

Abstract
Sodium-dependent glutamate uptake is essential for limiting excitotoxicity, and dysregulation of this process has been implicated in a wide array of neurological disorders. The majority of forebrain glutamate uptake is mediated by the astroglial glutamate transporter, GLT-1. We and others have shown that this transporter undergoes endocytosis and degradation in response to activation of protein kinase C (PKC), however, the mechanisms involved remain unclear. In the current study, transfected C6 glioma cells or primary cortical cultures were used to show that PKC activation results in incorporation of ubiquitin into GLT-1 immunoprecipitates. Mutation of all 11 lysine residues in the amino and carboxyl-terminal domains to arginine (11R) abolished this signal. Selective mutation of the 7 lysine residues in the carboxyl terminus (C7K-R) did not eliminate ubiquitination, but it completely blocked PKC-dependent internalization and degradation. Two families of variants of GLT-1 were prepared with various lysine residues mutated to Arginine. Analyses of these constructs indicated that redundant lysine residues in the carboxyl terminus were sufficient for the appearance of ubiquitinated product and degradation of GLT-1. Together these data define a novel mechanism by which the predominant forebrain glutamate transporter can be rapidly targeted for degradation.
Keywords: GLT-1, endocytosis, protein kinase C, sodium-dependent, ubiquitin, uptake
Introduction
As the predominant excitatory neurotransmitter in the mammalian central nervous system, glutamate is essential for synaptic depolarization and neurotransmission (Monaghan et al., 1989). However, extracellular levels of glutamate must be tightly controlled to limit glutamate-mediated excitotoxicity and neuronal cell death (Choi, 1992). Since there is no system to metabolize glutamate in the extracellular space, the only way to terminate glutamatergic transmission is through glutamate uptake (reviewed in Schousboe, 1981). A family of Na+-dependent, high affinity glutamate transporters is responsible for this task (for reviews, see Sims and Robinson, 1999; Danbolt, 2001). The majority of glutamate uptake is mediated by the glial glutamate transporter, GLT-1, which is located predominantly on astrocytes throughout the cortex and hippocampus (reviewed in Robinson, 1998; Danbolt, 2001).
GLT-1 deletion in mice results in lethal seizures, emphasizing the importance of this transporter for glutamate uptake and cell survival (Tanaka et al., 1997). Decreases in GLT-1 protein levels are observed prior to neuronal death in area CA1 of the hippocampus after an ischemic insult, suggesting that decreased uptake of glutamate via GLT-1 may contribute to later phases of damage caused by these insults (Bruhn et al., 2000). GLT-1 protein begins to decrease in CA1 within 3-6 h post-reperfusion, indicating that it can be rapidly regulated (Torp et al., 1995; Chen et al., 2005; Yeh et al., 2005). Likewise, GLT-1 protein is decreased after 1 h of oxygen-glucose deprivation in astrocytic cultures isolated from CA1 (Ouyang et al., 2007). As the levels GLT-1 protein are stable for at least 24 h in the presence of inhibitors of transcription or translation in primary astrocyte cultures (Zelenaia and Robinson, 2000), it appears that the half-life of GLT-1 is likely longer than 24 h. This might be expected given the importance of this transporter to limiting excitotoxicity. Together these studies suggest that the loss of GLT-1 observed after ischemic insults is due to accelerated degradation of GLT-1 rather than decreased transcription.
As is observed for many other neurotransmitter transporters, including those for dopamine, norepinephrine, serotonin, γ-aminobutyric acid (GABA), and glycine (Gomeza et al., 1995; Qian et al., 1997; Zhang et al., 1997; Apparsundaram et al., 1998; Beckman et al., 1999; Melikian and Buckley, 1999; Jayanthi et al., 2004; Jayanthi et al., 2005), activation of protein kinase C (PKC) rapidly (within min) decreases GLT-1-mediated uptake and decreases cell surface GLT-1 (Fang et al., 2002; Kalandadze et al., 2002; Zhou and Sutherland, 2004; Guillet et al., 2005; Susarla and Robinson, 2008). In the case of the dopamine transporter (DAT), PKC activation results in ubiquitination of lysine (Lys) residues in the amino terminus, leading to internalization and degradation (Miranda et al., 2005; Miranda et al., 2007). Ubiquitination of cell surface proteins is a common mechanism underlying internalization, endosomal sorting, and either recycling back to the cell surface or sorting to the proteosome or lysosome for degradation (reviewed in Haglund et al., 2003; Kim and Rao, 2006; Madshus, 2006; Miranda and Sorkin, 2007).
Recently, we showed that more prolonged activation of PKC decreases the levels of GLT-1 protein by a mechanism consistent with lysosomal degradation (Susarla and Robinson, 2008). Since GLT-1 is important for cell survival and prevention of excitotoxicity, we were interested in defining the molecular mechanisms that trigger internalization and loss of GLT-1. Therefore, in the present study we investigated mechanisms underlying PKC-mediated down-regulation of GLT-1. We present evidence that redundant carboxyl-terminal Lys residues of GLT-1 are ubiquitinated and that carboxyl-terminal ubiquitination of GLT-1 is required for internalization and degradation of the transporter. The data from this manuscript were presented at the Annual Society for Neuroscience Meeting and were partially described in an abstract for this meeting (Sheldon et al., 2007).
Materials and Methods
Materials
C6 glioma cells were obtained from the American Type Culture Collection (Rockville, MD). 10-cm cell culture plates were purchased from Corning (Cambridge, MA). Dulbecco’s modified Eagle’s medium (DMEM), L-glutamine, trypsin-EDTA, penicillin-streptomycin, geneticin, and Protein A-agarose were from Invitrogen (Grand Island, NY). Fetal bovine serum and Ham’s F-12 with glutamine was purchased from HyClone (Logan, UT). Bovine serum albumin (BSA), phorbol 12-myristate 13-acetate (PMA), rabbit anti-actin antibody, and rabbit anti-Flag antibody were obtained from Sigma (St. Louis, MO). Bisindolylmaleimide II (Bis II) was purchased from Calbiochem (La Jolla, CA). GenePorter transfection reagent was obtained from Gene Therapy Systems (San Diego, CA). N-hydroxysulfosuccinimidobiotin (NHS-biotin), UltraLink immobilized monomeric avidin, and bicinchoninic acid (BCA) protein assay kit were from Pierce (Rockford, IL). Mouse monoclonal anti-ubiquitin antibody P4D1 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-GLT-1 antibody was the kind gift of Dr. Jeffrey D. Rothstein (Rothstein et al., 1994). Anti-rabbit and anti-mouse horseradish peroxidase IgG, enhanced chemiluminescence kits (ECL), Hyperfilm ECL, and rainbow molecular weight marker were purchased from Amersham (Arlington Heights, IL). Immobilon P (polyvinylidene fluoride membrane) was from Millipore (Bedford, MA).
Cell Culture
C6 glioma cells were grown in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2/95% air at 37°C. Cells were used for experiments up to passage 60, and no changes in experimental effects or gross morphological changes were observed due to passage number. Primary rat cortical co-cultures of neurons and astrocytes were dissociated as described previously (Wilcox et al., 1994; Cummings et al., 1996). Briefly, E18 rat embryos were removed from anesthetized pregnant Sprague-Dawley rats. The cortices were dissected and trypsinized in DMEM containing 0.027% trypsin at 37°C for 20 min. The tissue was then triturated in media containing DMEM, 10% fetal bovine serum, 10% Ham’s F-12 with glutamine and 50 U/ml penicillin-streptomycin. Cells were plated at 400,000 cells per ml in 6 cm plastic tissue culture dishes with 3 ml per dish in media consisting of DMEM, 10% fetal bovine serum, 10% Ham’s F-12 and penicillin-streptomycin. The cultures were maintained in an incubator at 5% CO2/95% air and 37°C for 16 days prior to experimentation.
Generation of mutant transporters
Lys to Arg (K-R) substitution mutants were created using the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) and pcDNA3.1+ rat GLT-1 templates (Kalandadze et al., 2002). After 11 Lys in the amino and carboxyl termini were mutated to Arg to make the 11R transporter, Arg to Lys (R-K) restoration mutants were also created with Quikchange, using the pcDNA3.1+ 11R template. All sequences were confirmed at the molecular biology core at the Children’s Hospital of Philadelphia.
Transient transfection of C6 glioma cells
50-60% confluent C6 cells grown in 10-cm plates were transfected with 12 μg of cDNA mixed with 60 μl GenePorter in a 1 μg: 5 μl ratio. The amount of cDNA and GenePorter used for transfection of C6 cells grown in 12-well plates was adjusted for surface area/number of cells (Kalandadze et al., 2002). Cells were used for experiments ~18h after transfection.
Immunoprecipitation
Immunoprecipitation of glutamate transporters was performed as described previously, with small changes (Sheldon et al., 2006). Cells were serum-starved in DMEM containing 0.5% BSA prior to treatment (C6 cells 1h and primary cultures 3h). After treatment, cell monolayers were washed twice with ice-cold PBS (pH 7.35) containing 0.1 mM CaCl2 and 1.0 mM MgCl2. Cells were then lysed (0.7 mL for C6 cells grown in 10 cm dishes and 0.4 mL for primary cultures grown in 6 cm dishes) in Triton/Glycerol/HEPES (TGH) buffer containing: 150 mM KCl, 2 mM MgCl2, 20 mM HEPES, 10% glycerol, (pH 7.2), 1% Triton X-100, 1% sodium deoxycholate, 1 mM dithiothreitol (DTT), 1mM ethylene glycol tetraacetic acid (EGTA), 1mM ethylenediamine tetraacetic acid (EDTA), 10 μg/ml leupeptin, 1 mM phenylmethanesulfonyl fluoride, 10 μg/ml aprotinin, 1 mM iodoacetamide, 10 mM sodium fluoride, 1 mM sodium orthovanadate, and 10 mM N-ethylmaleimide (Huang et al., 2006a). Lysates were cleared of cellular debris by centrifugation at 17,000 × g for 20 min at 4°C. The resulting supernatant was pre-cleared by shaking with 40 μl protein-A agarose beads at 4°C for 1 h. This slurry was centrifuged at 17,000 × g for 15 min and then equal amounts of subsequent supernatant (reflecting 450 μg of protein from the lysate) were mixed overnight at 4°C with 15 μL GLT-1 antibody or a comparable amount of rabbit IgG antibody. Antibody bound proteins were isolated using 30 μl protein-A agarose beads. After incubating for 2 h at 4°C, the material was centrifuged and the pellets (beads) were washed three times: once with TGH buffer containing 500 mM NaCl, once with TGH buffer containing 100 mM NaCl, and once with TGH buffer containing no NaCl. Immunoprecipitates were eluted from the agarose beads by incubation at 95°C for 5 min in 30 μl SDS-PAGE loading buffer.
