

Abstract
Apoptosis is an important mechanism for maintaining tissue homeostasis and for preventing the proliferation of cells with mutations that could result in malignancy. Barrett's epithelium has been reported to be more resistant to apoptosis than normal esophageal squamous epithelium. We have explored the contribution of the NF-κB pathway to apoptotic resistance in non-neoplastic, telomerase-immortalized esophageal squamous (NES) and Barrett's (BAR-T) epithelial cell lines. We exposed these cells to UV-B irradiation in doses known to cause DNA damage and to induce apoptosis in normal cells, and studied apoptosis as well as the expression of phospho-H2AX, NF-κB, Bcl-2, XIAP, cIAP-1 and survivin proteins. We also used Bay 11-7085 and siRNAs to NF-κB and Bcl-2 to assess the effects of NF-κB and Bcl2 inhibition on apoptosis. UV-B irradiation at low doses (50 and 100 J/m2) caused DNA damage in both NES and BAR-T cells, but significantly increased apoptosis only in NES cells. UV-B irradiation caused a decrease in the levels of NF-κB, Bcl-2, cIAP-1, XIAP, and survivin in NES cells, but increased the levels of those proteins in BAR-T cells. The resistance of BAR-T cells to apoptosis induced by low-dose UV-B irradiation was abolished by Bay 11-7085 and by siRNA for NF-κB and was decreased significantly by siRNA for Bcl-2. We conclude that the ability of Barrett's epithelial cells to activate the NF-κB pathway when they have sustained DNA damage allows them to resist apoptosis. This capacity to avoid apoptosis despite genotoxic damage may underlie the persistence and malignant predisposition of Barrett's metaplasia.
Keywords: Barrett's esophagus, esophageal squamous cell, DNA damage, apoptosis, NF-κB, Bcl-2
Introduction
Gastroesophageal reflux disease (GERD) often causes peptic injury and inflammation of the esophageal squamous epithelium, a condition called reflux esophagitis. In most cases, reflux esophagitis heals through the regeneration of new squamous cells. In some patients, however, reflux esophagitis heals through a metaplastic process in which intestinal-type columnar cells replace reflux-damaged squamous cells (1). This condition is called Barrett's esophagus and, for reasons that are not clear, metaplastic Barrett's epithelium is predisposed to develop neoplasia. GERD and Barrett's esophagus are strong risk factors for esophageal adenocarcinoma, a tumor whose incidence has increased substantially in the United States over the past 30 years (1; 2).
Apoptosis is an important mechanism for maintaining tissue homeostasis and for preventing the proliferation of cells with mutations that could result in malignancy. Normally, mild genomic injury induces p53 expression that triggers cell cycle arrest, which enables the cell to repair its damaged DNA. If the genomic injury is severe and irreparable, however, apoptosis ensues. Failure to undergo apoptosis in the setting of severe DNA injury allows dangerous mutations to persist and contribute to cancer formation. Indeed, resistance to apoptosis is an essential physiological hallmark of cancer cells (3). Pro-apoptotic signals can be counterbalanced by the expression of anti-apoptotic proteins whose synthesis is mediated by the Nuclear Factor-κB (NF-κB) gene, a member of the Rel family of transcription factors (4). There are five members of the mammalian Rel family of proteins, with NF-κB/p65 (Rel A) being one of the most important.
Metaplasia appears to be a protective response to inflammation because, in a number of organs, the metaplastic tissue is more resistant to the noxious agents causing the chronic inflammation than the native tissue. For example, chronic gastritis due to Helicobacter pylori results in intestinal metaplasia, which is far less susceptible to H. pylori infection than the normal gastric epithelium. Similarly, Barrett's metaplasia appears to be more resistant to acid-peptic injury than the native esophageal squamous mucosa (5; 6). Bile also is present in refluxed gastric juice, and Barrett's metaplasia has been found to be more resistant to bile acid-induced apoptosis than normal esophageal squamous epithelium (6). This resistance to apoptosis may be one mechanism whereby Barrett's cells survive despite continuous exposure to refluxed gastric juice.
Clinical data suggest that alterations in the susceptibility to apoptosis underlie the neoplastic progression of Barrett's esophagus (7-9). In biopsy samples of Barrett's epithelium, for example, altered staining for the anti-apoptotic proteins Bcl-2 and Bcl-xL (downstream NF-κB target genes) has been found as the metaplasia progresses through dysplasia to adenocarcinoma (7; 9; 10). These data suggest a role for the NF-κB pathway in conferring an anti-apoptotic phenotype during the neoplastic progression of Barrett's esophagus (11).
Earlier studies found expression of NF-κB in only 13% of biopsy specimens of esophageal squamous epithelium (12). In contrast, NF-κB expression has been found in biopsy specimens of benign Barrett's epithelium in 40-60% of cases, and in esophageal adenocarcinoma in 61% to 80% of cases (12; 13). This suggests the possibility that reflux-mediated activation of the NF-κB pathway, which can increase the expression of NF-κB survival pathway proteins, may enable the metaplastic columnar cells to resist apoptosis and survive while the esophageal squamous cells succumb to GERD. If noxious agents in refluxed gastric juice simultaneously cause DNA injury and NF-κB activation in the Barrett's cells, furthermore, this might enable the survival of cells with cancer-promoting mutations. The present study was designed to explore the contribution of the NF-κB pathway to apoptotic resistance in metaplastic Barrett's epithelial cells.
