

Abstract
The RB tumor suppressor pathway is likely important in primitive neuroectodermal tumors (PNET) of the brain. In fact, 10-15% of children born with RB mutations develop brain PNETs, commonly in the pineal gland. Cyclin D1, which in association with Cyclin-dependent kinases (Cdk) 4 and 6 phosphorylates and inactivates the RB protein, is expressed in 40% of sporadic medulloblastoma, a PNET of the cerebellum. To understand tumorigenic events cooperating with RB pathway disruption in brain PNET, we generated a transgenic mouse where Cyclin D1 was expressed in pineal cells. Cyclin D1 enhanced pinealocyte proliferation, causing pineal gland enlargement. However, proliferation ceased beyond 2 weeks of age with reversal of Cdk4-mediated Rb phosphorylation despite continued expression of the transgene, and the pineal cells showed heterochromatin foci suggestive of a senescent-like state. In the absence of the p53 tumor suppressor, cell proliferation continued, resulting in pineal PNET that limited mouse survival to ~ 4 months. Interestingly, the Cdk-inhibitor p18Ink4c was induced in the transgenic pineal glands independently of p53, and transgenic mice that lacked Ink4c developed invasive PNET, though at an older age than those lacking p53. Analogous to our mouse model, we found that children with heritable retinoblastoma often had asymptomatic pineal gland enlargement that only rarely progressed to PNET. Our finding that the Cdk4-inhibitor p18Ink4c is a tumor suppressor in Cyclin D1-driven PNET suggests that pharmacological interventions to inhibit Cdk4 activity may be a useful chemoprevention or therapeutic strategy in cancer driven by primary Rb pathway disruption.
Keywords: Cyclin D1, p53, Ink4c, PNET, brain
Introduction
As a tumor suppressor, the retinoblastoma (RB) protein primarily controls entry into S phase of the cell cycle (1). D-type Cyclins and their catalytic partners Cdk4 and Cdk6 phosphorylate RB to foster the G1 to S phase transition. As might be expected, Cyclin D1 is highly expressed and its gene is amplified in a variety of human cancers (reviewed in ref. #s 2,3). Somewhat paradoxically, though, several mouse models have shown Cyclin D1 to be rather weakly oncogenic, promoting cancer only after long latency or cooperatively with other genetic changes (4-7). Understanding the secondary genetic changes will provide important insight into the biology of cancer driven by primary RB pathway inactivation.
We focused our studies on primitive neuroectodermal tumors (PNETs) originating in the pineal gland of the brain (8). It is clear that the RB tumor suppressor pathway is important in their biology because 10-15% of children with heritable retinoblastoma develop brain PNETs, most commonly originating from the pineal gland. Deregulated Cyclin D1 is detectable in over 40% of sporadic infratentorial PNET (called medulloblastoma) (9). Furthermore, medulloblastoma is often driven by deregulation of the Sonic hedgehog (Shh) pathway (10); Cyclin D1 is induced by Shh signaling (11) and its absence strongly impedes Shh-driven medulloblastoma (12). We employed a transgenic mouse in which human Cyclin D1 is driven by the Interphotoreceptor retinoid binding protein (Irbp) promoter, which is selectively expressed in the retina and pineal gland (13). Previous studies showed that ectopic Cyclin D1 fosters excess proliferation in developing photoreceptor cells, altering retina development, but it does not cause retinoblastoma (14).
We used the Irbp-Cyclin D1 transgenic mouse to explore the tumorigenic effects of primary deregulation of the Rb pathway in the pineal gland. We focused our studies on potential interactions between this oncogene and two tumor suppressive mechanisms involving (a) p53 and a key regulator and effector of its function, and (b) p18Ink4c, which primarily acts to control Cyclin D-associated kinase activity. Further, we investigated whether the model we developed was pertinent to pineal gland changes in children with heritable retinoblastoma.
Materials and Methods
Mouse Studies
Two different lines of Irbp-Cyclin D1 transgenic mice with indistinguishable phenotypes were used (14). Transgenic mice were bred with p53 -/- (Jackson Laboratory, Maine), p21 -/- (Jackson Laboratory, Maine), Arf lacZ/lacZ (15), or Ink4c -/- mice (16) and maintained in a mixed C57BL/6 × 129/Sv genetic background. PCR for relevant targeted alleles was used to verify mouse genotypes as previously described (14,16). Animals were examined at least twice weekly and were euthanized at defined experimental time points or when obviously ill in accordance with St. Jude Children’s Research Hospital (SJCRH) Animal Care and Use Committee guidelines; all studies were approved by this committee.