Biotinylation of cell surface transporters
C6 cells were serum-starved in DMEM containing 0.5% BSA for one hour at 37°C. Following vehicle (DMSO) or PMA (100 nM) treatment for 30 min, cell surface proteins were biotinylated as described previously (Davis et al., 1998; Fournier et al., 2004; Sheldon et al., 2006). Briefly, C6 cells were washed twice with ice-cold PBS Ca/Mg. The cells were then incubated in 2 ml biotinylation solution (1 mg/ml NHS-biotin in PBS Ca/Mg) for 25 min at 4°C with gentle shaking. The solution was aspirated and un-reacted biotin was quenched by incubating cells with PBS Ca/Mg containing 100 mM glycine for 25 min at 4°C with gentle agitation. Cells were lysed in 0.7 ml of radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors (1 μg/ml leupeptin, 250 μM phenylmethanesulfonyl fluoride, 1 μg/ml aprotinin, and 1 mM iodoacetamide) (Gonzalez et al., 2007). Cellular debris was removed by centrifugation at 17,000 × g for 20 min at 4°C, and then biotinylated proteins were batch-extracted using UltraLink immobilized monomeric avidin beads. SDS-PAGE sample buffer was added to cell lysate, biotinylated proteins (cell surface proteins), and non-biotinylated proteins (intracellular proteins). These three fractions were diluted so that the sum of the immunoreactivity in the biotinylated and non-biotinylated fractions would equal that observed in the lysate if the yield from extraction was 100%.
Measurement of Na+-dependent transport activity
Transport activity in C6 glioma was measured in 12-well plates as previously described (Dowd and Robinson, 1996; Kalandadze et al., 2002). C6 cells were assayed in a 37°C water bath, and wells were washed twice with either 1 ml of warm Na+- or choline-containing buffer and then incubated with 0.5 μM [3H]-glutamate for five min. After halting uptake of radioactive glutamate, the cells were solubilized, and samples were taken for analysis of radioactivity in a scintillation counter. Na+-dependent uptake was defined as the difference in radioactivity accumulated in the presence and absence of Na+.
Degradation assay
C6 cells were pretreated with 0.5% BSA in DMEM for one hour before incubation with vehicle (DMSO) or PMA (100 nM) for 2 h at 37°C. Next, cells were rinsed twice with PBS Ca/Mg and then lysed in 0.7 ml of RIPA buffer containing protease inhibitors (listed above). Cellular debris was removed by centrifugation at 17,000 × g for 20 min at 4°C. Cell lysates were then mixed 1:1 with SDS sample buffer.
Western blot analysis
Proteins were resolved using 8% SDS-polyacrylamide gels, transferred to polyvinylidene fluoride membranes and blocked overnight in TBS-T (50 mM Tris, pH 8.0, 150 mM NaCl, 0.2% Tween 20) containing 5% nonfat dry milk. Membranes were then probed with the appropriate primary antibody: anti-ubiquitin (1:200), anti-GLT-1 (1:10,000), anti-Flag (1:1000), or anti-actin (1:5,000). Membranes were washed in TBS-T containing 1% nonfat dry milk and then incubated with anti-rabbit or anti-mouse horseradish peroxidase IgG (1:5,000). Protein bands were visualized with ECL.
Immunoreactivity was quantified using NIH Image software. Immunoreactive bands are routinely observed at ~66 kDa, which corresponds to GLT-1 monomer, and at ~200 kDa, which corresponds to irreversible GLT-1 multimers/aggregates (Haugeto et al., 1996). We routinely quantitate and analyze the monomers, the multimers, and the sum of these bands (Sims and Robinson, 1999). In some of the current experiments, the multimer bands were more abundant, resulting in slight saturation of the signal for the multimer bands when the exposure was long enough to quantitate the monomer band. Therefore, it may appear that the monomer band changes more than the multimer band. In all cases, the changes in multimer and monomer were qualitatively similar. We also quantitated monomer and multimer bands for the data presented in figure 5 performed with wild type GLT-1 using different exposures of the film under conditions where the signal was not saturated. Under these conditions, the change in monomer and multimer were not statistically different. The data presented are the sum of the immunoreactivity found in the monomer and multimer bands under these conditions; therefore, if anything the average data presented represent a slight underestimate in change in transporter immunoreactivity. Transporter immunoreactivity in each fraction (lysate, cell surface, or intracellular fraction) was normalized to the amount of actin in the lysate fraction. The amount of transporter observed under treatment conditions was expressed as a percentage of transporter in the corresponding vehicle. In addition to acting as a loading control, actin immunoreactivity was measured to ensure that cells were not permeabilized during the biotinylation procedure. The percentage of biotinylated actin in untreated controls was 3% or less and did not change with treatment. Data are presented as the mean ± standard error of the mean (SE). For each individual construct, the effects of PMA (as a percentage) were compared to the effects of vehicle (100%) using a one-sample t-test. In addition, data were compared using one-way ANOVA with Bonferroni post-hoc analysis. These tests were employed to compare the effect of PMA on mutant transporters to the effect of PMA on wild-type GLT-1.
Figure 5. Effect of 2 h PMA treatment on protein levels of GLT-1 or GLT-1 (K-R) mutants.
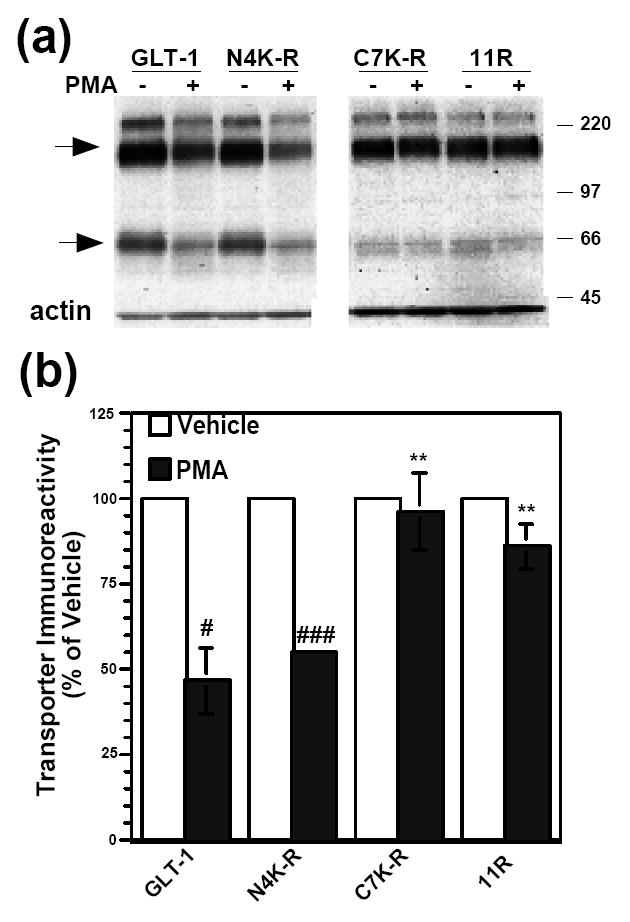
(a) C6 cells were transiently transfected with GLT-1 or GLT-1 (K-R) mutant transporter cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 2 h. Cell lysates were analyzed by Western blot. Representative Western blots probed with GLT-1 and actin antibodies. Arrows point to transporter monomers and multimers. (b) Summary of results of a minimum of three independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control; ### indicates a p<0.001 compared to the corresponding vehicle control (one-sample t-test). PMA had no significant effect on C7K-R or 11R expression compared to their vehicle controls (one-sample t-test). The effects of PMA on the different constructs were compared by one-way ANOVA with Bonferroni post-hoc analysis; ** indicates a p<0.01 compared to % change in lysate expression of GLT-1 due to PMA (one-way ANOVA with Bonferroni post-hoc analysis).
Results
GLT-1 protein levels decrease after activation of PKC
GLT-1-mediated activity is decreased by PKC activation, and this effect is associated with a redistribution of GLT-1 from the plasma membrane to an intracellular compartment (Sims et al., 2000; Fang et al., 2002; Kalandadze et al., 2002; Zhou and Sutherland, 2004; Guillet et al., 2005). More recently, we showed that prolonged treatment with the phorbol ester, PMA, decreases total GLT-1 protein levels within 2 to 4 h in C6 glioma cells transiently transfected with GLT-1, consistent with accelerated degradation (Susarla and Robinson, 2008). In the present study, we confirmed that PMA caused a loss in total GLT-1 protein levels within 2 h to ~50% of control (Fig 1a). To determine if this loss of GLT-1 is related to activation of PKC, the effects of the selective PKC antagonist, Bis II, were examined. Bis II completely blocked the PMA-induced loss of GLT-1 (Fig. 1a), suggesting that the degradation of GLT-1 is PKC-dependent. In our previous study, we demonstrated that two different inhibitors of lysosomal degradation partially attenuated this loss of GLT-1 immunoreactivity (Susarla and Robinson, 2008). More recently Gibb and colleagues demonstrated that caspase-3 selectively cleaves the carboxyl-terminal domain of GLT-1 (Gibb et al., 2007). Since this domain contains the epitope recognized by the GLT-1 antibody used in the current study and the loss of GLT-1 was only partially blocked by lysosomal inhibitors, it was important to determine if some of this loss of GLT-1 immunoreactivity might be attributed to partial proteolysis of the carboxyl terminus. To address this possibility, C6 glioma were transfected with an epitope-tagged variant of GLT-1 with a Flag epitope at the amino terminus, and the effects PMA (2 h) on transporter levels were examined (Fig. 1b, c). In these experiments, the decreases in GLT-1 immunoreactivity with the Flag-tagged variant of GLT-1 was not different from that observed with wild-type GLT-1. Importantly, the decrease in amino-terminal Flag immunoreactivity (Fig. 1c) was not significantly different from that observed for the carboxyl-terminal GLT-1 immunoreactivity (Fig. 1b) (p = 0.68 by Student’s paired t-test). Since Gibb and colleagues showed that there was no loss of the amino terminus after activation of caspase-3 (Gibb et al., 2007), these data are consistent with the hypothesis that this loss of GLT-1 immunoreactivity cannot be attributed to partial proteolysis; instead it is likely due to degradation of the transporter. To determine if transient treatment with PMA causes a loss of GLT-1 immunoreactivity, transfected C6 glioma were treated with PMA for 5 min followed by an extensive wash and continued incubation for 2h in the absence of PMA. This transient treatment with PMA caused the same decrease in GLT-1 as that observed with continuous treatment with PMA for 2 h (Fig. 1d). As PMA is relatively lipophillic one cannot rule out the possibility that these washes do not completely remove PMA, however, these data do suggest that transient activation of PKC may also cause a loss of GLT-1 protein under these circumstances.
Figure 1. Effect of PMA on the protein levels of GLT-1 or flag-tagged GLT-1.