Materials and Methods
Cell Culture
We used two telomerase-immortalized, esophageal squamous cell lines created from endoscopic biopsy specimens of normal esophageal squamous mucosa taken from the distal esophagus of patients who had GERD with (NES-B3T) and without (NES-G2T) Barrett's esophagus. We also used three telomerase-immortalized, Barrett's epithelial cell lines (BAR-T, BAR-T 9, and BAR-T 10) that were created from endoscopic biopsy specimens of non-dysplastic Barrett's specialized intestinal metaplasia taken from three patients with long segment (>3 cm) Barrett's esophagus (14-16). As controls, we used normal rat intestinal epithelial cells (IEC-6) and normal mouse intestinal epithelial cells (MSIE) (17; 18). All cell lines were cultured in their respective growth medias as previously described (14; 15; 17; 18). MSIE cells are derived from the Immortomouse and, when cultured at 33°C with 5% CO2, are immortal (18; 19). All other cell lines were maintained in monolayer culture at 37°C in humidified air with 5% CO2. The telomerase-immortalized NES and Barrett's cell lines were co-cultured with a fibroblast feeder layer as previously described (20). Prior to UV-B irradiation, sub-confluent cells were placed in minimal media (no serum and lacking growth factors) overnight at 37°C and 5% CO2.
UV-B Irradiation
Genomic damage caused by UV-B irradiation is a well established inducer of apoptosis and p53 expression (21). We used UV-B irradiation to induce apoptosis in our cells which was carried out with a homemade box built with four ultraviolet-B bulbs (280-320nm wavelength, Philips Lighting, USA). The applied UV-B dose was measured using a radiometer (IL-1400A Radiometer, International/Photometer, MA). Equally seeded wells of cells at 70% confluence were irradiated with 50, 100, 200, and 400 J/m2 of UV-B. Twenty-four hours later, cells were collected for analysis.
Detection of Apoptosis
Apoptosis rates were assessed quantitatively using Annexin V (BD Biosciences, San Diego, CA) as previously described (22). Briefly, cells were harvested by trypsinization, stained with Annexin V-FITC, and 50 μg/ml of propidium iodide (PI), then immediately analyzed by flow cytometry (FACScaliber, Becton Dickson, Franklin Lake, NJ); cells staining only with annexin V were considered to be apoptotic (22). Experiments were performed in triplicate and a total of 10,000 cells were analyzed in each individual experiment.
Western Blotting
The cells were harvested by scraping into lysis buffer containing 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH 7.4), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and one Protease Inhibitor Cocktail Tablet per 50 ml of lysis buffer (Roche Applied Science, Indianapolis, IN). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis; protein concentrations were determined using the BCA-200 Protein Assay kit (Pierce, Rockford, IL). After separation and transfer to nitrocellulose membrane, membranes were incubated with 1:1000 dilutions of rabbit polyclonal antibodies anti-human, anti-phospho-H2AX, anti-NF-κB, anti-Bcl2, and antibodies against the cIAPS: survivin, XIAP, and cIAP-1 (Cell Signaling Inc, Beverley, MA). Horseradish peroxidase secondary antibody (Cell Signaling) was used at 1:2000, and chemiluminescence was determined using the ECL detection system (Pierce, Rockford, IL). Membranes were then stripped using Restore Stripping Buffer (Pierce) and were re-probed with β-actin (Sigma, St Louis, MO) to confirm equal loading.
Inhibition of the NF-κB Signaling Pathway
Bay11-7085 (Sigma), is a pharmacologic inhibitor which blocks IκBα phosphorylation. Cells were treated with Bay11-7085 at concentrations ranging from 1-10 μM for 1 hour as described, followed by UV-B irradiation (23). Since pharmacologic agents can have non-specific effects, we confirmed the role of NF-κB/p65 in mediating effects on apoptosis using a specific NF-κB/p65 siRNA. We also used a specific siRNA against Bcl-2, a downstream NF-κB target gene implicated in apoptotic resistance of Barrett's esophagus (9; 24). Cells were plated at 50% confluence in 6-well tissue culture plates. After 24 hours, the cells were transfected using the SignalSilence® NF-κB p65 or Bcl-2 siRNA (Cell Signaling Technology), per the manufacturer's instructions. 48 hrs after transfection, cells were exposed to UV-B irradiation. As a control, cells were transfected with an siRNA oligonucleotide against lamin A/C (Millipore, St. Charles, MO).
Statistical Analyses
The data were collected from at least three independent experiments. Quantitative data are expressed as the mean plus (±) the standard error of the mean (SEM). Statistical analysis was performed using ANOVA and the Student-Newman-Keuls multiple-comparison test with the Instat for Windows statistical software package (GraphPad Software, San Diego, CA). P values <0.05 were considered significant for all analyses.
Results
Low-Dose UV-B Irradiation (50 and 100 J/m2) Induces DNA Damage in Normal Esophageal Squamous (NES) Cells and in Barrett's Epithelial (BAR-T) Cells, but Induces Apoptosis Only in NES Cells
Clinical studies suggest that Barrett's cells are more resistant to apoptosis than normal esophageal squamous cells. We used UV-B irradiation, a well established inducer of DNA damage and apoptosis, to verify those clinical data in our benign, telomerase-immortalized cells (21). NES-G2T, NES-B3T, BAR-T, BAR-T9, and BAR-T10 cells all were grown to 70% confluence and placed in minimal media overnight. Cells were then exposed to UV-B irradiation at doses known to cause DNA damage and to induce apoptosis in normal cells (21). Twenty-four hours later, DNA damage was assessed by expression of phospho-H2AX; apoptosis was determined by Annexin V staining. As expected, we found that UV-B irradiation induced a dose-dependent increase in DNA damage in BAR-T, NES-G2T, and NES-B3T cells (Figure 1).
Figure 1.