Analyses of protein expression in the pineal gland
Protein extracts were prepared from pineal tissue by lysis in RIPA buffer supplemented with protease and phosphatase inhibitors (50mM NaF, 20mM β-glycerophosphate, 1mM NaOVD, 5mM NaPPO4, 1mM PMSF, 1mM DTT, 10μg/ml leupeptin, 1μg/ml pepstatin, 10μg/ml aprotinin), sonication, and centrifugation. Electrophoresis was performed using NuPage 4-12% Bis-Tris or 3-8% Tris-Acetate gradient gels. Proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA), and detected using antibodies to p21Waf1; Cdk2; HSC70 (Santa Cruz Biotechnology, Santa Cruz, CA); p18Ink4c (Zymed, San Francisco, CA); and the hemagglutinin (HA) epitope (Covance, Trenton, NJ). Rb was immunoprecipitated with anti-Rb antibody G3-245 (BD Pharmingen, San Jose, CA) and detected using either the same antibody or the anti-phospho-S780 (corresponding to serine 773 in the mouse) antibody (Cell Signaling Technology, Danvers, MA). Genomic DNA was extracted from paraffin blocks containing eyes or brain tumor as previously described (17). Amplification of certain transcripts from the pineal gland was accomplished by RTPCR with the following primers: transgenic human Cyclin D1: 5’-GGTCCACCTCCTCCTCCTCCTCTT (forward) and 5’-GTCCTACTACCGCCTCACACGCTT (reverse); Retinal S-antigen (Arrestin): 5’-GCGCGATGCCTCTTCTTTCTCTGT (forward) and 5’-AGCCCATCCCCGTGACTGTGAC (reverse) was used as a pineal gland-specific marker (18); Gapdh primers have been described (19). Genomic DNA was extracted from paraffin blocks containing eyes or brain tumor as previously described (17); p53 and Gapdh were amplified from DNA as described above.
Histological studies
Mice were sacrificed using CO2 or isoflurane followed by decapitation. The skull was immediately denuded, the mandible was removed, and the base of the skull was removed. After several holes were bored near the pineal gland through the cranium using an 18g needle, the skull/brain was fixed in ~25 ml of 4% paraformaldehyde in phosphate-buffered saline (PBS) for 48 (P14 and less) or 72 hours (> P14). Following equilibration in 70% ethanol, tissue was embedded in paraffin using an automated processor. For mice older than 10 days, skulls were decalcified using TBD-2 decalcifier (ThermoElectron Corporation, Waltham, MA) following fixation, prior to embedding. Pinealocyte morphology was assessed by light microscopy analysis of hematoxylin and eosin (H&E) stained coronal sections. Pineal gland size was determined in comparable sections using an eye-piece reticule. Cell density was calculated by counting DAPI-stained nuclei in an area defined by using the eye-piece reticule, to assure that the same nucleus was not counted twice.
For immunohistochemical and immunofluorescence studies, 4-8 μm sections cut from formalin-fixed paraffin-embedded tissues were baked at 60°C for 30 minutes then deparaffinized. Antigen retrieval was performed in a steamer at >95°C for 30 minutes in target retrieval buffer (pH 6.0, cat # S1699, DAKO, Carpinteria, CA) for Ki67; high pH target retrieval buffer (cat # S13307, DAKO, Carpinteria, CA) for Cyclin D1; and citrate antigen retrieval buffer (pH 6.0) for SNP and HP1γ. Slides were incubated with anti-Ki67 antibody (NovoCastra, Newcastle, UK), anti-Cyclin D1 (Santa Cruz Biotechnology, Santa Cruz, CA), or anti-SNP (DAKO, Carpinteria, CA), followed by biotinylated secondary antibody; bound antibody was detected using streptavidin conjugated to horseradish peroxidase and DAB substrate (DAKO, Carpinteria, CA). Anti-HP1γ and anti-H3K9-M3 (Upstate Laboratories, Syracuse, NY) were detected with FITC and Cyanine Cy2 secondary antibodies, respectively. TUNEL labeling was done using the Dead-End Cell Death Labeling Kit (Roche, Indianapolis, IN). The total number of Ki67 and of TUNEL positive cells was determined by counting representative 200x fields using a fluorescence microscope equipped with an eye-piece reticule. At this magnification, one reticule box equals 2.5 × 103 μm2; the total area in the counted samples ranged from 4 to 412 reticule boxes (0.01 mm2-1 mm2), owing to the differences in size of wild type versus transgenic pineal glands and pineal PNET. The number of positive nuclei/area was divided by the total number of DAPI-positive nuclei/area (because the cellular density varied with the genotype) to normalize the number of Ki67- or TUNEL-positive nuclei to total cell number. Digital photomicrographs of stained sections were obtained by using an Olympus BX60 microscope equipped with a SPOT RT Slider camera (Diagnostic Instruments, Sterling Heights, MI), or with a Zeiss 510 NLO multiphoton/confocal laser scanning microscope. Composite images were constructed using Photoshop CS2 software (Adobe Systems, Mountain View, CA).