(a) C6 cells were transiently transfected with GLT-1 cDNA. After 16-20 h, cells were pre-treated with Bis II (10 μM) for 5 min and then with vehicle (DMSO) or PMA (100 nM) for 2 h. In addition, GLT-1-transfected C6 cells were pre-treated with Bis II (10 μM) for 5 min before either vehicle or PMA treatment. Cell lysates were analyzed by Western blot. Representative Western blots probed with GLT-1 and actin antibodies. Summary of results of three independent experiments (mean ± SE). The effects of PMA were examined by ANOVA; ## indicates a p<0.01 compared to the vehicle control. (b) C6 cells were transiently transfected with GLT-1 or amino-terminal tagged Flag-GLT-1 cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 2 h. Cell lysates were analyzed by Western blot. Representative Western blots probed with GLT-1 and actin antibodies. The lower panel represents the summary of results of three independent experiments (mean ± SE) with the levels of GLT-1 expressed as % of that observed Flag-GLT-1 transfected cells that were treated with vehicle. The effects of PMA were examined by one-way ANOVA; # indicates a p<0.05 compared to the corresponding vehicle control (one-sample t-test). (c) These same samples were probed with an anti-Flag antibody. Summary of results of three independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; ## indicates a p<0.01 compared to the vehicle control (one-sample t-test). (d) C6 cells were transiently transfected with GLT-1 cDNA and treated with vehicle (DMSO) or PMA (100 nM) for 2 h or 5 min followed by washout and a 2 h incubation without PMA. Cell lysates were analyzed by Western blot. Representative Western blots probed with GLT-1 and actin antibodies and summary of results of three independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control (one-sample t-test).
Ubiquitin is incorporated into GLT-1 immunoprecipitates after PKC activation
To investigate whether ubiquitination of GLT-1 is involved in PKC-induced internalization or degradation, we tested for the presence of ubiquitin in GLT-1 immunoprecipitates after PKC activation. C6 glioma cells were transiently transfected with GLT-1 cDNA and treated with PMA for 30 min. Transfected C6 cells were also pre-treated with or without the PKC-selective inhibitor, Bis II, for 5 min. Following treatment, GLT-1 was immunoprecipitated and analyzed by Western blot using an anti-ubiquitin antibody. PKC activation resulted in broad ubiquitin signals around ~100 kDa and ~250 kDa; both bands are ~30 kDa larger than the GLT-1 monomers and multimer bands, respectively (Fig. 2a). In parallel experiments, no ubiquitin signal was observed in cells transfected with empty vector (mock) that were treated with vehicle or PMA and immunoprecipitated with an anti-GLT-1 antibody. In addition, no signal was observed in GLT-1 transfected cells that were treated and immunoprecipitated with a control antibody (IgG). The appearance of the ubiquitin bands was blocked by the PKC inhibitor, Bis II. These Western blots were stripped and reprobed with GLT-1 antibody to test for equal immunoprecipitation of GLT-1 (Fig. 2b). Somewhat surprisingly, no GLT-1 immunoreactivity was detected as diffuse bands of ~100 or 250 kDa similar in size to those observed for ubiquitin immunoreactivity (Fig. 2b). In some of these blots, a sharp band at ~100 kDa is observed. This band is observed in mock transfected cells or after immunoprecipitation with control serum (IgG). We frequently observe this band with other immunoprecipitations and suspect that it represents dimers of antibody. Since the molecular weight shift was similar for both monomers and multimers and the agarose beads used for immunoprecipitation underwent relatively stringent high salt washes, the simplest explanation is that ubiquitin is directly incorporated into GLT-1 (see discussion for other possible explanations).
Figure 2. Effects of PMA on incorporation of ubiquitin into GLT-1 immunoprecipiates from transiently transfected C6 glioma or primary cultures.
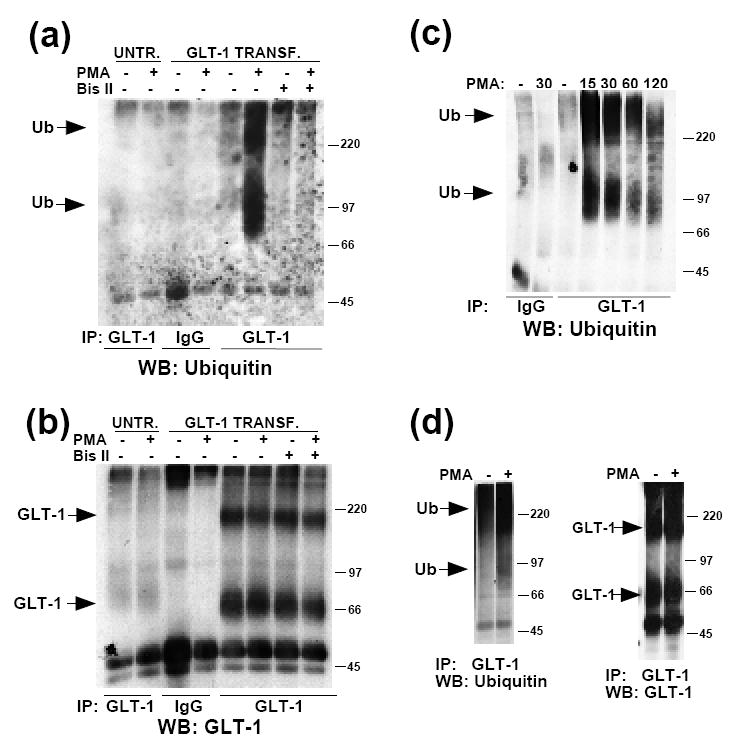
(a) Untransfected or GLT-1 transfected C6 cells were treated with vehicle (DMSO) or PMA (100 nM) for 30 min at 37°C. In addition, GLT-1-transfected C6 cells were pre-treated with Bis II (10 μM) for 5 min before either vehicle or PMA treatment. Following treatment, cells were lysed, and proteins were immunoprecipitated with control rabbit IgG or anti-GLT-1 antibody at 4°C, as described in the Materials and Methods. Immunoprecipitates were analyzed by Western blot. Representative Western blot probed with ubiquitin antibody, showing the effects of PKC activation or inhibition on ubiquitination of GLT-1 (n=3). Arrows point to ubiquitin immunoreactive signal. (b) The Western blot was stripped and reprobed with GLT-1 antibody to test for equal immunoprecipitation of GLT-1 in samples. (c) GLT-1 transfected cells were treated with PMA for varying amounts of time (min). Representative Western blot showing the time-course of ubiquitination of GLT-1 (n=2). (d) Primary neuron/astrocyte co-cultures were treated with vehicle or PMA for 30 min. GLT-1 was immunoprecipitated following treatment. Immunoprecipitates were analyzed by Western blot and probed with ubiquitin antibody. Representative Western blot showing ubiquitination of GLT-1 after PMA treatment (3 out of 5 experiments displayed ubiquitin immunoreactivity). The blot was stripped and reprobed for GLT-1.
The time-course for incorporation of ubiquitin into GLT-1 immunoprecipitates was investigated by treating GLT-1-transfected C6 cells with PMA for up to 2 h. There was robust ubiquitination of GLT-1 at short time points, but the signal declined at longer time points (Fig. 2c). Based on a comparison with our previously published time-course for the loss in total GLT-1 immunoreactivity (Susarla and Robinson, 2008), this time-course is consistent with ubiquitination preceding degradation. To determine if ubiquitination of GLT-1 is selective for C6 cells, GLT-1 was immunoprecipitated from primary cortical co-cultures of astrocytes and neurons treated with PMA for 30 min. Ubiquitin immunoreactivity was also observed in this system, indicating that ubiquitination of GLT-1 can occur in a more physiologically relevant system (Fig. 2d).
Mutation of 11 Lys residues in the carboxyl and amino termini abolishes the incorporation of ubiquitin into GLT-1 immunoprecipitates
Based on the crystal structure of a bacterial glutamate transporter and cysteine mutagenesis scanning of mammalian GLT-1, there are 8 transmembrane domains with cytoplasmic carboxyl and amino termini (Grunewald and Kanner, 2000; Yernool et al., 2004). Using these analyses, we predicted that there were up to 16 intracellular Lys residues, four on the amino terminus, seven on the carboxyl terminus, and up to five in the intracellular loops. We began by mutating the 4 Lys residues of the amino terminus to Arg (N4K-R), the 7 Lys residues of the carboxyl terminus to Arg (C7K-R), and by mutating all 11 Lys residues in both termini to Arg (11R). All mutants were tested for glutamate transport function. C6 cells were transfected with GLT-1, empty vector (pcDNA3.1+), or K-R mutant transporter cDNAs, and glutamate uptake activity was measured. Since C6 cells endogenously express the neuronal glutamate transporter, EAAC1, glutamate uptake for each construct was expressed as a percentage of uptake observed with naïve (control) cells (Kalandadze et al., 2002). GLT-1 and these K-R mutant transporters significantly increased glutamate uptake compared to naïve or mock-transfect cells (Fig. 3a), providing strong evidence that these mutations do not cause such dramatic changes in protein conformation that they affect transporter function.
Figure 3. Activity and ubiquitination of wild-type GLT-1 or Lys to Arg (K-R) mutants.