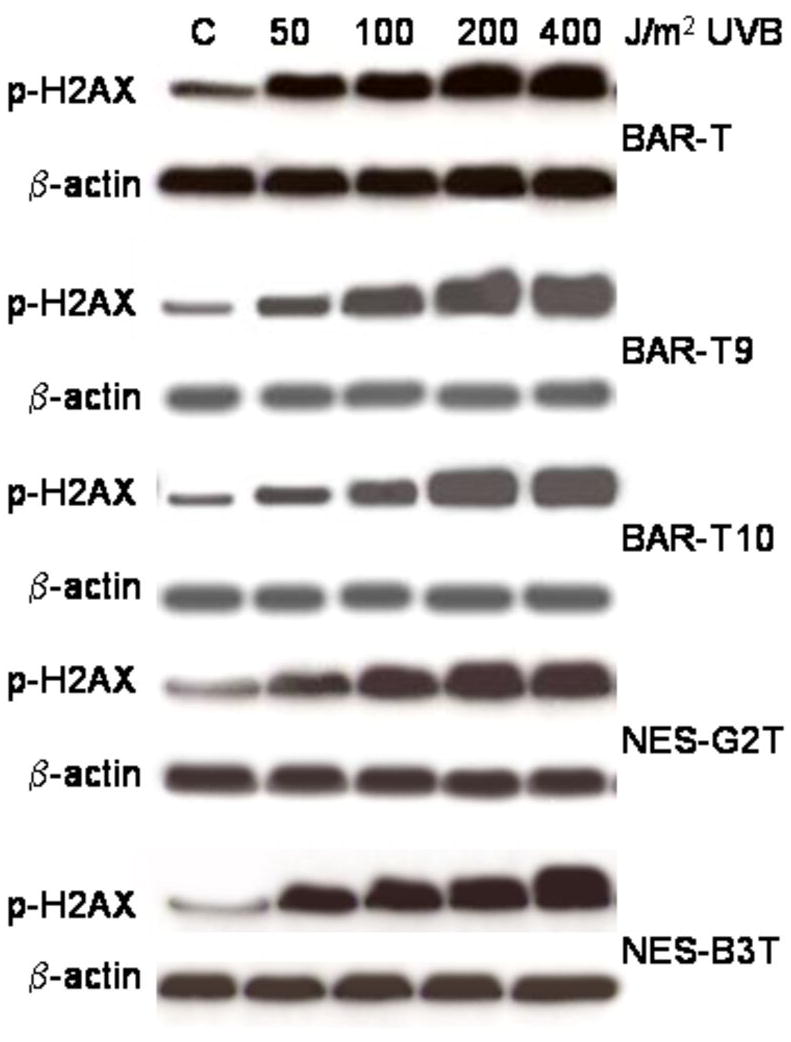
Representative Western blots demonstrating a dose-dependent increase in phospho-H2AX expression following UV-B irradiation with 50-400 J/m2 in BAR-T and NES cells.
We previously reported that UV-B irradiation at doses of 50 and 100 J/m2 did not induce apoptosis in BAR-T cells, although apoptosis was observed with higher doses (200 and 400 J/m2) (22). In NES-G2T and NES-B3T cells, we found that UV-B irradiation induced apoptosis in a dose-dependent manner and, in contrast to the BAR-T cells, even doses as low as at 50 J/m2 caused apoptosis (Figure 2). Moreover, the level of apoptosis induced by just 100 J/m2 in our NES cell lines (NES-G2T 57.8 ± 2.5%; NES-B3T 59.5 ± .50%) was significantly higher than that induced by 400 J/m2 in the BAR-T cells (41.2 ± 2.7% SEM; p<0.05) (22).
Figure 2.
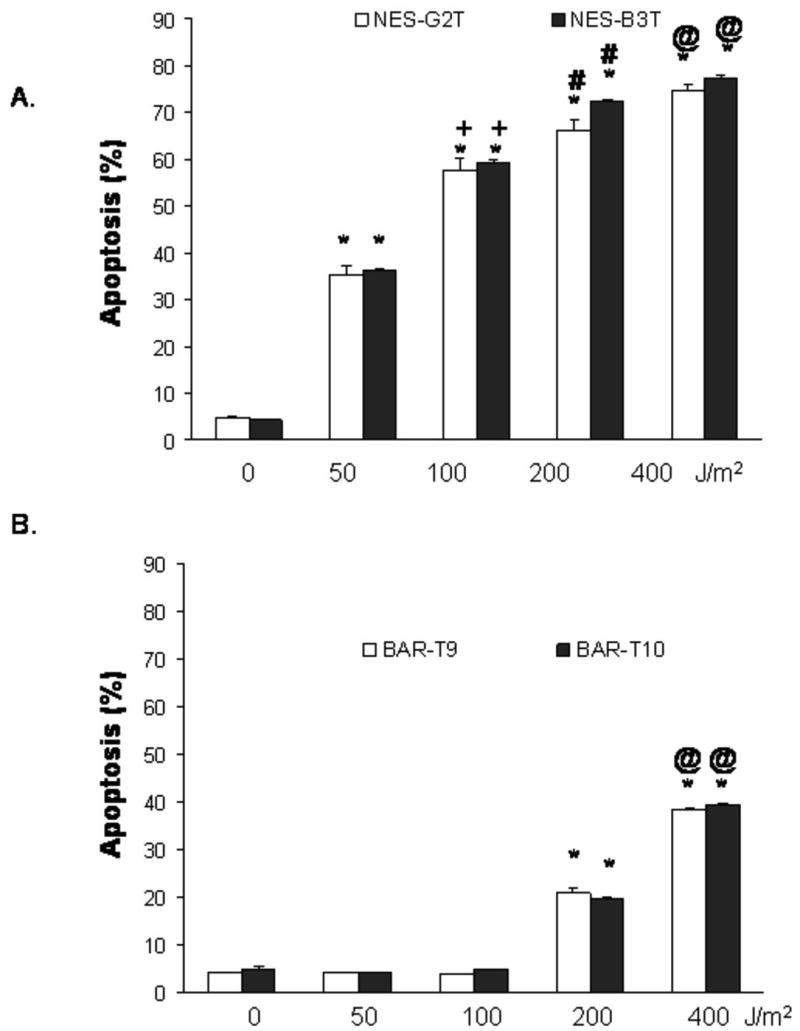
Results of UV-B irradiation on apoptosis in (A) NES-G2T and NES-B3T squamous cells and (B) BAR-T9 and BAR-T10 cells as determined by Annexin V staining. Note that UV-B irradiation at doses of 50 and 100 J/m2 did not induce apoptosis in the Barrett's cells. The bar graphs depict the mean + SEM of at least 3 individual experiments. (NES-G2T: *, p<0.001 compared to non-irradiated controls; +, p<0.001 compared to 50 J/m2; #, p<0.01 compared to 100 J/m2, @, p<0.01 compared to 200 J/m2; NES-B3T: *, p<0.001 compared to non-irradiated controls; +, p<0.001 compared to 50 J/m2; #, p<0.001 compared to 100 J/m2, @, p<0.001 compared to 200 J/m2; BAR-T9 and BAR-T10: *, p<0.001 compared to non-irradiated controls; @, p<0.001 compared to 200 J/m2)
To confirm that these findings were not unique to the BAR-T Barrett's cell line, we exposed two additional telomerase-immortalized Barrett's epithelial cell lines (BAR-T9 and BAR-T10) to UV-B irradiation and determined the effects on DNA damage and apoptosis. Similar to the BAR-T cells, we found that low dose UV-B irradiation increased phospho-H2AX expression in the BAR-T9 and BAR-T10 cells (Figure 1). In addition, UV-B irradiation at 200 and 400 J/m2, but not at 50 or 100 J/m2, induced apoptosis in BAR-T9 and BAR-T10 cells (Figure 2). Similar to the BAR-T cells, the level of apoptosis induced induced by just 100 J/m2 in our NES cell lines (NES-G2T 57.8 ± 2.5%; NES-B3T 59.5 ± .50%) was significantly higher than that induced by 400 J/m2 in the BAR-T cell lines (BAR-T9 38.3 ± .30 % SEM; BAR-T10 39.4 ± .21 % SEM; p<0.05 for BAR-T9 and BAR-T10) cells. This demonstrates that apoptotic resistance to low dose UV-B irradiation is not restricted to a single cell line, but rather appears to be a common feature of Barrett's epithelial cells.
