel, an accepted chemotherapy in prostate cancer, or chloroquine, an inhibitor of autophagy. These results are likely to be generally applicable to other prostate cancer cells, since virtually all prostate cancer specimens examined in this report as well as that by Clark, et al (12) expressed undetectable levels of ASS. By depletion of arginine, ADI-PEG20 causes metabolic stress on auxotrophic cells, complimenting conventional therapies largely based on genotoxic stress. Although arginine deprivation therapy based on bovine arginase has seen limited applications clinically, ADI-PEG20 has 1000-fold greater affinity for arginine (33) with fewer side effects. Our work described here thus offers a new treatment option for prostate cancer. In addition, we uncover novel cellular responses of arginine depletion, including autophagy and caspase-independent cell death.
The delayed onset of apoptosis suggests the possibility of compensation mechanisms after arginine depletion. Here, we present evidence for the first time that single amino acid starvation through arginine degradation by ADI-PEG20 is sufficient to trigger autophagy in prostate cancer cells. LC3 translocation and cleavage occur within hours of ADI-PEG20 treatment, indicating that autophagy is an early response. AMPK senses cellular AMP/ATP ratio, and in its phosphorylated form, signals the lack of nutrients in the environment to the mTOR complex via TSC2 (34). Inhibition of mTOR leads to suppression of S6K activity. Consistent with our findings, Feun, et al. have reported the effects of ADI-PEG20 on mTOR signaling which include dephosphorylation of mTOR downstream effectors S6K and 4E-BP and increased phosphorylation of AMPK in ASS-negative melanoma cell lines (35). This chain of events has been shown to promote autophagy (36). There are various signaling cascades that regulate mTOR/S6K including the PI3K (class I)/Akt pathway; inhibition of which has been shown to induce autophagy in malignant gliomas (32, 37). While we did not specifically examine the activation of the PI3K (class I)/Akt pathway, ADI-PEG20 inhibited mTOR events associated with a rapid activation of AMPK, suggesting this mechanism in arginine deprivation-induced autophagy. Furthermore, we observed ADI-PEG20 induced ERK1/2 activation, which has been shown to regulate autophagy under a variety of stimuli (32, 38).
What is the biological function of ADI-PEG20 induced autophagy? Autophagy can be pro-survival or pro-death, depending on cellular context and duration of treatment. To study whether ADI-PEG20 induced autophagy contributes to or attenuates cell death, we chose to block ADI-PEG20 induced autophagy with the inhibitor chloroquine, which inhibits late stage autophagy by alkalinizing lysosomes and disrupting the autophagolysosome (39). Since chloroquine itself may have functions other than inactivating lysosomes (40), we also employed siRNA targeting an essential component of autophagy, Beclin1, a component of the class III PI3 kinase complex that nucleates autophagosomes (29). Our data show inhibition of early stage autophagy by chloroquine or Beclin1 knockdown accelerates and enhances cell death following ADI-PEG20, strongly suggesting ADI-PEG20 induced autophagy triggers a protective response during early stages of treatment. At present, we cannot rule out that prolonged ADI-PEG20 treatment (>96 hours) may trigger autophagic cell death (programmed cell death type II), which is usually caspase-independent. In our study, we found chloroquine itself had little effect on the cell killing of CWR22Rv1, unless ADI-PEG20 is present and autophagy is induced. In addition, co-administration of chloroquine with ADI-PEG20 did not activate caspase-3. This again suggests that the major effect of chloroquine is to block autophagy, enhancing the underlying mechanism of caspase-independent apoptosis. Consistent with this result, PC3 cells with reduced ASS levels also underwent autophagy after ADI-PEG20 treatment. The inhibition of autophagy with 3-methyladenine significantly reduced cell proliferation in the presence of ADI-PEG20. Both chloroquine and ADI-PEG20 have no effect on LNCaP cells, which express ASS. Interestingly, ASS positive hepatocellular carcinomas resistant to ADI-PEG20 responded to arginine deprivation by pegylated recombinant arginase (41), providing a potential alternative to ADI-PEG20-resistant tumors and cell lines such as LNCaP.
In cancer, an autophagy paradox has emerged in which survival and death are context specific, particularly due to complex interactions between autophagic and apoptotic pathways. Accordingly, cancer therapies have been reported to have opposing effects on cell death. Photodynamic therapy promotes autophagic cell death in apoptosis-deficient cancer cells (42) while sulforaphane-induced autophagy in PC3 and LNCaP is protective (43). Furthermore, manipulation of autophagy can sensitize tumor cells to subsequent treatments. Induction of autophagy by an mTOR inhibitor increased prostate cancer cell susceptibility to irradiation (44). Conversely, chloroquine is a highly promising autophagy inhibitor for clinical use. Although it is extensively used to treat malaria (20), its uses against cancer are only recently emerging. In a myc-induced lymphoma model, autophagic inhibition by chloroquine enhanced the ability of alkylating agents to suppress tumor growth (45). This underscores the importance of autophagy to fundamental cell processes and its ability to modulate the effect of chemotherapies across a wide variety of cancers.