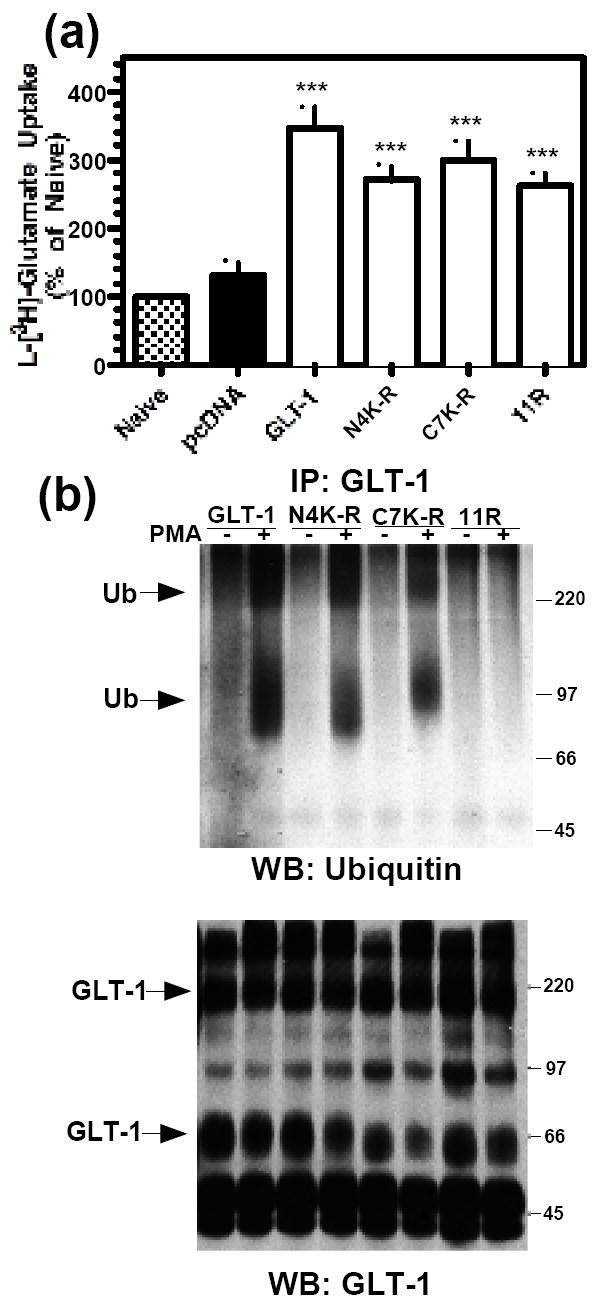
C6 cells were transiently transfected with GLT-1, N4K-R, C7K-R or 11R cDNAs. (a) Na+-dependent L-[3H]-glutamate transport was measured as described in the Materials and Methods. A summary of results of four independent experiments performed in triplicate (mean ± S.E.) is shown. Data are expressed as a percentage of activity observed in naïve cells (control). *** indicates a p<0.001 compared to naïve control (one-way ANOVA with Bonferroni post-hoc analysis). (b) C6 cells were treated with vehicle (DMSO) or PMA (100 nM) for 30 min. Following treatment, transporters were immunoprecipitated with GLT-1 antibody. Immunoprecipitates were analyzed by Western blot. Representative Western blot probed with ubiquitin antibody (n=3). Arrows point to ubiquitin immunoreactive signal. Note the lack of ubiquitin signal in the 11R samples. Western blots were stripped and reprobed with GLT-1 antibody.
To determine if these lysine residues are required for incorporation of ubiquitin, C6 cells were transfected with GLT-1 or K-R mutant transporter cDNAs. After activation of PKC with PMA for 30 min, anti-GLT-1 antibodies were used to immunoprecipitate these transporters, and the resulting immunoprecipitates were analyzed for ubiquitin immunoreactivity by Western blot analysis. Although we observed evidence for ubiquitin immunoreactivity of similar sizes to that observed for wild-type GLT-1 with both the N4K-R and C7K-R constructs, no ubiquitin immunoreactivity was observed in the immunoprecipitates from cells transfected with the 11R mutant variant (Fig. 3b). The amount of GLT-1 immunoreactivity observed in these immunoprecipitates was comparable (Fig 3b, lower panel). These studies provide strong evidence that the appearance of ubiquitin immunoreactivity in these immunoprecipitates can occur when Lys residues are preserved in either the amino or the carboxyl termini of the transporter, but that elimination of all 11 lys residues abolishes the incorporation of ubiquitin immunoreactivity into GLT-1 immunoprecipitates.
Carboxyl-terminal Lys residues are required for internalization and degradation of GLT-1
To determine if these same Lys residues are required for internalization, C6 cells were transfected with GLT-1, N4K-R, C7K-R, or 11R cDNAs. After activation of PKC with PMA (30 min), a membrane impermeant biotin reagent was utilized to covalently label and separate biotinylated (cell surface) from non-biotinylated (intracellular) proteins (Daniels and Amara, 1998; Davis et al., 1998; Sheldon et al., 2006). The percentage of transporter at the cell surface under basal conditions did not differ between constructs and was essentially 100% (data not shown). As shown previously, activation of PKC caused a significant decrease in the amount of biotinylated GLT-1 to 73 ± 7% of control (Fig. 4a,e). This effect was associated with a slight increase in non-biotinylated transporter, but was also associated with a significant decrease in the amount of total GLT-1 immunoreactivity to 82 ± 5% of control (Fig 4e). Although PMA caused a significant decrease in the amount of biotinylated N4K-R transporter that was not significantly different from that observed for wild-type GLT-1, it had no effect on either the C7K-R or the 11R mutant variants of GLT-1 (Fig. 4c,d,e). These results suggest that although Lys residues on both the amino and carboxyl termini are required for incorporation of ubiquitin into immunoprecipitates, only Lys residues on the carboxyl terminus are required for regulated internalization of the transporter.
Figure 4. Effect of PMA on cell surface expression of GLT-1 (K-R) mutant transporters.
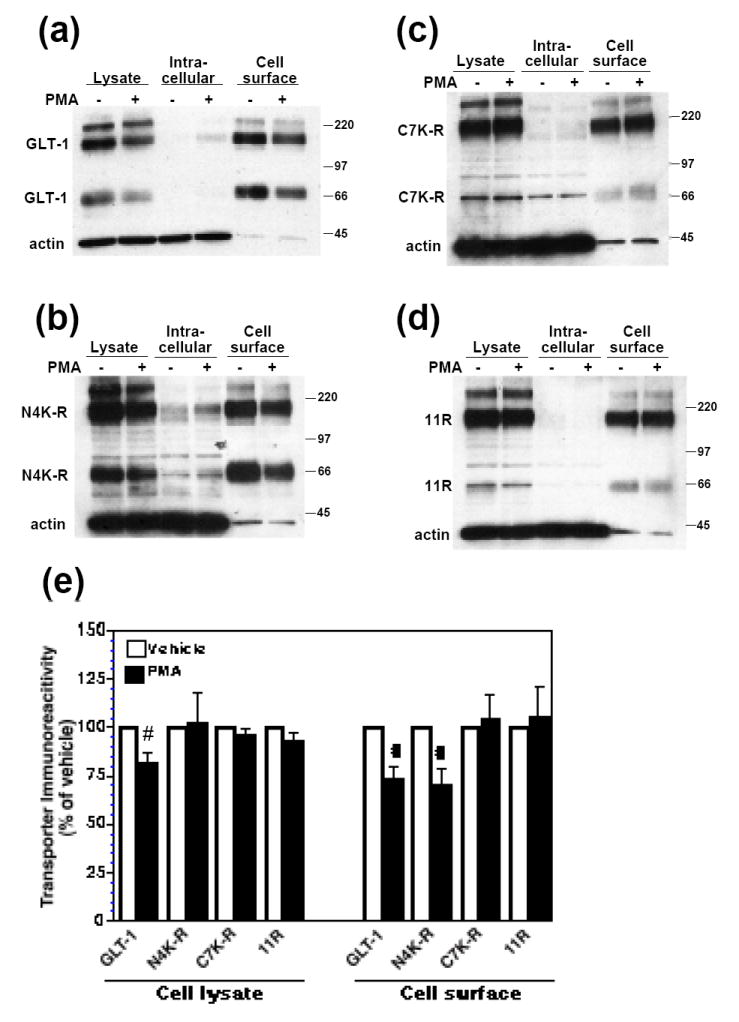
C6 cells were transiently transfected with GLT-1 or GLT-1 (K-R) mutant transporter cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 30 min. Following treatment, cell surface proteins were biotinylated and batch extracted with avidin beads at 4°C, as described in the Materials and Methods. Cellular fractions were analyzed by Western blot. (a) Representative Western blot for GLT-1 samples probed with GLT-1 and actin antibodies, showing decreased cell surface expression of GLT-1 after PMA treatment. (b) Representative Western blot for N4K-R samples probed with GLT-1 and actin antibodies, showing decreased cell surface expression of N4K-R after PMA treatment. (c) Representative Western blot for C7K-R samples probed with GLT-1 and actin antibodies. In this experiment most of the transporter migrated as a multimer. (d) Representative Western blot for 11R samples probed with GLT-1 and actin antibodies. In this experiment most of the transporter migrated as a multimer. (e) Summary of results of five independent experiments (mean ± SE). Immunoreactivity observed with PMA treatment was expressed as a percentage of that observed with vehicle treatment (control). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control (one-sample t-test). PMA had no significant effect on C7K-R or 11R cell surface expression (one-sample t-test). The percentage of biotinylated actin in untreated controls was 3% or less for all constructs and did not change with treatment.
Although the effects of PMA on total transporter were not significantly different for wild-type GLT-1 and the N4K-R construct, PMA did not decrease total N4K-R transporter immunoreactivity compared to vehicle treated control after 30 min (Fig. 4e). Since the decreases in total GLT-1 are somewhat small and therefore more difficult to detect with relatively small sample sizes, the effects of longer-term activation of PKC on total transporter immunoreactivity were compared for the three mutant variants. C6 cells were transfected with GLT-1, N4K-R, C7K-R, or 11R and treated with PMA for 2 h. Lysates were then analyzed by Western blot. PMA caused a decrease in total GLT-1 protein (to 47 ± 10% of control) or N4K-R (to 55 ± 1% of control), but had no effect on the levels of the C7K-R or 11R mutant variants of GLT-1 (Fig. 5a, b). The effect of PMA on the N4K-R mutant variant did not differ significantly from the effect of PMA on wild-type GLT-1, and there was no effect of PMA on the C7K-R or 11R constructs. Together, these studies suggest that the carboxyl-terminal Lys residues are required for internalization and degradation of GLT-1.
In order to narrow down which Lys residues in the carboxyl terminus were required for degradation of GLT-1, sequential K-R mutations were made within the carboxyl terminus of GLT-1 (Fig. 6a). PMA (2h) caused a decrease in immunoreactivity of the mutant transporters, C2K-R and C4K-R (to 52 ± 7% of and 48 ± 2% of control, respectively). However, there was no decrease in immunoreactivity of C6K-R with PMA treatment (Fig. 6b,c). This suggests that either the two additional residues mutated in the C6K-R mutant (K517 and K526) are involved and/or that a certain number of lysine residues are required for internalization/degradation.
Figure 6. Effect of 2 h PMA treatment on the protein levels of wild-type GLT-1 or carboxyl-terminal K-R mutants.