Resistance to UV-B Induced Apoptosis is not due to the Intestinal Phenotype of Barrett's Epithelial Cells
To determine whether the differences we observed in resistance to apoptosis in response to low dose UV-B irradiation were due primarily to differences in cell phenotype (i.e. intestinal-type columnar cells vs. squamous cells), we treated normal rat intestinal epithelial cells (IEC-6) and normal mouse intestinal epithelial cells (MSIE) with UV-B irradiation, and assessed DNA damage and apoptosis in response to genotoxic injury. As expected, both columnar cell lines exhibited an increase in DNA damage after UV-B irradiation (Figure 3A). Unlike the Barrett's cells, however, the IEC-6 and MSIE intestinal cell lines demonstrated a significant increase in apoptosis even after low dose UV-B irradiation (Figure 3B).
Figure 3.
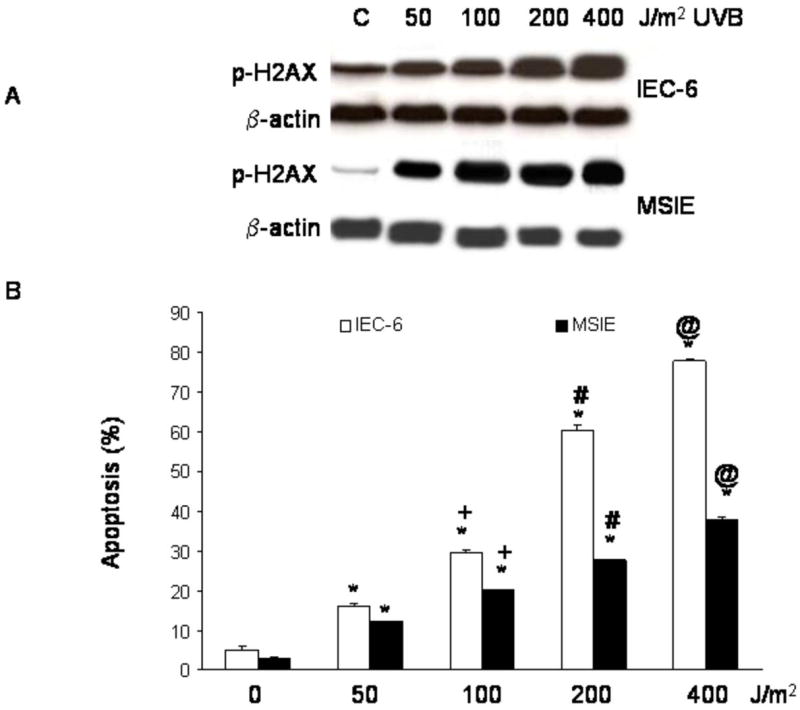
(A) Representative Western blots demonstrating a dose-dependent increase in phospho-H2AX expression following UV-B irradiation with 50-400 J/m2 in IEC-6 and MSIE intestinal cells. (B) Results of UV-B irradiation on apoptosis in IEC-6 and MSIE cells as determined by Annexin V staining. The bar graphs depict the mean + SEM of at least 3 individual experiments. (*, p<0.001 compared to non-irradiated controls; +, p<0.001 compared to 50 J/m2; #, p<0.001 compared to 100 J/m2, @, p<0.001 compared to 200 J/m2)
The NF-κB Pathway is Activated by Low Dose UV-B Irradiation in BAR-T Cells, but Not in NES Cells
Having found a difference in apoptosis induction by low-dose UV-B irradiation between esophageal squamous and metaplastic Barrett' cells, we next sought to determine the contribution of the NF-κB pathway to that difference. We performed Western blotting for NF-kB/p65 and its target genes products (Bcl-2, cIAP-1, XIAP, and survivin) 24 hours after UV-B irradiation. As shown in Figure 4, low-dose UV-B irradiation (50 and 100J/m2) increased the expression of NF-κB, Bcl-2, XIAP, cIAP2 and survivin proteins in a dose-dependent manner in BAR-T cells, but decreased their expression in NES-G2T and NES-B3T cells. We also found that low-dose UV-B irradiation increased NF-κB expression in BAR-T9 and BAR-T10 (data not shown). These data suggest that the NF-κB pathway is activated by low dose UV-B irradiation in BAR-T, but not in NES cells. Moreover, we found that 50 J/m2 UV-B irradiation virtually abolished NF-κB and XIAP protein expression in NES-B3T cells, but not in NES-G2T cells, which continued to express these proteins at lower levels.