Collection of clinical data
This retrospective study was approved by the SJCRH Institutional Review Board. Imaging studies were retrieved for 32 children with bilateral retinoblastoma diagnosed at or before 2 years of age at SJCRH between 1997 and 2003. MR images were reviewed from the 26 in whom the scan was (a) obtained within 6 months of diagnosis and (b) judged to have adequate imaging of the pineal gland. Maximum pineal gland axial and antero-posterior dimensions were compared to published size normal values (20).
Statistical analysis
Kaplan-Meier survival curves were estimated for 4 to 17 mice of each genotype. Pair-wise comparisons were made in the Irbp-Cyclin D1 mice that were p53 +/+ (n = 15), +/- (n = 17), and -/- (n = 7) by using exact log rank tests. The two-sample t-test was utilized to compare mean human pineal gland sizes between children with bilateral retinoblastoma and normal children <2 years of age (20), and to assess differences in quantitative measurements among mouse pineal gland samples.
Results
Transgenic expression of Cyclin D1 causes pineal gland hyperplasia
We first verified that the human Cyclin D1 transgene was selectively expressed in the pineal gland but not in the adjacent brain (Figure 1A and Supplemental Figure 1A); mouse Cyclin D1 was undetectable at this stage (Supplemental Figure 1B). Although the transgenic mice appeared healthy, the pineal gland was visibly enlarged at two months of age (Figure 1A and 1B, top). Quantitative analysis showed 5- and 6-fold enlargement over the wild-type pineal size at postnatal day (P) 10 and P49, respectively (Figure 1B, bottom). At earlier points, the wild type and transgenic pineal glands were similar in size.
Figure 1. Pineal gland hyperplasia develops in Irbp-Cyclin D1 transgenic mice.
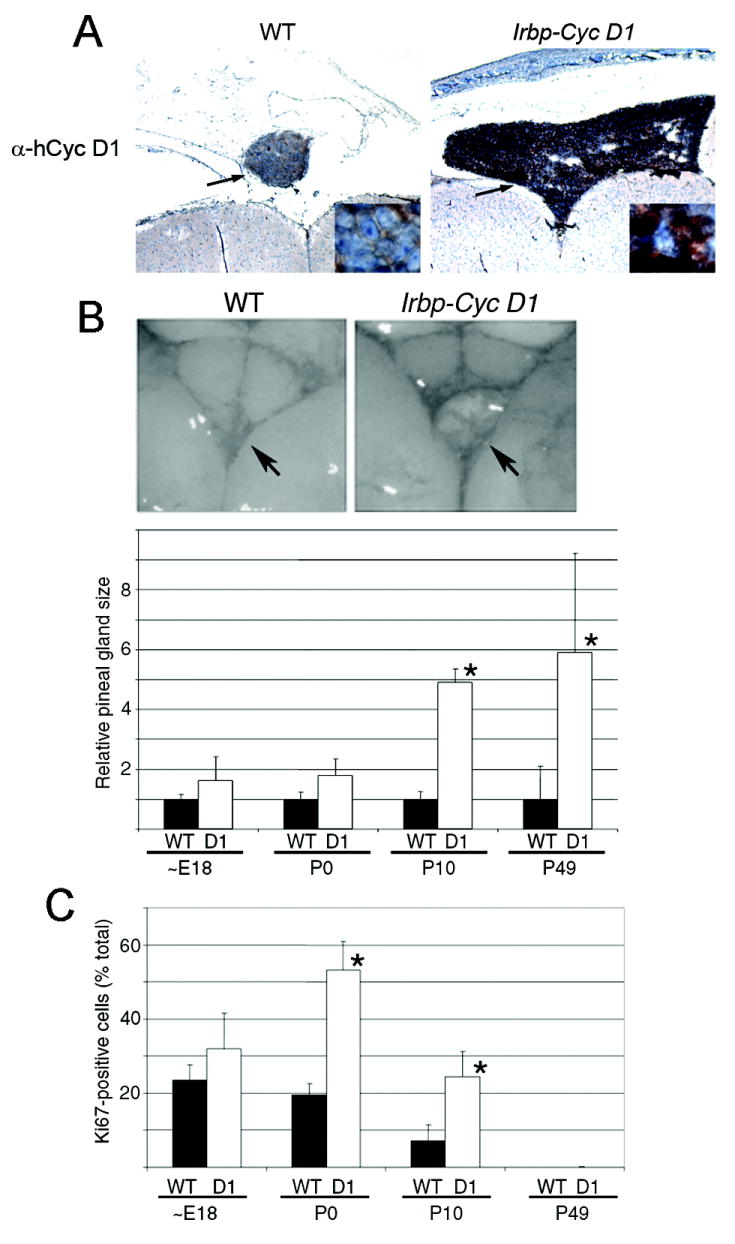
(A) Representative photomicrographs of the pineal gland (arrow) from P10 wild type and transgenic mice following immunostaining for human Cyclin D1. Original magnification: 40x and 400x (inset). (B) Representative photographs of the pineal gland (arrow) on the dorsal surface of brain (top) and quantitative analysis of pineal gland size (expressed relative to wild type mice at each age)(bottom) from wild type and transgenic mice. (C) Quantitative analysis of Ki67-positive pinealocytes in wild type and transgenic mice of the indicated ages. Values represent mean and standard deviation of measurements from 3 – 6 separate mice. * signifies p < 0.05 as compared to wild type.