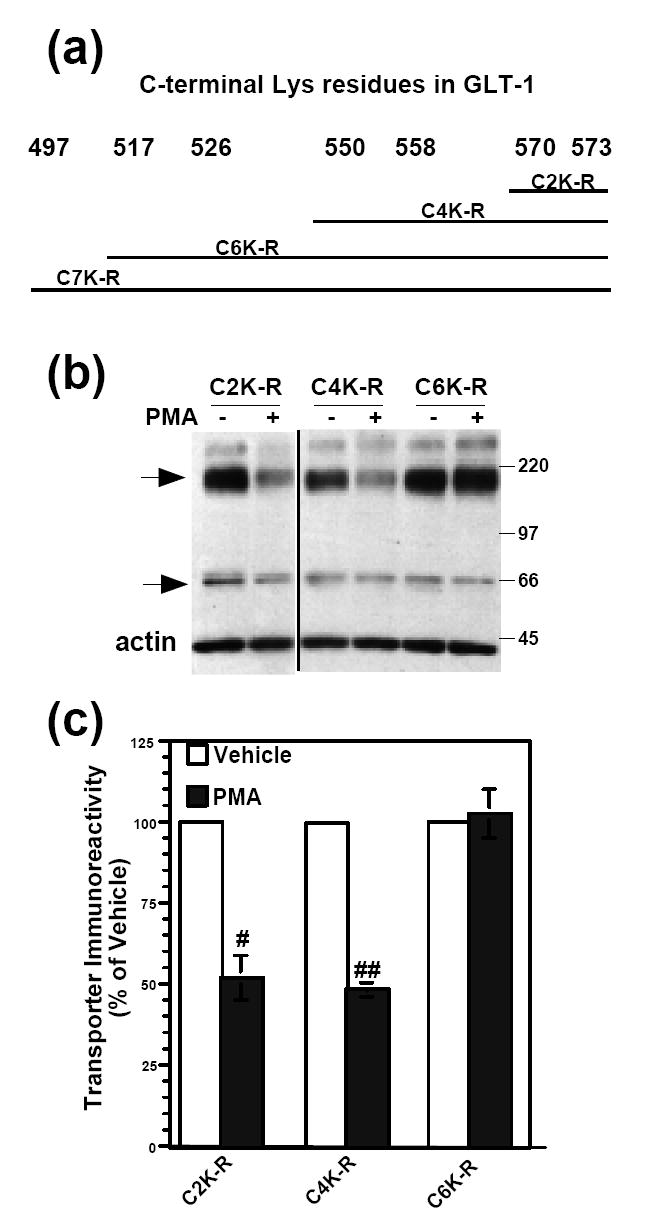
(a) Schematic representation of transporters with increasing numbers of carboxyl-terminal Lys-Arg (K-R) mutations in GLT-1. (b) C6 cells were transiently transfected with carboxyl-terminal K-R mutant cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 2 h. Cells were lysed and then analyzed by Western blot. Representative Western blot probed with GLT-1 and actin antibodies. Arrows point to transporter monomers and multimers. All lanes are from different parts of the same gel. (c) Summary of results of three independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control; ## indicates a p<0.01 compared to the corresponding vehicle control (one-sample t-test). PMA had no significant effect on C6K-R expression compared to control (one-sample t-test).
Single, double, and triple Lys-Arg mutations do not abolish the incorporation of ubiquitin or degradation of GLT-1
Since C6K-R was not degraded following a 2h PMA treatment, we focused on mutation of K517, K526 and the nearby residue, K550. To determine if single K-R mutations abolished incorporation of ubiquitin into transporter immunoprecipitates, these residues were mutated in the N4K-R backbone to eliminate the signal that occurs when these residues are present (see Fig 3b). N4K-R(517R), N4K-R(526R), N4K-R(517,526R), or N4K-R(517,526,550R) mutant transporters were expressed in C6 cells. GLT-1 was used as a positive control for ubiquitination and 11R was used as a negative control. As was observed in our earlier studies, no PMA-induced increase in ubiquitin signal was observed in the immunoprecipiates from cells transfected with the 11R mutant variant. Although the sequential mutagenesis of the C-terminal tail implicated lysine residues 517 and 526 (see Fig. 6), mutation of these residues alone or in combination with lysine 550 did not abolish the PMA-induced incorporation of ubiquitin into transporter immunoprecipitates (Fig. 7a).
Figure 7. Effect of PMA on incorporation of ubiquitin and protein levels of carboxyl-terminal single, double, or triple K-R mutations in the N4K-R or GLT-1 backbone.
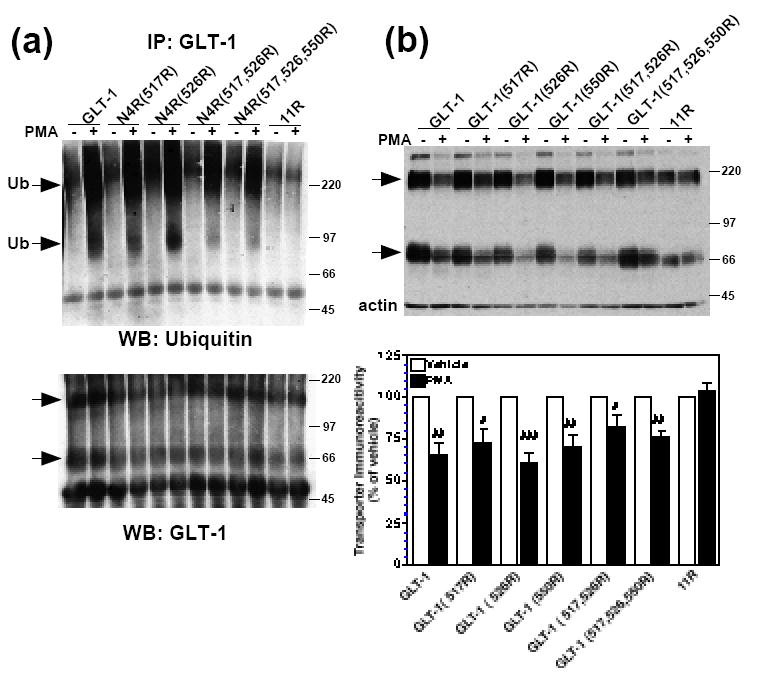
(a) C6 cells were transiently transfected with GLT-1, N4K-R(517R), N4K-R(526R), N4K-R(517,526R), N4K-R(517,526,550R), or 11R cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 30 min. Following treatment, transporters were immunoprecipitated with GLT-1 antibody, as described in the Materials and Methods. Immunoprecipitates were analyzed by Western blot. Representative Western blot probed with ubiquitin antibody (n=3). Arrows point to ubiquitin immunoreactive signal. Western blots were stripped and reprobed with GLT-1 to confirm immunoprecipitation of transporters from all samples. Arrows point to transporter monomers and multimers. (b) C6 cells were transiently transfected with GLT-1, GLT-1(517R), GLT-1(526R), GLT-1(550R), GLT-1(517,526R), GLT-1(517,526,550R), or 11R cDNAs and treated with vehicle (DMSO) or PMA (100 nM) for 2 h. Following treatment, cells were lysed, and lysates were analyzed by Western blot. Arrows point to transporter monomers and multimers. Summary of results of a minimum of six independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control; ## indicates a p<0.01 compared to the corresponding vehicle control; ### indicates a p<0.001 compared to the corresponding vehicle control (one-sample t-test). PMA had no significant effect on 11R expression compared to control (one-sample t-test).
To determine if mutation of these residues is sufficient to abolish PMA-induced degradation of GLT-1 single, double, and triple mutants of K517R, K526R, and K550R were made in the GLT-1 backbone. C6 cells were transfected with these mutant transporters, and after ~18 h the cells were treated with PMA for 2 h and analyzed by Western blot. Wild type GLT-1 or the 11R mutant variant were transfected in parallel as positive and negative controls, respectively. Compared to their corresponding vehicle-treated control, PMA significantly decreased the levels of total GLT-1 and all of the mutant variants that contained any lysine residues but had no effect on the 11R mutant variant (Fig. 7b). When the effects of PMA on these mutant variants were compared, there was no significant differences except for the comparisons to the 11R transporter variant. These data provide strong evidence that mutation of K517, K526, and K550 is not sufficient to abolish ubiquitination and degradation of GLT-1. Together with the data described in Fig. 6, these studies suggest that redundant lysine residues in the carboxyl terminus are sufficient for the incorporation of ubiquitin into transporter immunoprecipitates and for internalization/degradation of GLT-1.
Single Lys residues in the carboxyl terminus are sufficient for incorporation of ubiquitin and for degradation of GLT-1
To further test the possibility that lysine residues might function in a redundant fashion, single, double, and triple reverse mutations were made in the 11R backbone (R517K, R526K, and R550K). C6 cells were transfected with these mutant variants and treated with PMA for 30 min. Again in parallel experiments, cells were also transfected with wild-type GLT-1 or the 11R mutant variant as positive and negative controls. Upon immunoprecipitation with anti-GLT-1 antibody, no PMA-induced appearance of a ubiquitin signal was observed with the 11R mutant variant. However, introduction of a single lysine residue was sufficient to observe PMA-induced incorporation of ubiquitin (Fig. 8a). Although the signal was consistently less intense with the transporter variants containing a single lysine residue than that observed with wild-type GLT-1, a PMA-induced incorporation of ubiquitin was observed when any one of three different single lysine residues (517, 526, or 550) were present (Fig. 8a). This provides clear evidence that redundant lysine residues are sufficient to observe incorporation of ubiquitin into transporter immunoprecipitates. The ubiquitin signal was consistently darker for 11R(517,526K) and 11R(517,526,550K) mutant transporters suggesting that the availability of more Lys residues in the carboxyl terminus increases the ubiquitination of the transporters. To determine if the presence of this ubiquitin signal is associated with PMA-induced degradation (i.e. whether single Lys residues were sufficient for degradation), C6 cells were transfected with the 11R(R-K) mutant transporters and treated with PMA for 2 h. Compared to their corresponding vehicle controls, PMA caused a significant decrease in transporter immunoreactivity for the following transporters: 11R(526K): to 64 ± 2%; 11R(517,526K): to 57 ± 4%; 11R(517,526,550K): 64 ± 4%; and GLT-1: 67 ± 4%, indicating that even a single Lys residue in the carboxyl terminus was sufficient to promote degradation (Fig. 8b). Although there was a trend towards decreased transporter immunoreactivity compared to vehicle-treated controls for 11R(517K) (to 79 ± 9% of control) and 11R(550K) (to 83 ± 7.5% of control), this effect was not quite statistically significant by one sample t-test (p = 0.08 and p = 0.07, respectively). The effects of PMA on all of the lysine containing mutant transporters were significantly different from that observed with the 11R construct by ANOVA. Therefore, it seems likely that both the 11R(517K) and the 11R(550K) are affected by PMA. Together these data strongly suggest that redundant single Lys residues in the carboxyl-terminus of 11R are sufficient for the PMA-induced appearance of ubiquitin in transporter immunoprecipitates and for the PMA-induced loss in total transporter expression.
Figure 8. Effect of PMA on incorporation of ubiquitin and protein levels of carboxyl-terminal single, double, or triple R-K mutations in the 11R backbone.