Figure 4.

Representative Western blots demonstrating expression of NF-κB, Bcl-2, cIAP-1, XIAP, and survivin following irradiation with 50-400 J/m2 UV-B in BAR-T, NES-G2T, and NES-B3T cell lines. Note the expression of these survival proteins increases in BAR-T cells, but decreases in the NES cell lines in response to UV-B. Note that low-dose UV-B irradiation (50 J/m2) virtually abolishes the expression of NF-κB and XIAP in the NES-B3T cells, but not in the NES-G2T cells.
Inhibition of NF-κB Sensitizes BAR-T cells to Apoptosis Induced by Low-dose UV-B Irradiation
Having found that low-dose UV-B irradiation increases NF-κB expression in BAR-T cells, we next sought to determine whether NF-κB contributes to their resistance to apoptosis. We inhibited NF-κB activity using both Bay 11-7085 (a pharmacological inhibitor) and a specific siRNA against NF-κB/p65. Cells were treated with concentrations of Bay 11-7085 ranging from 0-10 μM and then irradiated with 50-400 J/m2 of UV-B. In BAR-T cells, the resistance to apoptosis in response to low dose UV-B irradiation was abolished by treatment with Bay 11-7085 in a dose-dependent fashion; Bay 11-7085 also induced a dose-dependent increase in the rates of apoptosis following 200 and 400 J/m2 of UV-B, with the maximal rate of apoptosis found to plateau with 5 μM of Bay 11-7085 (Figure 5A). We next confirmed our findings by transfecting the cells with a NF-κB/p65 siRNA and determining the rates of apoptosis following low dose UV-B irradiation. To determine the efficiency of NF-κB/p65 siRNA on inhibiting NF-κB/p65 expression, Western blotting was performed at baseline and following irradiation with 100 J/m2, a condition which we found increases NF-κB expression. We found complete loss of NF-κB/p65 expression following transfection with siRNA at both conditions (Figure 5B). Transfection of the BAR-T cells with siRNA against the p65 subunit of NF-κB resulted in a significant increase in apoptosis after low dose UV-B irradiation; the siRNA against NF-κB/p65 also significantly increased the rates of apoptosis following 200 and 400 J/m2 of UV-B (Figure 5B). In contrast, low-dose UV-B irradiation did not cause apoptosis in control cells that were transfected with a siRNA targeted against lamin A/C, suggesting that the apoptosis induced by low dose UV-B was indeed due to inhibition of NF-κB and not due to a non-specific effect of the siRNA (Figure 5B).
Figure 5.
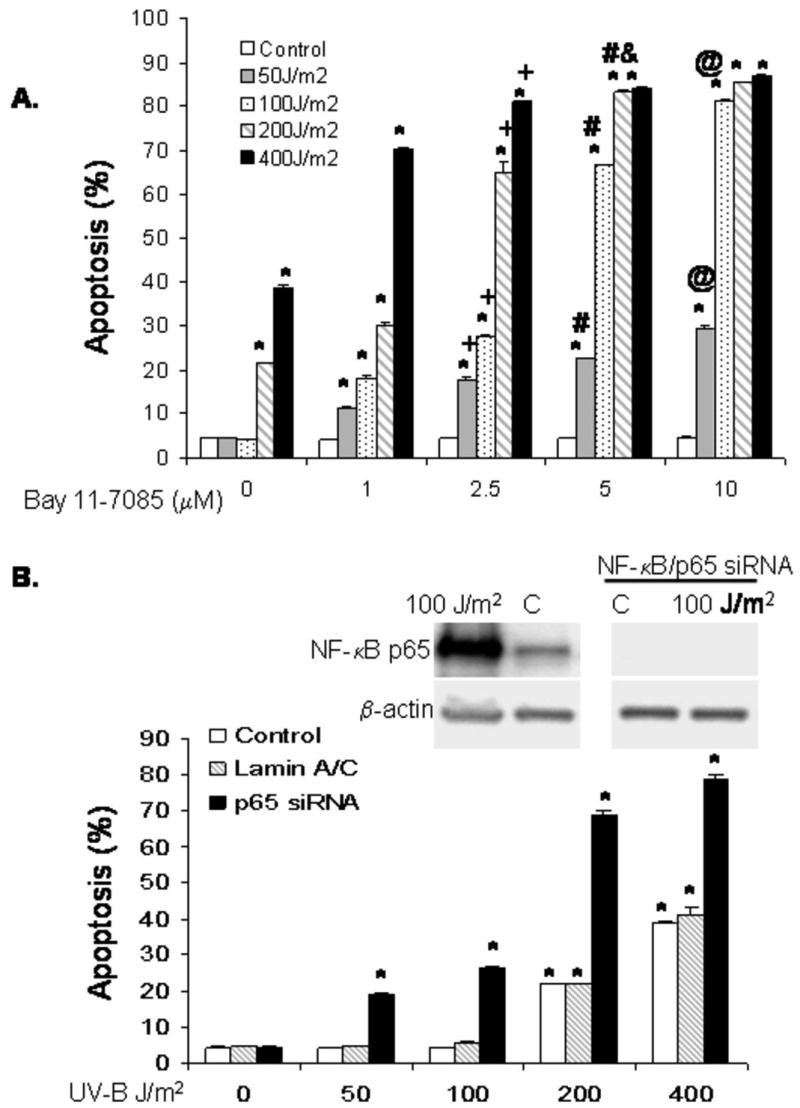
(A) Results of UV-B irradiation and Bay 11-7085 on apoptosis as determined by Annexin V staining. Inhibition of NF-κB activity with Bay11-7085 sensitizes BAR-T cells to apoptosis induced by low dose UV-B in a dose dependent manner. The bar graphs depict the mean + SEM of at least 3 individual experiments. (*, p<0.001 compared to non-irradiated corresponding controls; +, p<0.001 compared to 1 μM; #, p<0.001 and &, p<0.01 compared to 2.5 μM, @, p<0.001 compared to 5 μM). (B) Western blots for NF-κB/p65 expression following siRNA infection and results of UV-B irradiation and NF-κB/p65 siRNA on apoptosis as determined by Annexin V staining. Note that inhibition of NF-κB/p65 sensitizes BAR-T cells to apoptosis induced by low dose UV-B irradiation. SiRNA against lamin A/C served as a control. The bar graphs depict the mean + SEM of at least 3 individual experiments. (*, p<0.001 compared to non-irradiated corresponding controls)
Inhibition of Bcl-2 Sensitizes BAR-T cells to Apoptosis Induced by Low-dose UV-B Irradiation