Cyclin D1 drives cell proliferation by functionally inactivating Rb and the related p107 and p130 (e.g., ref. # 21). Consistent with this fact, pineal gland enlargement was preceded by increased pinealocyte density (Supplemental Figure 1C), and increased proportion of cells expressing the proliferation marker Ki67 at P0 and P10 (Figure 1C and Supplemental Figure 2). Interestingly, the number of Ki67-positive cells decreased over time, and both wild type and Irbp-Cyclin D1 pinealocytes had exited the cell cycle by P49 (Figure 1C). Hence, ectopic expression of Cyclin D1 transiently enhanced cell proliferation to cause pineal gland enlargement that did not appear to have untoward consequences.
Cessation of proliferation in adult Irbp-Cyclin D1 mice correlates with Rb hypophosphorylation despite continued Cyclin D1 expression
Cyclin D1 promotes cell proliferation by blocking Rb function by two generally-accepted mechanisms: inducing Cdk4- and Cdk6-dependent Rb hyperphosphorylation to disrupt its functional “pocket”, or titrating Cdk inhibitors like p21Cip1 and p27Kip1 from Cdk2-containing complexes (reviewed in ref. #s 1,22). By immunoprecipitating Rb and immunoblotting for Cdk4(6)-dependent phosphorylated serine 773 and 778 (23,24), we found that excess proliferation in the transgenic pineal gland at P10 correlated with enhanced Cyclin D1-directed phosphorylation that was not observed at two months when proliferation had ceased (Figure 2A and data not shown). Despite the reversal of Rb phosphorylation, both immunoblotting (Figure 2B) and immunohistochemical staining (Figure 2C) showed that transgenic Cyclin D1 expression was maintained and endogenous Cdk4 was only slightly decreased at 2 months. Hence, Rb dephosphorylation and arrested pinealocyte proliferation in the aging transgenic mice did not merely result from repression of the transgene or its catalytic subunit.
Figure 2. Rb hypophosphorylation despite continued Cyclin D1 expression in aging Irbp-Cyclin D1 mice.
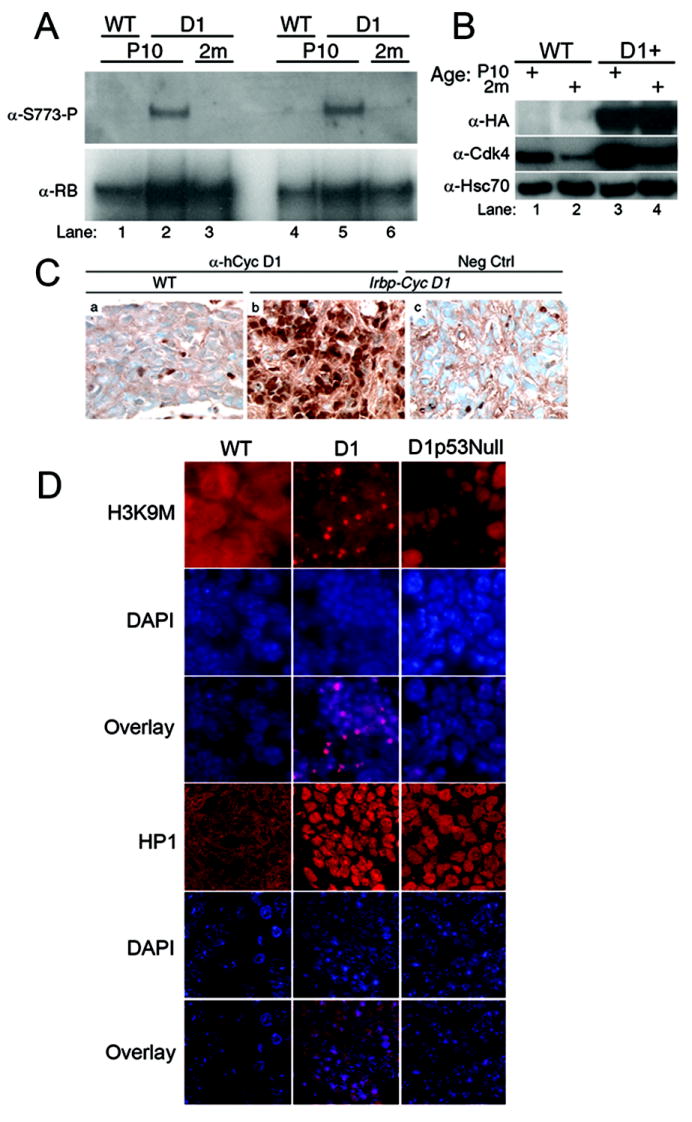
(A) Immunoprecipitation and western blotting for total Rb and phospho-Ser773 in mouse Rb from two representative pineal glands of wild type (lanes 1 and 4) and transgenic (lanes 2, 3, 5, 6) mice at P10 (lanes 1, 2, 4, 5) and 2 months (lanes 3 and 6). (B) Western blotting reveals the indicated proteins in pineal gland lysates from wild type (lanes 1, 2) and transgenic (lanes 3, 4) mice at P10 (lanes 1, 3) and 2 months (lanes 2, 4). HA denotes hemagglutinin epitope tag of transgenic Cyclin D1. (C) Immunohistochemical staining for human Cyclin D1 at P49 in wild type (a) and transgenic (b, c) pineal gland. Negative control was stained without the primary antibody. (D) Immunofluorescence staining for H3-K9M and HP1γ with corresponding DAPI staining in wild type, Irbp-Cyclin D1 and Irbp-Cyclin D1, p53 -/- pineal glands at P49. H3K9-M3 images are taken at 400x; HP1γ at 1000x magnification with a confocal microscope.