The absence of ASS as a biomarker for ADI-PEG20 efficacy has previously been established in hepatoma and melanoma cell lines. Phase I/II clinical trials with ADI-PEG20 led to a 47% response rate in patients with unresectable hepatocellular carcinomas and a 25% response rate in metastatic melanoma patients (46, 47). In this study, we show ADI-PEG20 can be effective against prostate cancer. ASS expression can be determined by immunohistochemistry and potentially be used as a clinical indicator for ADI-PEG20 use. The absence of ASS protein in all examined prostate tumor samples makes ADI-PEG20 a promising therapeutic avenue to follow. The combination of ADI-PEG20, which induces caspase-independent apoptosis, and taxanes, which are caspase-dependent, is more effective than monotherapy. This concept of synergistic interaction between cancer therapies is an active area of research. In particular, combining therapies that target different mechanisms of cell death may increase efficacy beyond either agent alone. Furthermore, the rise of advanced imaging for tumor assessment and staging may allow clinical monitoring of tumor responsiveness to ADI-PEG20 by PET. Finally, arginine deprivation by ADI-PEG20 induces autophagy as a protective mechanism. Co-administration with an autophagic inhibitor such as chloroquine can potentially enhance cell death in prostate tumors. The intricate link between autophagy and apoptosis points to autophagy as an additional target for anti-cancer treatments. Thus, ADI-PEG20 is a novel prostate cancer therapy whose mechanism of action can be complemented by other chemotherapies to maximize cell death.
Acknowledgments
We are grateful to the support and reagents provided by Dr. Jenny Wei-Jen Kung, Dr. Liang Xia, Dr. Bor-Wen Wu, and Dr. John Bomalaski. We thank Ms. Subbulakshmi Virudachalam for technical advice, Dr. Ai-Hong Ma for initially testing the avidity of ASS antibody, and Dr. Ralph DeVere White for providing total RNA from primary prostate tissue. This work was supported by NIH DK52659 and CA114575 (to HJK), 5TL1RR024145-02 (to RHK); DOD W81XWH-08-1-0167 (to RHK), and a research agreement by DesigneRx Pharmaceuticals, Inc (to RJB). HJK also acknowledges the support of the Auburn Community Cancer Endowment Fund.
Footnotes
Chen, Pei-Jer, National Taiwan University, Taipei, Taiwan. peijerchen@ntu.edu.tw. ASS mRNA assay. Personal communication, 2008.
References
- 1.Miyazaki K, Takaku H, Umeda M, et al. Potent growth inhibition of human tumor cells in culture by arginine deiminase purified from a culture medium of a Mycoplasma-infected cell line. Cancer research. 1990;50:4522–7. [PubMed] [Google Scholar]
- 2.Takaku H, Matsumoto M, Misawa S, Miyazaki K. Anti-tumor activity of arginine deiminase from Mycoplasma argini and its growth-inhibitory mechanism. Jpn J Cancer Res. 1995;86:840–6. doi: 10.1111/j.1349-7006.1995.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takaku H, Takase M, Abe S, Hayashi H, Miyazaki K. In vivo anti-tumor activity of arginine deiminase purified from Mycoplasma arginini. International journal of cancer. 1992;51:244–9. doi: 10.1002/ijc.2910510213. [DOI] [PubMed] [Google Scholar]
- 4.Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI-SS PEG 20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer research. 2002;62:5443–50. [PubMed] [Google Scholar]
- 5.Holtsberg FW, Ensor CM, Steiner MR, Bomalaski JS, Clark MA. Poly(ethylene glycol) (PEG) conjugated arginine deiminase: effects of PEG formulations on its pharmacological properties. J Control Release. 2002;80:259–71. doi: 10.1016/s0168-3659(02)00042-1. [DOI] [PubMed] [Google Scholar]
- 6.Szlosarek PW, Klabatsa A, Pallaska A, et al. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin Cancer Res. 2006;12:7126–31. doi: 10.1158/1078-0432.CCR-06-1101. [DOI] [PubMed] [Google Scholar]
- 7.Yoon CY, Shim YJ, Kim EH, et al. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. International journal of cancer. 2007;120:897–905. doi: 10.1002/ijc.22322. [DOI] [PubMed] [Google Scholar]
- 8.Bowles TL, Kim R, Galante J, et al. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. International journal of cancer. 2008;123:1950–5. doi: 10.1002/ijc.23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. European journal of biochemistry/FEBS. 2003;270:1887–99. doi: 10.1046/j.1432-1033.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 10.Lind DS. Arginine and cancer. The Journal of nutrition. 2004;134:2837S–41S. doi: 10.1093/jn/134.10.2837S. discussion 53S. [DOI] [PubMed] [Google Scholar]
- 11.Shen LJ, Beloussow K, Shen WC. Modulation of arginine metabolic pathways as the potential anti-tumor mechanism of recombinant arginine deiminase. Cancer letters. 2006;231:30–5. doi: 10.1016/j.canlet.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Dillon BJ, Prieto VG, Curley SA, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer. 2004;100:826–33. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- 13.Gong H, Zolzer F, von Recklinghausen G, et al. Arginine deiminase inhibits cell proliferation by arresting cell cycle and inducing apoptosis. Biochemical and biophysical research communications. 1999;261:10–4. doi: 10.1006/bbrc.1999.1004. [DOI] [PubMed] [Google Scholar]