Since Bcl-2 is a downstream target gene of NF-κB and clinical data suggest a role for Bcl-2 expression during the early stages of carcinogenesis in Barrett's esophagus, we next sought to determine whether Bcl-2 expression contributes to resistance to apoptosis of metaplastic Barrett's epithelial cells (9; 24). We transfected BAR-T cells with Bcl-2 siRNA oligonucleotides and determined the rates of apoptosis following UV-B irradiation. To determine the efficiency of siRNA for inhibiting Bcl-2 expression, Western blotting was performed at baseline and following irradiation with 400 J/m2, a condition which we found increases Bcl-2 protein expression. We noted complete loss of Bcl-2 expression following transfection with the siRNA both at baseline and following UV-B irradiation (Figure 6A). We found that transfection of the BAR-T cells with siRNA against Bcl-2 resulted in only a small, but statistically significant increase in apoptosis after low dose UV-B irradiation; the siRNA against Bcl-2 also slightly increased the rates of apoptosis following 200 and 400 J/m2 of UV-B (Figure 6B). Low-dose UV-B irradiation did not cause apoptosis in control cells that were transfected with a siRNA targeted against lamin A/C.
Figure 6.
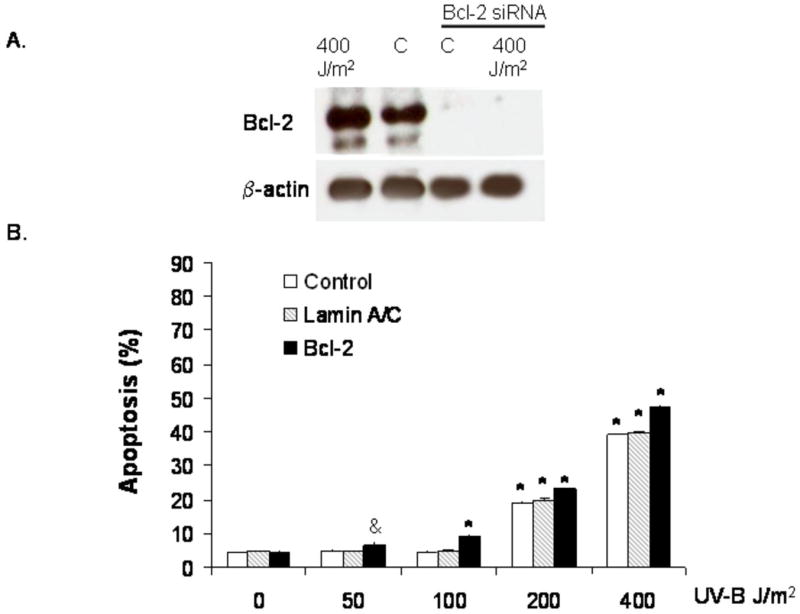
(A) Western blots for Bcl-2 expression following siRNA infection. Note that Bcl-2 protein expression is inhibited following infection with Bcl-2 siRNA at baseline and after treatment with 400 J/m2. (B) Results of UV-B irradiation and Bcl-2 siRNA on apoptosis as determined by Annexin V staining; siRNA against lamin A/C served as a control. Note that inhibition of Bcl-2 causes a small, but statistically significant increase in apoptosis in response to low dose UV-B irradiation. The bar graphs depict the mean + SEM of at least 3 individual experiments. (&, p<0.01 compared to non-irradiated corresponding control; *, p<0.001 compared to non-irradiated corresponding controls)
Discussion
Studies using esophageal biopsy specimens have suggested that Barrett's epithelial cells are more resistant to apoptosis than normal squamous epithelial cells. Biopsy specimens contain inflammatory cells and stromal cells in addition to epithelial cells and, in biopsy specimens, it can be difficult to distinguish direct effects of an agent on epithelial cells from indirect effects that are mediated by inflammatory and stromal cells. Conceivably, the apoptotic resistance reported for Barrett's epithelial biopsy specimens might be mediated by inflammatory and stromal cells rather than by the epithelial cells. To eliminate the potential confounding effects of these non-epithelial cells, we have used cultures of benign, telomerase-immortalized esophageal epithelial cells to study the direct effects of a toxic agent on apoptosis.
To determine whether Barrett's epithelial cells are more resistant to apoptosis than esophageal squamous epithelial cells, we exposed benign, telomerase-immortalized squamous (NES) and Barrett's (BAR-T) cell lines to UV-B irradiation. Although UV-B irradiation clearly is not a physiologically relevant agent in the esophagus, UV-B is an extensively studied and well-established inducer of DNA damage and apoptosis in a number of different cell types (21; 25). As expected, we found that UV-B irradiation in all doses studied (50, 100, 200 and 400 J/m2) induced DNA damage (detected by phospho-H2AX expression) in Barrett's and squamous epithelial cells alike. In the esophageal squamous cell lines (NES-G2T, and NES-B3T), we found that UV-B irradiation in all doses studied increased the rate of apoptosis in a dose-dependent fashion. A similar UV-B dose-response on apoptosis has been observed in human keratinocytes (21). In three Barrett's epithelial cell lines, in contrast, we found that low-dose UV-B irradiation (50 and 100 J/m2) did not increase apoptosis, although higher doses (200 and 400 J/m2) did have pro-apoptotic effects (22). Taken together, these studies demonstrate that the epithelial cells of Barrett's esophagus are more resistant to apoptosis than esophageal squamous epithelial cells, and that Barrett's epithelial cells can resist apoptosis in the face of genotoxic injury.