We considered whether the cell cycle arrest induced at 2 months might represent cellular senescence, a tumor suppressive mechanism in which functionally-active Rb is known to play a role (reviewed in ref. #s 25-27). It co-localizes with heterochromatin-associated proteins in subnuclear foci (senescence-associated heterochromatin foci) to silence the expression of certain genes like those activated by E2Fs (28). Immunofluorescence staining showed that the heterochromatin-associated protein 1-gamma (HP1γ) and histone H3 trimethylated at lysine 9 (H3-K9M) accumulated in foci in pinealocyte nuclei in 2 month old Irbp-Cyclin D1 mice, but not in P10 transgenic mice (not shown) nor in 2 month old wild type mice (Figure 2D). We conclude that decreased pinealocyte proliferation in 2 month old Irbp-Cyclin D1 mice correlates with loss of Cdk4(6)-dependent Rb phosphorylation and with the formation of heterochromatin changes suggesting a state of cellular senescence.
p53 suppresses tumor progression in the Irbp-Cyclin D1 pineal gland
The p53 tumor suppressor induces cellular senescence in response to a variety of stimuli, including oncogene expression (for example, ref. #s 29,30); as such we used a genetic approach to define its importance in the senescent-like state we observed in the pineal gland. The pineal gland was only slightly larger at P10 in Irbp-Cyclin D1, p53 -/- mice as compared to Irbp-Cyclin D1 mice (Figure 3A); this was associated with a trend toward more Ki67-positive cells at P0 and P10, but no measurable difference in the small amount of apoptosis (Supplemental Figures 3A and B). In contrast to these minor effects, loss of p53 altered pinealocyte proliferation dramatically at P49: The fraction of Ki67-positive cells approached 41.7% (SD 4.2%, n = 560 nuclei counted from 3 samples) in transgenic mice lacking p53 whereas proliferation had essentially ceased in Irbp-Cyclin D1, p53 +/+ mice (Figure 3B, panel d versus b) and non-transgenic p53 -/- mice (Supplemental Figure 4). Further, senescence-associated heterochromatin foci were not observed in the pineal gland in the absence of p53 (Figure 2D, right panels). These findings indicate that p53 is required for cell proliferation arrest and the senescence-like state that occurs as Irbp-Cyclin D1 transgenic mice age.
Figure 3. Cyclin D1-driven cell proliferation ceases in a p53-dependent manner.