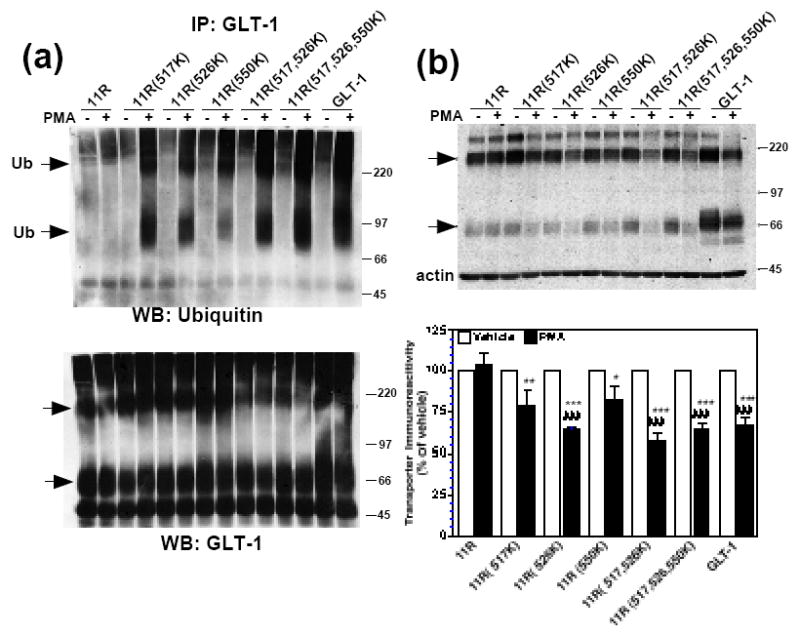
C6 cells were transiently transfected with 11R, 11R(517K), 11R(526K), 11R(550K), 11R(517,526K), 11R(517,526,550K), or GLT-1 cDNAs. (a) C6 cells were treated with vehicle or PMA for 30 min, immunoprecipitated with GLT-1 antibody, and analyzed by Western blot, as described in the Materials and Methods. Representative Western blot probed with ubiquitin antibody (n=3). Arrows point to ubiquitin immunoreactive signal. Western blots were stripped and reprobed with GLT-1 to confirm immunoprecipitation of transporters. Arrows point to transporter monomers and multimers. (b) C6 cells were treated with vehicle or PMA for 2 h, lysed, and analyzed by Western blot. Arrows point to transporter monomers and multimers. Summary of results of six independent experiments (mean ± SE). The effects of PMA were examined by one-sample t-tests; # indicates a p<0.05 compared to the corresponding vehicle control; ## indicates a p<0.01 compared to the corresponding vehicle control; ### indicates a p<0.001 compared to the corresponding vehicle control (one-sample t-test). PMA had no significant effect on 11R, 11R(517K), or 11R(550K) expression compared to control (one-sample t-test), although there was a trend towards decreased expression for 11R(517K) and 11R(550K). The effects of PMA on the different constructs were compared by one-way ANOVA with Bonferroni post-hoc analysis. All 11R(R-K) mutants displayed significantly different % changes in response to PMA compared to 11R. * indicates a p<0.05 compared to % change due to PMA for 11R; ** indicates a p<0.01 compared to % change due to PMA for 11R; *** indicates a p<0.001 compared to % change due to PMA for 11R (one-way ANOVA with Bonferroni post-hoc analysis).
Discussion
Others and we have shown that activation of PKC decreases GLT-1-mediated uptake and that this effect is associated with a movement of transporters off the cell surface. This effect is observed in both transfected cells and primary co-cultures of neurons and astrocytes (Fang et al., 2002; Kalandadze et al., 2002; Zhou and Sutherland, 2004; González et al., 2005; Guillet et al., 2005; Susarla and Robinson, 2008) and appears to be dependent upon clathrin-mediated endocytosis (Zhou and Sutherland, 2004; Susarla and Robinson, 2008). It is associated with a decrease in total transporter immunoreactivity that is attenuated by inhibitors of lysosomal degradation (Susarla and Robinson, 2008). However, the mechanisms mediating this internalization and loss of GLT-1 have not been elucidated. Since ubiquitination has been implicated in endocytosis and degradation of many different membrane proteins including other neurotransmitter transporters such as the dopamine transporter (Miranda et al., 2005; Miranda et al., 2007), the goal of the present study was to determine if activation of PKC caused an incorporation of ubiquitin into GLT-1 immunoprecipitates and to determine if we could implicate this event in internalization/degradation of GLT-1. We show that incubation with the PKC activator, PMA, results in incorporation of ubiquitin in GLT-1 immunoprecipitates in both transfected C6 cells and primary cortical cultures (mixture of astrocytes and neurons). Using C6 cells as a model system, we found that this PMA-induced incorporation of ubiquitin and the decrease in total GLT-1 immunoreactivity were blocked by the PKC inhibitor, Bis II, suggesting that these effects are dependent upon activation of PKC. Mutation of the 4 amino-terminal Lys residues or the 7 carboxyl-terminal Lys residues of GLT-1 was not sufficient to abolish the incorporation of ubiquitin into transporter immunoprecipitates, but mutation of all 11 lysine residues in the amino and carboxyl-termini eliminated this signal. Mutation of the amino-terminal lysine residues did not have a significant effect on internalization or degradation of GLT-1, but mutation of the carboxyl terminal residues abolished PMA-induced internalization and degradation of GLT-1. We found that nearly all of the lysine residues in the carboxyl terminus need to be mutated to arginine to eliminate the incorporation of ubiquitin or internalization/degradation. Finally, we provide evidence that the presence of single lysine residues in several different locations within the carboxyl terminus were sufficient to observe incorporation of ubiquitin or internalization/degradation, suggesting that redundant lysine residues are sufficient for ubiquitination and degradation of GLT-1. While the current manuscript was in preparation for submission, González-González et al. (2008) reported similar findings supporting the role of ubiquitination of carboxyl-terminal lysine residues in PMA-induced internalization. Some of the results from this other study are discussed in the context of the current study.
Increasing evidence gathered over the last decade has implicated ubiquitination in the trafficking and/or degradation of a number of cell surface proteins, including the neutral amino acid transporter (ATA2), DAT, sodium-potassium ATPase, glycine receptor, NMDA receptor, aquaporin channels, amiloride-sensitive epithelial sodium channel (eNaC), KCNQ2/3 and KCNQ3/5 voltage-gated potassium channels, and growth factor receptors (Alwan et al., 2003; Colledge et al., 2003; Geetha et al., 2005; Miranda et al., 2005; Arevalo et al., 2006; Huang et al., 2006a; Kamsteeg et al., 2006; Huang et al., 2007; Miranda et al., 2007 Miranda and Sorkin, 2007; Zhou et al., 2007). Ubiquitination is a three-step process; first ubiquitin is activated and conjugated to an E1 enzyme, then transferred to an E2 enzyme, and finally an E3 enzyme mediates the transfer of ubiquitin to a Lys residue of the substrate protein (for reviews, see Di Fiore et al., 2003; Haglund et al., 2003; Kim and Rao, 2006; Madshus, 2006; Woelk et al., 2007). There is evidence that the third step of ubiquitination may be somewhat promiscuous. For example, single mutations and different combinations of two or three mutations of nine Lys residues in the epidermal growth factor receptor (EGFR) do not alter ubiquitination levels of the EGFR; however, ubiquitination and lysosomal degradation are inhibited when five, six, or nine Lys residues are all mutated to Arg (Huang et al., 2006a). Similarly, mutation of individual Lys residues in the amino terminus does not affect ubiquitination or endocytosis, but mutation of a combination of three Lys residues reduces ubiquitination and blocks internalization of DAT (Miranda et al., 2007). In our studies, we found that the presence of K517, K526, and K550 are sufficient to support PMA-induced ubiquitination and degradation of the transporter, but that mutation of these sites does not abolish these effects. Similarly, González-González et al. (2008) found that the presence of several different single lysine residues (K517, K526, K550, or K570) were sufficient to support PMA-induced internalization of GLT-1 and PMA-induced decreases in transport activity. Together these studies suggest that the sites required for ubiquitination may be redundant. It is also possible that there are preferred sites in wild-type proteins with alternative sites serving the same purpose when the preferred sites are mutated.
In many of these cases, evidence similar to that described in the current study (incorporation of ubiquitin into immunoprecipitates combined with mutagenesis of lysine residues) has been considered sufficient to suggest that that ubiquitin is directly attached to the target protein. However, there is evidence that various components of the endocytic machinery can be ubiquitinated, including the E3 ligases themselves (for review, see Gao and Karin, 2005). In fact, from our data we cannot formally rule out this possibility, and we acknowledge that we do not observe a PKC-dependent increase in GLT-1 immunoreactivity that is the same molecular weight as that observed for the ubiquitin bands when the immunoprecipitates are re-probed with an anti-GLT-1 antibody (for example, see Fig. 2b, 3b, 7a, and 8a). We favor direct modification of GLT-1 by ubiquitin for the following reasons: First, these immunoprecipitates were washed with high (500 mM) NaCl and no salt to reduce the likelihood that interacting proteins would remain associated with GLT-1. Second, it would seem a remarkable co-incidence that ‘putative’ associated ubiquitinated proteins involved in endocytosis would migrate approximately 30 kDa larger than both the monomer and the multimer bands. Third, the fact that the size of lower molecular weight bands shifts somewhat upon mutation of either the amino or carboxyl terminal lysine residues (see Fig. 3b) would seem to decrease the likelihood that these bands were due to co-immunoprecipitation of associated proteins. Since the amount of total GLT-1 immunoreactivity decreases (even with the short term activation of PKC) and the decrease in the biotinylated/cell surface fraction is not significantly different from that observed in lysate (see Fig. 4), it seems possible that only a very small percentage of GLT-1 is ubiquitinated at any one time and that ubiquitination results in rapid degradation. Consistent with this idea, we do not normally see much accumulation of non-biotinylated transporter after activation of PKC, except for when ammonium chloride or chloroquine are used to inhibit lysosomal function (see Fig 4 in the current study and Fig. 8 in Susarla and Robinson, 2008). Finally, it seems unlikely that an ubiquitinated interacting protein would interact flexibly with so many different lysine residues in the amino and carboxyl termini. Although at present we suspect that the transporter is directly ubiquitinated, we acknowledge that a direct demonstration of ubiquitination will require mass spectrometry analysis of GLT-1 to demonstrate a direct covalent modification, as has been performed with proteins like DAT (Miranda et al., 2005) and the EGFR (Huang et al., 2006a).