To determine whether the differences we observed in resistance to apoptosis of Barrett's epithelial cells were due primarily to differences in cell phenotype (i.e. intestinal-type columnar cells vs. squamous cells), we treated normal rat intestinal epithelial cells (IEC-6) and normal mouse intestinal epithelial cells (MSIE) with UV-B irradiation, and assessed DNA damage and apoptosis. Both of these columnar cell lines exhibited DNA damage after UV-B irradiation. Unlike the BAR-T intestinal-type columnar cells, however, the IEC-6 and MSIE intestinal cell lines demonstrated apoptosis after low dose UV-B irradiation. Thus the apoptotic resistance of Barrett's epithelial cells is not due solely to their intestinal phenotype.
Pro-apoptotic signals can be counterbalanced by the expression of anti-apoptotic proteins whose synthesis is mediated by NF-κB. Clinical studies have suggested a role for NF-κB and its downstream targets Bcl-2 and Bcl-xL in the apoptotic resistance of Barrett's metaplasia (7; 9; 12; 13). Another important family of NF-κB target genes are the inhibitors of apoptosis (IAPs), which block the activities of the caspases (26; 27). To our knowledge, there are no published data on the expression of IAP family members in Barrett's esophagus. We found that low-dose UV-B irradiation (50 and 100J/m2) increased the expression of NF-κB, Bcl-2, XIAP, cIAP-1, and survivin proteins in a dose-dependent manner in BAR-T cells, but decreased the expression of those same proteins in NES-G2T and NES-B3T cells. These findings suggest that activation of the NF-κB pathway contributes to apoptotic resistance in Barrett's epithelial cells.
To demonstrate a mechanistic link between NF-κB activation and resistance to UV-B-induced apoptosis in our metaplastic Barrett's cells, we treated them with Bay 11-7085, a pharmacological inhibitor of NF-κB activity, or with NF-κB/p65 siRNA prior to UV-B irradiation. We found that both of these inhibitors abolished resistance to apoptosis induced by low dose UV-B irradiation in BAR-T cells, suggesting that the NF-κB pathway mediates the apoptotic resistance of metaplastic Barrett's cells. Using an siRNA against Bcl-2, we also explored whether this NF-κB downstream anti-apoptotic protein played a role in this apoptotic resistance. We found that inhibition of Bcl-2 caused a small, but statistically significant increase in apoptosis following low dose UV-B irradiation. This suggests that Bcl-2 may contribute to apoptotic resistance, but that other NF-κB-dependent anti-apoptotic proteins (perhaps the IAP family members) play a role as well. Further studies are warranted to determine the specific proteins involved in this phenomenon.
Using our esophageal squamous cell lines, we found that 50 J/m2 UV-B irradiation virtually abolished NF-κB and XIAP protein expression in NES-B3T cells (from a patient with Barrett's esophagus), but not in NES-G2T cells (from a patient who had GERD without Barrett's esophagus). This suggests that the esophageal squamous epithelium of Barrett's patients may be more susceptible to apoptosis induced by genotoxic injury than the squamous epithelium of GERD patients who do not develop Barrett's esophagus. We and others have previously reported differences in reflux-induced activation of pro-proliferative molecular signaling pathways between esophageal squamous cells from GERD patients with and without Barrett's esophagus that might play a role in determining whether reflux esophagitis heals through metaplasia or through squamous cell regeneration (15; 28; 29). If esophageal squamous cells have a similar response to toxic agents in refluxed gastric juice as they do to UV-B irradiation, then it is conceivable that differences in the expression of anti-apoptotic proteins might determine whether the squamous epithelium would remain intact or would succumb to apoptosis and require repair, perhaps by metaplastic Barrett's epithelial cells that express NF-κB. Further investigations are needed in this area as well.
In conclusion, we have shown that Barrett's epithelial cells are more resistant to apoptosis than normal esophageal squamous epithelial cells in the face of genotoxic injury induced by low dose UV-B irradiation. Moreover, this apoptotic resistance is a unique property of benign Barrett's epithelial cells, not merely a feature of their intestinal phenotype. We have also found that low dose UV-B irradiation increases the expression of NF-κB, Bcl-2, cIAP-1, XIAP, and survivin proteins in a dose-dependent fashion in Barrett's cells, but decreases the expression of those proteins in esophageal squamous cells, suggesting that activation of the NF-κB pathway mediates resistance to UV-B-induced apoptosis in Barrett's epithelial cells. In addition, we have found differences in expression of NF-κB and XIAP in response to low dose UV-B irradiation between our esophageal squamous cell lines from patients with and without Barrett's esophagus, suggesting that differences in apoptotic resistance pathways might contribute to the pathogenesis of Barrett's metaplasia. Finally, the resistance to low-dose UV-B-induced apoptosis in Barrett's cells could be abolished by treatment with inhibitors of NF-κB and Bcl-2. These findings demonstrate that activation of the NF-κB pathway plays a mechanistic role in the resistance to apoptosis of Barrett's epithelial cells and suggest that this pathway may be a novel target at which to direct therapies to prevent the development and neoplastic progression of Barrett's esophagus.
Acknowledgments
This work was supported by the Office of Medical Research, Department of Veteran's Affairs, Dallas, TX (R.F.S.), the Harris Methodist Health Foundation, Dr. Clark R. Gregg Fund (R.F.S., K.H-C.), the National Institutes of Health (R01-DK63621 to R.F.S and R01-HL067256 and R01-HL61897 to L.S.T.), and the Vanderbilt University Medical Center's Digestive Disease Research Center (NIH DK058404 to R.H.W.)