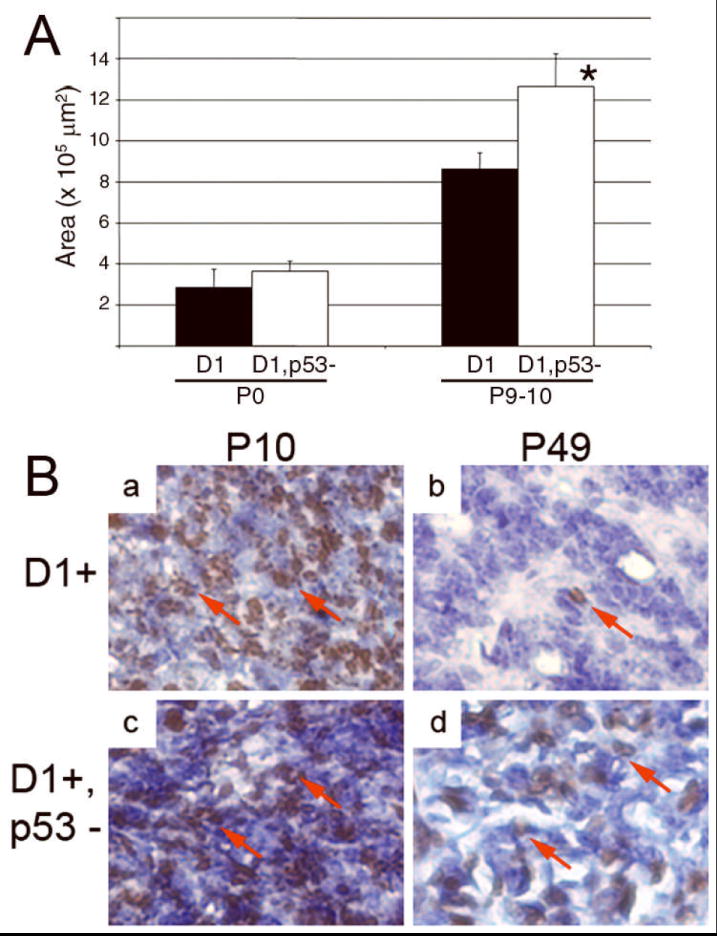
(A) Quantitative analyses of pineal gland size in transgenic mice of the indicated ages that have (black bars) or lack (white bars) p53. * signifies p < 0.05 as compared to wild type mice. (B) Representative photomicrographs of pineal gland sections stained for Ki67 in transgenic mice that either retain (a, b) or lack (c, d) p53, at the ages indicated. Arrows show Ki67-positive cells.
Irbp-Cyclin D1, p53 +/- and Irbp-Cyclin D1, p53 -/- mice had markedly decreased survival as compared to transgenic mice retaining wild type p53 alleles (Figure 4A) [Irbp-Cyclin D1, p53 +/+ vs. Irbp-Cyclin D1, p53 +/- (p = 0.0156); Irbp-Cyclin D1, p53 +/- vs. Irbp-Cyclin D1, p53 -/- (p = 0.0049); and Irbp-Cyclin D1, p53 +/+ vs. Irbp-Cyclin D1, p53 -/- (p = 0.0001)]. They often developed a visibly enlarged skull not evident in Irbp-Cyclin D1, p53 +/+ mice or in p53-deficient mice without the Irbp-Cyclin D1 transgene. This finding suggested the presence of a brain tumor, which almost never occurs in p53 -/- mice (31,32). Whenever possible, the ill-appearing mice were euthanized; in all cases (n = 7 in the survival study and additional mice subsequently), a large, hemorrhagic tumor was noted in the midline dorsal region of the brain (Figure 4B, panel a). The tumor cells expressed human Cyclin D1 and the neuronal marker synaptophysin, confirming pineal PNET, while glial fibrillary acidic protein was only detected in infiltrating astrocytes (Figure 4B, b – d and Supplemental Figure 5). In two PNETs examined from Irbp-Cyclin D1, p53 +/- mice, we could not amplify the remaining p53 allele from tumor-derived genomic DNA, but it was detectable in ocular tissue and in a spontaneous soft tissue tumor arising in an Arf -/- mouse processed in parallel (Figure 4C; some data not shown). This indicated that the remaining p53 allele was lost in the progression to PNET.
Figure 4. Pineoblastoma progression in Irbp-Cyclin D1, p53 -/- mice.