Based on sequence similarities in the catalytic domains, it has been estimated that there are only one, maybe two E1 ligases, less than sixty E2 ligases, and more than 400 E3 ligases (for review, see Li et al., 2005). The number of E3 ligases implies that the specificity for ubiquitination of a protein most likely occurs at the level of the E3 ligase. A protein may be monoubiquitinated (ubiquitin on a single Lys residue), multiubiquitinated (ubiquitin molecules attached to multiple Lys residues), or polyubiquitinated (ubiquitin conjugated to Lys residues in ubiquitin itself resulting in ubiquitin chains). The type of ubiquitination a cell surface protein undergoes may determine whether it is recycled back to the cell surface or degraded (for reviews, see DiAntonio and Hicke, 2004; Hegde, 2004). Although it was originally thought that ubiquitination was selectively involved in proteosomal degradation, there is evidence that ubiquitinated membrane proteins can transit through the proteosome and ultimately be targeted for lysosomal degradation (Alwan et al., 2003 or see Huang et al., 2006b; Mukhopadhyay and Riezman, 2007 for discussions). Based on the fact that the ubiquitin immunoreactive bands are ~30 kDa larger than the monomers and multimers of GLT-1 and the fact that ubiquitin is 8.6 kDa, it would seem that three or four ubiquitin molecules may be conjugated to GLT-1. However, we cannot discern at this time whether GLT-1 is multi- or polyubiquitinated. Since the molecular weight of ubiquitinated monomers and multimers of the various mutant transporters does not appear to differ from that of ubiquitinated wild-type GLT-1, we might postulate that GLT-1 can be polyubiquitinated on one of the redundant Lys residues in the carboxyl terminus.
Previously, the laboratory has used reciprocal domains of EAAC1 and GLT-1 to identify amino acids 475 to 517 of GLT-1 as necessary and sufficient for PKC-regulated endocytosis (Kalandadze et al., 2002). Mutation of the five Ser residues within this domain partially attenuates PMA-induced internalization of GLT-1 and incorporation of radiolabeled phosphate into GLT-1 immunoprecipitates. In the carboxyl terminus only two lysine residues are conserved in GLT-1 and EAAC1 (497 and 550 of GLT-1). Therefore, if one assumes that GLT-1 is the direct target of ubiquitination, these two studies suggest a couple of different possibilities that are based on precedent (for reviews, see Gao and Karin, 2005; Snyder, 2005; Miranda and Sorkin, 2007). First, as observed with other proteins, it is possible that GLT-1 is phosphorylated in this domain and this phosphorylation event triggers binding of the E3 ligase and subsequent ubiquitination. Second, it is possible that the E3 ligase is directly or indirectly regulated by PKC activation and amino acid residues 475 thru 517 contain a combination of the domain required for ligase docking and lysine residues that can be ubiquitinated. In our earlier study, we had found that mutation of Ser-486 was also sufficient to partially attenuate PKC-dependent internalization of GLT-1 (Kalandadze et al., 2002). Therefore, we transfected C6 glioma with the S486A variant of GLT-1 or the variant with all 5 serine residues mutated to alanine and tested for incorporation of ubiquitin. In preliminary studies, the incorporation of ubiquitin was not different with either of these mutant variants (unpublished observations, n=1 or 2, respectively). These GLT-1(S-A) mutant variants were not tested for amino versus carboxyl-terminal ubiquitination. Therefore, it would be interesting to determine whether these Ser residues are required more specifically for carboxyl-terminal ubiquitination. In any case, the GLT-1 domain previously identified includes both K497 and K517 of GLT-1. Since individual Lys residues in the carboxyl terminus appear sufficient for ubiquitination and degradation of the transporter, the presence of these two Lys residues within the previously identified domain may be sufficient for endocytosis.
For many of the receptors, transporters, and channels that are known to be ubiquitinated, the E3 ubiquitin ligase is still unknown, although some key E3 ligases, such as Cbl and members of the Nedd4, HECT domain containing, E3 ligase family have been identified for some of these proteins. Boehmer et al. recently found that co-expression of Nedd4-2 (a member of the Nedd4 family of E3 ligases) with human homolog of GLT-1 in Xenopus oocytes decreases glutamate uptake and transporter surface expression, suggesting that Nedd4-2 can target GLT-1 (Boehmer et al., 2006). However, the effect may be indirect, as EAAT2 does not contain the PPXY recognition motif for Nedd4-2 binding.
Since ubiquitinated GLT-1 has been observed in synaptosomes, in various cell lines, and in primary cultures (current study and see Gonzalez-Gonzalez et al., 2008), it seems somewhat unlikely that this effect is unique to C6 glioma, but one might be predict that this process will be tightly controlled in normal brain tissue. One example that has emerged as a nice model over the last few years involves Nedd4-2-dependent ubiquitination and inhibition of the epithelial Na+-channel (ENaC). The activity and surface expression of ENaC is regulated by several different hormones and signaling molecules, including protein kinase A (PKA), serum glucocorticoid-inducible kinase (SGK), Akt kinase (protein kinase B) and AMP-activated kinase (AMPK). Many of these signals favor binding or unbinding of Nedd4-2 to 14-3-3. Upon activation of the right combination of signals, Nedd4-2 is released from 14-3-3 and becomes available to interact with and ubiquitinate ENaC (Bhalla et al., 2005; Ichimura et al., 2005; Bhalla et al., 2006; Nagaki et al., 2006; Lee et al., 2007; Zhou et al., 2007 for review, see Snyder, 2005). Similar inhibitory interactions are being identified for other E3 ligases (for a recent example, see Oberst et al., 2007). We suspect that ubiquitination of GLT-1 will be similarly regulated, and it seems possible that this event may be robust under pathologic conditions. For example, GLT-1 levels are lower in patients who die of amyotrophic lateral sclerosis (ALS) and in rodent models of ALS (Gurney et al., 1994; Rothstein et al., 1995; Howland et al., 2002). Gibb et al. recently observed incorporation of ubiquitin immunoreactivity in GLT-1 immunoprecipitates from end-stage G93A-SOD1 mice (Gibb et al., 2007). The levels of GLT-1 decrease between 20 and 50% within 3-6 h of an ischemic insult (Torp et al., 1995; Pow et al., 2004; Chen et al., 2005; Yeh et al., 2005; Ouyang et al., 2007, for review, see Sheldon and Robinson, 2007). If this effect were due to decreased transcription, one would expect that the half-life of the GLT-1 would be on the order of 6-10 h. Although the half-life has not been examined in vivo, cell culture studies suggest that the half-life is in excess of 24 h (Zelenaia et al., 1999). Therefore it seems likely that this effect of oxygen/glucose deprivation is due, at least in part, to accelerated degradation of GLT-1. Interestingly, PKC is trafficked to the plasma membrane during cerebral ischemia in rats (Cardell et al., 1990), and PKC activity increases during oxygen-glucose deprivation in rat hippocampal CA1 cultures (Larsen et al., 2004), providing a potential linkage between PKC activation and GLT-1 degradation.
In summary, we have found evidence that ubiquitin is incorporated into GLT-1 immunoprecipitates after PKC activation in both transfected C6 cells and primary cortical co-cultures. We also demonstrate that lysine residues in the carboxyl terminus of GLT-1 are essential for regulated endocytosis and degradation of GLT-1. Within the carboxyl terminus, Lys residues required for ubiquitination and degradation of the transporter appear redundant. In addition, even a single Lys residue in the carboxyl terminus of a normally non-responsive transporter (11R) is sufficient to mediate this effect. GLT-1 is known to limit excitotoxicity, and loss of GLT-1 has been implicated in a host of pathological conditions, although the mechanisms underlying the loss of GLT-1 under those circumstances remains unclear. We hypothesize that ubiquitination and targeted degradation of GLT-1 could be one mechanism whereby GLT-1 is downregulated during pathological conditions, as well.
Acknowledgments
The authors are supported by grants from the National Institutes of Health (RO1s NS29868/HD060132 and NS36465 to MBR and ALS was partially supported by NRSA MH071008). The authors would like to thank Margie Maronski for preparation of rat primary cortical cultures and Dr. Jeffrey Rothstein (Johns Hopkins) for his generous provision of the anti-GLT-1 antibody.
Abbreviations
- GLT-1
glial glutamate transporter 1
- PKC
protein kinase C
- Bis II
bisindolylmaleimide II
- DAT
dopamine transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alwan HA, van Zoelen EJ, van Leeuwen JE. Ligand-induced lysosomal epidermal growth factor receptor (EGFR) degradation is preceded by proteasome-dependent EGFR deubiquitination. J Biol Chem. 2003;278:35781–35790. doi: 10.1074/jbc.M301326200. [DOI] [PubMed] [Google Scholar]
- Apparsundaram S, Schroeter S, Giovanetti E, Blakely RD. Acute regulation of norepinephrine transport: II. PKC-modulated surface expression of human norepinephrine transporter proteins. J Pharmacol Exp Ther. 1998;287:744–751. [PubMed] [Google Scholar]
- Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Beckman ML, Bernstein EM, Quick MW. Multiple G protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. J Neurosci. 1999;19(9RC):1–6. doi: 10.1523/JNEUROSCI.19-11-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem. 2006;281:26159–26169. doi: 10.1074/jbc.M606045200. [DOI] [PubMed] [Google Scholar]
- Bhalla V, Daidie D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol. 2005;19:3073–3084. doi: 10.1210/me.2005-0193. [DOI] [PubMed] [Google Scholar]
- Boehmer C, Palmanda M, Rajamanickam J, Schneipp R, Amara S, Lang F. Post-translational regulation of EAAT2 function by co-expressed ubiquitin ligase Nedd4-2 is impacted by SGK kinases. J Neurochem. 2006;97:911–921. doi: 10.1111/j.1471-4159.2006.03629.x. [DOI] [PubMed] [Google Scholar]
- Bruhn T, Levy LM, Nielsen M, Christensen T, Johansen FF, Diemer NH. Ischemia induced changes in expression of the astrocyte glutamate transporter GLT1 in hippocampus of the rat. Neurochem Int. 2000;37:277–285. doi: 10.1016/s0197-0186(00)00029-2. [DOI] [PubMed] [Google Scholar]
- Cardell M, Bingren H, Wieloch T, Zivin J, Saitoh T. Protein kinase C is translocated to cell membranes during cerebral ischemia. Neurosci Lett. 1990;119:228–232. doi: 10.1016/0304-3940(90)90840-6. [DOI] [PubMed] [Google Scholar]
- Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology. 2005;49:703–714. doi: 10.1016/j.neuropharm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium-dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Daniels GM, Amara SG. Selective labeling of neurotransmitter transporters at the cell surface. Methods Enzymol. 1998;296:307–318. doi: 10.1016/s0076-6879(98)96023-2. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB. Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci. 1998;18:2475–2485. doi: 10.1523/JNEUROSCI.18-07-02475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore PP, Polo S, Hofmann K. When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol. 2003;4:491–497. doi: 10.1038/nrm1124. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Hicke L. Ubiquitin-dependent ragulation of the synapse. Annu Rev Neurosci. 2004;27:223–246. doi: 10.1146/annurev.neuro.27.070203.144317. [DOI] [PubMed] [Google Scholar]