Reference List
- 1.Spechler SJ. Clinical practice. Barrett's Esophagus. N Engl J Med. 2002;346:836–42. doi: 10.1056/NEJMcp012118. [DOI] [PubMed] [Google Scholar]
- 2.Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:142–6. doi: 10.1093/jnci/dji024. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 5.Jovov B, Van Itallie CM, Shaheen NJ, et al. Claudin-18: a dominant tight junction protein in Barrett's esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1106–13. doi: 10.1152/ajpgi.00158.2007. [DOI] [PubMed] [Google Scholar]
- 6.Dvorakova K, Payne CM, Ramsey L, et al. Apoptosis resistance in Barrett's esophagus: ex vivo bioassay of live stressed tissues. Am J Gastroenterol. 2005;100:424–31. doi: 10.1111/j.1572-0241.2005.40932.x. [DOI] [PubMed] [Google Scholar]
- 7.van der Woude CJ, Jansen PL, Tiebosch AT, et al. Expression of apoptosis-related proteins in Barrett's metaplasia-dysplasia-carcinoma sequence: a switch to a more resistant phenotype. Hum Pathol. 2002;33:686–92. doi: 10.1053/hupa.2002.124908. [DOI] [PubMed] [Google Scholar]
- 8.Raouf AA, Evoy DA, Carton E, Mulligan E, Griffin MM, Reynolds JV. Loss of Bcl-2 expression in Barrett's dysplasia and adenocarcinoma is associated with tumor progression and worse survival but not with response to neoadjuvant chemoradiation. Dis Esophagus. 2003;16:17–23. doi: 10.1046/j.1442-2050.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- 9.Katada N, Hinder RA, Smyrk TC, et al. Apoptosis is inhibited early in the dysplasia-carcinoma sequence of Barrett esophagus. Arch Surg. 1997;132:728–33. doi: 10.1001/archsurg.1997.01430310042007. [DOI] [PubMed] [Google Scholar]
- 10.Iravani S, Zhang HQ, Yuan ZQ, et al. Modification of insulin-like growth factor 1 receptor, c-Src, and Bcl-XL protein expression during the progression of Barrett's neoplasia. Hum Pathol. 2003;34:975–82. doi: 10.1053/s0046-8177(03)00354-x. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 12.O'Riordan JM, Abdel-latif MM, Ravi N, et al. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol. 2005;100:1257–64. doi: 10.1111/j.1572-0241.2005.41338.x. [DOI] [PubMed] [Google Scholar]
- 13.Abdel-latif MM, O'Riordan J, Windle HJ, et al. NF-kappaB activation in esophageal adenocarcinoma: relationship to Barrett's metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann Surg. 2004;239:491–500. doi: 10.1097/01.sla.0000118751.95179.c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaiswal KR, Morales CP, Feagins LA, et al. Characterization of telomerase-immortalized, non-neoplastic, human Barrett's cell line (BAR-T) Dis Esophagus. 2007;20:256–64. doi: 10.1111/j.1442-2050.2007.00683.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang HY, Zhang X, Chen X, et al. Differences in activity and phosphorylation of MAPK enzymes in esophageal squamous cells of GERD patients with and without Barrett's esophagus. Am J Physiol Gastrointest Liver Physiol. 2008;295:G470–78. doi: 10.1152/ajpgi.90262.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang HY, Zhang X, Hormi-Carver K, Feagins LA, Spechler SJ, Souza RF. In Non-neoplastic Barrett's Epithelial Cells, Acid Exerts Early Antiproliferative Effects through Activation of the Chk2 Pathway. Cancer Res. 2007;67:8580–7. doi: 10.1158/0008-5472.CAN-07-2023. [DOI] [PubMed] [Google Scholar]
- 17.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–65. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A. 1993;90:587–91. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jat PS, Noble MD, Ataliotis P, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci U S A. 1991;88:5096–100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez RD, Morales CP, Herbert BS, et al. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cotton J, Spandau DF. Ultraviolet B-radiation dose influences the induction of apoptosis and p53 in human keratinocytes. Radiat Res. 1997;147:148–55. [PubMed] [Google Scholar]
- 22.Hormi-Carver K, Feagins LA, Spechler SJ, Souza RF. All trans-retinoic acid induces apoptosis via p38 and caspase pathways in metaplastic Barrett's cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G18–27. doi: 10.1152/ajpgi.00237.2006. [DOI] [PubMed] [Google Scholar]
- 23.Huerta-Yepez S, Vega M, Jazirehi A, et al. Nitric oxide sensitizes prostate carcinoma cell lines to TRAIL-mediated apoptosis via inactivation of NF-kappa B and inhibition of Bcl-xl expression. Oncogene. 2004;23:4993–5003. doi: 10.1038/sj.onc.1207655. [DOI] [PubMed] [Google Scholar]
- 24.Rioux-Leclercq N, Turlin B, Sutherland F, et al. Analysis of Ki-67, p53 and Bcl-2 expression in the dysplasia-carcinoma sequence of Barrett's esophagus. Oncol Rep. 1999;6:877–82. doi: 10.3892/or.6.4.877. [DOI] [PubMed] [Google Scholar]
- 25.Latonen L, Laiho M. Cellular UV damage responses--functions of tumor suppressor p53. Biochim Biophys Acta. 2005;1755:71–89. doi: 10.1016/j.bbcan.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–62. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiraki K, Takase K, Nakano T. The emerging role of caspase inhibitors in gastrointestinal cancers. J Gastroenterol. 2002;37:323–31. doi: 10.1007/s005350200045. [DOI] [PubMed] [Google Scholar]
- 28.Souza RF, Shewmake KL, Shen Y, et al. Differences in ERK Activation in Squamous Mucosa in Patients Who Have Gastroesophageal Reflux Disease with and without Barrett's Esophagus. Am J Gastroenterol. 2005;100:551–9. doi: 10.1111/j.1572-0241.2005.41122.x. [DOI] [PubMed] [Google Scholar]
- 29.Ali I, Rafiee P, Hogan WJ, et al. Dickkopf homologs in squamous mucosa of esophagitis patients are overexpressed compared with Barrett's patients and healthy controls. Am J Gastroenterol. 2006;101:1437–48. doi: 10.1111/j.1572-0241.2006.00584.x. [DOI] [PubMed] [Google Scholar]