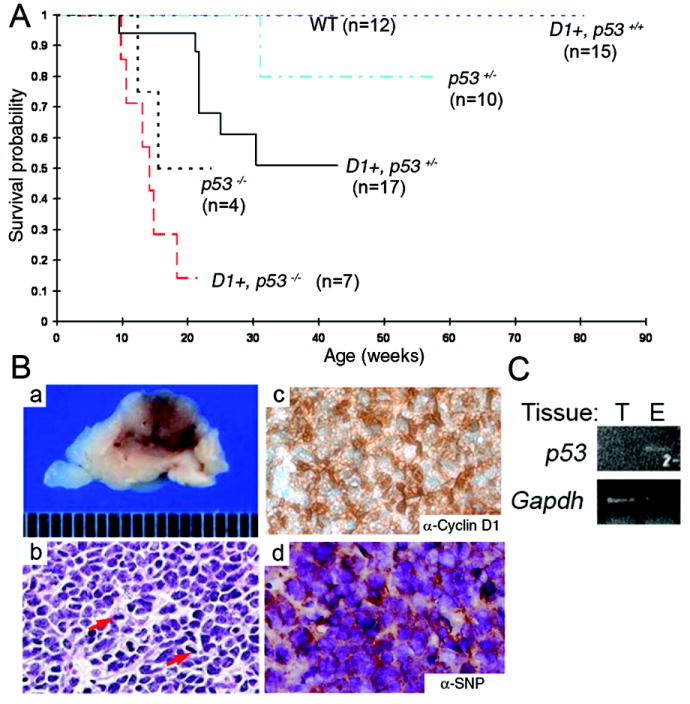
(A) Kaplan-Meier survival curves for wild type (n = 12), Irbp-Cyclin D1 (n = 15), Irbp-Cyclin D1, p53 +/- (n = 17), Irbp-Cyclin D1, p53 -/- (n = 7), p53 +/- (n = 10) and p53 -/- (n = 4) mice. The survival distributions among the Irbp-Cyclin D1 mice that were p53 +/+, +/-, and -/- were significantly different. (B) Representative photograph of midline sagittal section of mouse brain shows large, hemorrhagic tumor (a). H&E staining (b) reveals numerous mitotic figures (arrows); immunohistochemical staining shows expression of human Cyclin D1 (c) and a neuronal protein, synaptophysin (SNP) (d). Original magnification: 200x. (C) Representative photo of ethidium bromide-stained agarose gel following electrophoresis of the indicated PCR products amplified from pineal gland tumor (T) or eye (E) DNA from an Irbp-Cyclin D1, p53 +/- mouse.
p53-dependent tumor suppression does not depend on p21Cip1 or p19Arf
Oncogenic signals can activate the p53 pathway in an Arf-dependent manner and the Cdk inhibitor p21Cip1 is a key downstream effector of p53 (reviewed in ref. # 33). Interestingly, Arf expression was not detectable by Western blotting or by using an Arf lacZ/lacZ reporter mouse (34) at P10 or at two months (negative data not shown). In contrast, p21Cip1 was detectable at P10 in the Irbp-Cyclin D1 pineal gland; relatively higher expression in the Irbp-Cyclin D1, p53 +/+ versus Irbp-Cyclin D1, p53 -/- pineal gland indicated that the induction at P10 was p53-dependent (Figure 5A and 5B, compare lane 3 with lane 1). At 2 months when proliferation had ceased in wild type and transgenic mice (Figure 1C), p21Cip1 was not evident (Figures 5A, lanes 2 and 4; Figure 5B, lane 2). The continued p21Cip1 expression in the pineal gland of 2 month old, Irbp-Cyclin D1, p53 -/- mice (Figure 5B, lane 4) indicated that it was not sufficient to arrest pinealocyte proliferation at that stage.
Figure 5. p18Ink4c preserves cell cycle arrest and blocks PNET progression in Irbp-Cyclin D1 mice.
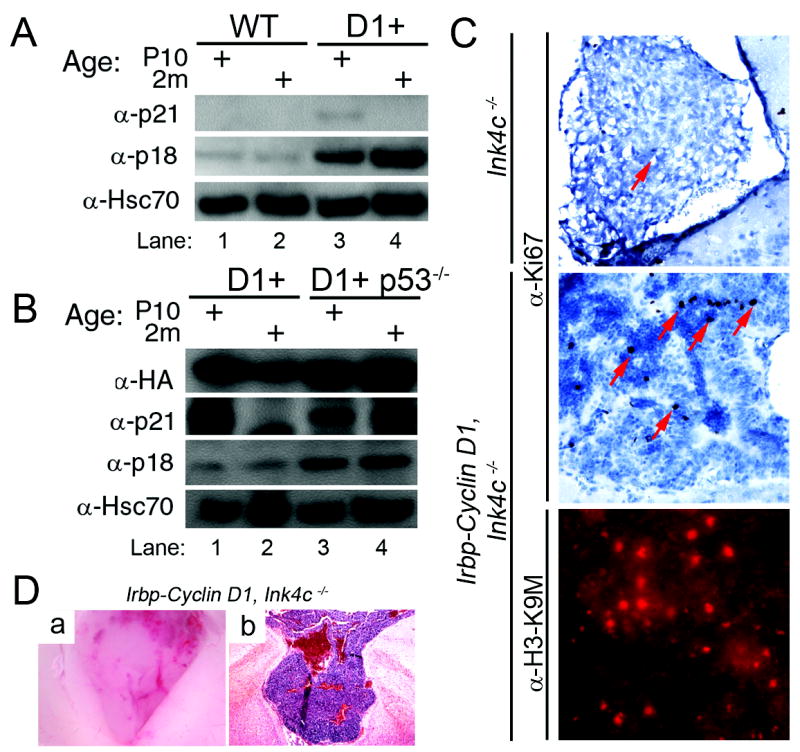
(A and B) Western blotting reveals the indicated proteins in pineal gland lysates from mice of the indicated genotypes at P10 and 2 months. HA denotes hemagglutinin epitope on the human Cyclin D1. (C) Immunohistochemical staining for the proliferation marker Ki67 (top two panels) and heterochromatin-associated H3-K9M (bottom panel) in the pineal gland of the indicated mice at P49. (D) Representative photomicrographs showing Irbp-Cyclin D1, Ink4c -/- pineal gland tumor in whole mount (a) and following H&E staining (b).