- Dowd LA, Robinson MB. Rapid stimulation of EAAC1-mediated Na+-dependent L-glutamate transport activity in C6 glioma by phorbol ester. J Neurochem. 1996;67:508–516. doi: 10.1046/j.1471-4159.1996.67020508.x. [DOI] [PubMed] [Google Scholar]
- Fang H, Huang Y, Zuo Z. The different responses of rat glutamate transporter type 2 and its mutant (tyrosine 403 to histidine) activity to volatile anesthetics and activation of protein kinase C. Brain Res. 2002;953:255–264. doi: 10.1016/s0006-8993(02)03299-7. [DOI] [PubMed] [Google Scholar]
- Fournier KM, González MI, Robinson MB. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J Biol Chem. 2004;279:34505–34513. doi: 10.1074/jbc.M404032200. [DOI] [PubMed] [Google Scholar]
- Gao M, Karin M. Regulating the regulators: control of protein ubiquitination and ubiquitin-like modifications by extracellular stimuli. Mol Cell. 2005;19:581–593. doi: 10.1016/j.molcel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Geetha T, Jiang J, Wooten MW. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell. 2005;20:301–312. doi: 10.1016/j.molcel.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Gibb SL, Boston-Howes W, Lavina SZ, Gustincich S, Brown RH, Jr, Pasinelli P, Trotti D. A caspase-3 cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1 linked ALS. J Biol Chem. 2007 doi: 10.1074/jbc.M704314200. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zafra F, Olivares L, Gimenez C, Aragon C. Regulation by phorbol esters of the glycine transporter (GLYT1) in glioblastoma cells. Biochim Biophys Acta. 1995;1233:41–46. doi: 10.1016/0005-2736(94)00249-o. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Susarla BT, Fournier KM, Sheldon AL, Robinson MB. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04881.x. [DOI] [PubMed] [Google Scholar]
- González MI, Susarla BTS, Robinson MB. Evidence that protein kinase Ca interacts with an regulates the glial glutamate tran sporter GLT-1. J Neurochem. 2005;94:1180–1188. doi: 10.1111/j.1471-4159.2005.03330.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gonzalez IM, Garcia-Tardon N, Gimenez C, Zafra F. PKC-dependent endocytosis of the GLT1 glutamate transporter depends on ubiquitylation of lysines located in a C-terminal cluster. Glia. 2008;56:963–974. doi: 10.1002/glia.20670. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Kanner BI. The accessibility of a novel reentrant loop of the glutamate transporter GLT-1 is restricted by its substrate. J Biol Chem. 2000;275:9684–9689. doi: 10.1074/jbc.275.13.9684. [DOI] [PubMed] [Google Scholar]
- Guillet BA, Velly LJ, Canolle B, Masmejean FM, Nieoullon AL, Pisano P. Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched cultures. Neurochem Int. 2005;46:337–346. doi: 10.1016/j.neuint.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci. 2003;28:598–603. doi: 10.1016/j.tibs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Haugeto Ø, Ullensveng K, Levy LM, Chaudhry FA, Honore T, Neilsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-proteosome-mediated local degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proceedings of the National Academy of Sciences USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci U S A. 2007;104:16904–16909. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell. 2006a;21:737–748. doi: 10.1016/j.molcel.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Huang Y, Niwa J, Sobue G, Breitwieser GE. Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J Biol Chem. 2006b;281:11610–11617. doi: 10.1074/jbc.M513552200. [DOI] [PubMed] [Google Scholar]
- Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem. 2005;280:13187–13194. doi: 10.1074/jbc.M412884200. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ, Ramamoorthy S. Regulated internalization and phosphorylation of the native norepinephrine transporter in response to phorbol esters. Evidence for localization in lipid rafts and lipid raft-mediated internalization. J Biol Chem. 2004;279:19315–19326. doi: 10.1074/jbc.M311172200. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ, Blakely RD, Ramamoorthy S. Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol Pharmacol. 2005;67:2077–2087. doi: 10.1124/mol.104.009555. [DOI] [PubMed] [Google Scholar]
- Kalandadze A, Wu Y, Robinson MB. Protein kinase C activation decreases cell surface expression of the GLT-1 subtype of glutamate transporter. Requirement of a carboxyl-terminal domain and partial dependence on serine 486. J Biol Chem. 2002;277:45741–45750. doi: 10.1074/jbc.M203771200. [DOI] [PubMed] [Google Scholar]
- Kamsteeg EJ, Hendriks G, Boone M, Konings IB, Oorschot V, van der Sluijs P, Klumperman J, Deen PM. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc Natl Acad Sci U S A. 2006;103:18344–18349. doi: 10.1073/pnas.0604073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rao H. What’s Ub chain linkage got to do with it? Sci STKE 2006. 2006:pe18. doi: 10.1126/stke.3302006pe18. [DOI] [PubMed] [Google Scholar]
- Larsen GA, Berg-Johnsen J, Moe MC, Vinje ML. Calcium-dependent protein kinase C activation in acutely isolated neurons during oxygen and glucose deprivation. Neurochem Res. 2004;29:1931–1937. doi: 10.1023/b:nere.0000042220.16373.29. [DOI] [PubMed] [Google Scholar]
- Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt Mediates the Effect of Insulin on Epithelial Sodium Channels by Inhibiting Nedd4-2. J Biol Chem. 2007;282:29866–29873. doi: 10.1074/jbc.M701923200. [DOI] [PubMed] [Google Scholar]
- Li W, Chanda SK, Micik I, Joazeiro CA. Methods for the functional genomic analysis of ubiquitin ligases. Methods Enzymol. 2005;398:280–291. doi: 10.1016/S0076-6879(05)98023-3. [DOI] [PubMed] [Google Scholar]
- Madshus IH. Ubiquitin binding in endocytosis--how tight should it be and where does it happen? Traffic. 2006;7:258–261. doi: 10.1111/j.1600-0854.2006.00393.x. [DOI] [PubMed] [Google Scholar]
- Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Sorkin A. Regulation of receptors and transporters by ubiquitination: new insights into surprisingly similar mechanisms. Mol Interv. 2007;7:157–167. doi: 10.1124/mi.7.3.7. [DOI] [PubMed] [Google Scholar]
- Miranda M, Dionne KR, Sorkina T, Sorkin A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol Biol Cell. 2007;18:313–323. doi: 10.1091/mbc.E06-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Wu CC, Sorkina T, Korstjens DR, Sorkin A. Enhanced ubiquitylation and accelerated degradation of the dopamine transporter mediated by protein kinase C. J Biol Chem. 2005;280:35617–35624. doi: 10.1074/jbc.M506618200. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: Their classes, pharmacology, and distinct properties in the function of the nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Nagaki K, Yamamura H, Shimada S, Saito T, Hisanaga S, Taoka M, Isobe T, Ichimura T. 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase Nedd4-2 and epithelial Na+ channels. Biochemistry. 2006;45:6733–6740. doi: 10.1021/bi052640q. [DOI] [PubMed] [Google Scholar]
- Oberst A, Malatesta M, Aqeilan RI, Rossi M, Salomoni P, Murillas R, Sharma P, Kuehn MR, Oren M, Croce CM, Bernassola F, Melino G. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proc Natl Acad Sci U S A. 2007;104:11280–11285. doi: 10.1073/pnas.0701773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV, Naidoo T, Lingwood BE, Healy GN, Williams SM, Sullivan RK, O’Driscoll S, Colditz PB. Loss of glial glutamate transporters and induction of neuronal expression of GLT-1B in the hypoxic neonatal pig brain. Brain Res Dev Brain Res. 2004;153:1–11. doi: 10.1016/j.devbrainres.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–57. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB. Examination of glutamate transporter heterogeneity using synaptosomal preprations. Methods Enzymol. 1998;296:189–202. doi: 10.1016/s0076-6879(98)96015-3. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Schousboe A. Transport and metabolism of glutamate and GABA in neurons and glial cells. Int Rev Neurobiol. 1981;22:1–45. doi: 10.1016/s0074-7742(08)60289-5. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007 doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Robinson MB. A carboxyl-terminal determinant of the neuronal glutamate transporter, EAAC1, is required for platelet-derived growth factor-dependent trafficking. J Biol Chem. 2006;281:4876–4886. doi: 10.1074/jbc.M504983200. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Gonzalez MI, Krizman-Genda EN, Susarla B, Robinson MB. Lysine residues in the carboxyl terminus of the glial glutamate transporter, GLT-1, are inved in PKC-mediated ubiquitination, internalization, and degradation of the transporter. Soc Neurosci Abs. 2007;37:682–687. [Google Scholar]
- Sims KD, Robinson MB. Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol. 1999;13:169–197. doi: 10.1615/critrevneurobiol.v13.i2.30. [DOI] [PubMed] [Google Scholar]
- Sims KD, Straff DJ, Robinson MB. Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporters through activation of phosphatidylinositol 3-kinase. J Biol Chem. 2000;274:5228–5327. doi: 10.1074/jbc.275.7.5228. [DOI] [PubMed] [Google Scholar]
- Snyder PM. Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology. 2005;146:5079–5085. doi: 10.1210/en.2005-0894. [DOI] [PubMed] [Google Scholar]
- Susarla BT, Robinson MB. Internalization and degradation of the glutamate transporter GLT-1 in response to phorbol ester. Neurochem Int. 2008;52:709–722. doi: 10.1016/j.neuint.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP. Reduced postischemic expression of a glial glutamate transporter, GLT, 1, in the rat hippocampus. Exp Brain Res. 1995;103:51–58. doi: 10.1007/BF00241964. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Buchhalter J, Dichter MA. Properties of inhibitory and excitatory synapses between hippocampal neurons in very low density cultures. Synapse. 1994;18:128–151. doi: 10.1002/syn.890180206. [DOI] [PubMed] [Google Scholar]
- Woelk T, Sigismund S, Penengo L, Polo S. The ubiquitination code: a signalling problem. Cell Div. 2007;2:11. doi: 10.1186/1747-1028-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TH, Hwang HM, Chen JJ, Wu T, Li AH, Wang HL. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis. 2005;18:476–483. doi: 10.1016/j.nbd.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Zelenaia OA, Robinson MB. Degradation of glial glutamate transporter mRNAs is selectively blocked by inhibition of cellular transcription. J Neurochem. 2000;75:2252–2258. doi: 10.1046/j.1471-4159.2000.0752252.x. [DOI] [PubMed] [Google Scholar]
- Zelenaia OA, Gochenauer GE, Robinson MB. Glutamate transporter mRNA stability in cultured astrocytes is regulated by a factor (s) dependent on RNA and protein synthesis. Society for Neuroscience Abstracts. 1999;25:427. [Google Scholar]
- Zhang L, Coffey LL, Reith MEA. Regulation of the functional activity of the human dopamine transporter by protein kinase C. Biochem Pharmacol. 1997;53:677–688. doi: 10.1016/s0006-2952(96)00898-2. [DOI] [PubMed] [Google Scholar]
- Zhou J, Sutherland ML. Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci. 2004;24:6301–6306. doi: 10.1523/JNEUROSCI.1404-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem. 2007;282:20207–20212. doi: 10.1074/jbc.M611329200. [DOI] [PubMed] [Google Scholar]