- 14.Beloussow K, Wang L, Wu J, Ann D, Shen WC. Recombinant arginine deiminase as a potential anti-angiogenic agent. Cancer letters. 2002;183:155–62. doi: 10.1016/s0304-3835(01)00793-5. [DOI] [PubMed] [Google Scholar]
- 15.Gong H, Pottgen C, Stuben G, Havers W, Stuschke M, Schweigerer L. Arginine deiminase and other antiangiogenic agents inhibit unfavorable neuroblastoma growth: potentiation by irradiation. International journal of cancer. 2003;106:723–8. doi: 10.1002/ijc.11298. [DOI] [PubMed] [Google Scholar]
- 16.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Developmental cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. The EMBO journal. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 21.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–23. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 22.Maiuri MC, Criollo A, Tasdemir E, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 23.Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 24.Gozuacik D, Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2:74–9. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- 25.Desai SJ, Ma AH, Tepper CG, Chen HW, Kung HJ. Inappropriate activation of the androgen receptor by nonsteroids: involvement of the Src kinase pathway and its therapeutic implications. Cancer research. 2006;66:10449–59. doi: 10.1158/0008-5472.CAN-06-2582. [DOI] [PubMed] [Google Scholar]
- 26.Parsons CM, Sutcliffe JL, Bold RJ. Preoperative evaluation of pancreatic adenocarcinoma. Journal of hepato-biliary-pancreatic surgery. 2008;15:429–35. doi: 10.1007/s00534-007-1240-7. [DOI] [PubMed] [Google Scholar]
- 27.Van Poppel H. Recent docetaxel studies establish a new standard of care in hormone refractory prostate cancer. The Canadian journal of urology. 2005;12 Suppl 1:81–5. [PubMed] [Google Scholar]
- 28.Mortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature. 1977;270:174–6. doi: 10.1038/270174a0. [DOI] [PubMed] [Google Scholar]
- 29.Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature cell biology. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 31.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell death and differentiation. 2005;12 Suppl 2:1509–18. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 32.Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635–7. doi: 10.4161/auto.4916. [DOI] [PubMed] [Google Scholar]
- 33.Dillon BJ, Holtsberg FW, Ensor CM, Bomalaski JS, Clark MA. Biochemical characterization of the arginine degrading enzymes arginase and arginine deiminase and their effect on nitric oxide production. Med Sci Monit. 2002;8:BR248–53. [PubMed] [Google Scholar]
- 34.Hardie DG. The AMP-activated protein kinase pathway--new players upstream and downstream. Journal of cell science. 2004;117:5479–87. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 35.Feun L, You M, Wu CJ, et al. Arginine deprivation as a targeted therapy for cancer. Current pharmaceutical design. 2008;14:1049–57. doi: 10.2174/138161208784246199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu ZX, Liang J, Haridas V, et al. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell death and differentiation. 2007;14:1948–57. doi: 10.1038/sj.cdd.4402207. [DOI] [PubMed] [Google Scholar]
- 37.Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Molecular pharmacology. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 38.Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. The Journal of biological chemistry. 2003;278:16667–74. doi: 10.1074/jbc.M210998200. [DOI] [PubMed] [Google Scholar]
- 39.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature reviews. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 40.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. The Journal of clinical investigation. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng PN, Lam TL, Lam WM, et al. Pegylated recombinant human arginase (rhArg-peg 5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer research. 2007;67:309–17. doi: 10.1158/0008-5472.CAN-06-1945. [DOI] [PubMed] [Google Scholar]
- 42.Xue LY, Chiu SM, Azizuddin K, Joseph S, Oleinick NL. Protection by Bcl-2 against apoptotic but not autophagic cell death after photodynamic therapy. Autophagy. 2008;4:125–7. doi: 10.4161/auto.5287. [DOI] [PubMed] [Google Scholar]
- 43.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer research. 2006;66:5828–35. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 44.Cao C, Subhawong T, Albert JM, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer research. 2006;66:10040–7. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 45.Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. The Journal of clinical investigation. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Izzo F, Marra P, Beneduce G, et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol. 2004;22:1815–22. doi: 10.1200/JCO.2004.11.120. [DOI] [PubMed] [Google Scholar]
- 47.Ascierto PA, Scala S, Castello G, et al. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol. 2005;23:7660–8. doi: 10.1200/JCO.2005.02.0933. [DOI] [PubMed] [Google Scholar]