Although the absence of p19Arf or p21Cip1 at two months in p53 +/+ transgenic mice suggested that neither was essential for cell cycle arrest and tumor suppression, we used a genetic strategy to formally evaluate their roles. As in transgenic mice retaining p21 (Figure 1C and 3B, b) pinealocyte proliferation had ceased in Irbp-Cyclin D1, p21 -/- mice at two months of age (Supplemental Figure 6A), with heterochromatin foci formation similar to that in Irbp-Cyclin D1, p21 +/+ mice (Supplemental Figure 6B). A small number (n = 3) of Irbp-Cyclin D1, p21 -/- mice have aged to ~5 months with no evidence of compromised survival. Similarly, 11 Irbp-Cyclin D1, Arf +/- and 6 Irbp-Cyclin D1, Arf -/- mice observed for an average of 9.2 (range 2 – 18) months showed no evidence of brain tumor. Hence, neither p19Arf nor p21Cip1 was essential for tumor suppression in Irbp-Cyclin D1 mice.
p18Ink4c maintains cell cycle arrest and blocks tumor formation
Recent work established that the Cdk4(6) inhibitor p18Ink4c prevents PNET in a mouse model of medulloblastoma driven by Ptc mutation (35). In our model, loss of Cdk4(6)-mediated Rb phosphorylation at P49 correlated with progressively higher expression of p18Ink4c in the transgenic pineal gland (Figure 5A), proceeding independently of p53 (Figure 5B). At two months of age, very rare Ki67 positive cells (~0.05% of total) were present in wild type, Irbp-Cyclin D1, and non-transgenic Ink4c -/- pineal glands (Figures 1C and 5C, top panel). In contrast, ~1.5% of cells in Irbp-Cyclin D1, Ink4c -/- pineal glands expressed Ki67 (Figure 5C, middle panel). This 30-fold increase in proliferating cells was important because Irbp-Cyclin D1, Ink4c -/- mice developed pineal gland PNET as they aged to ~7 months (n = 3 of 3 mice examined) (Figure 5D). In contrast, six Irbp-Cyclin D1, Ink4c +/- mice euthanized between 8 and 18 months of age had no evidence of pineal gland PNET on gross or microscopic examination. Despite the continued proliferation in a fraction of cells in the Irbp-Cyclin D1, Ink4c -/- pineal gland at 2 months, immunostaining for H3-K9M showed that Ink4c was not essential for formation of senescence-associated heterochromatin changes (Figure 5C, bottom panel). These findings show that Ink4c, a bona fide tumor suppressor in this model, was required for the maintenance of cell cycle arrest in Irbp-Cyclin D1 pinealocytes but it was not needed for formation of the heterochromatin foci associated with an initial cell cycle exit.
Pineal gland hypertrophy in children with heritable retinoblastoma
We considered whether sub-clinical pineal enlargement also occurred in children with bilateral retinoblastoma, who have heritable RB deficiency and are prone to developing PNET in the pineal gland (36,37). Review of magnetic resonance images of the brain in 26 such children showed that the pineal gland size exceeded normal in one dimension in 19 (73%) and in two dimensions in 10 (38%) (two-tailed T-tests: p < 0.001 and p = 0.004, respectively) (Figure 6A). Only one of these patients developed frank pineoblastoma. Tissue was not available for sequencing of the p53 gene in the tumor tissue, but immunohistochemical staining indicated that p53 was detectable (Figure 6B) and, therefore, likely mutated (38,39). Coupled with our mouse model, these findings suggested that haploinsufficiency for RB may result in a relative excess of Cyclin D1/Cdk4(6) activity that fosters pineal gland enlargement but only rarely progresses to PNET.
Figure 6. Pineal gland enlargement is observed in children with heritable retinoblastoma.

(A) Graphical representation of pineal gland size in children with bilateral retinoblastoma. Individual points correspond to antero-posterior (AP) and axial (Ax) dimensions of pineal gland from MR images at time of retinoblastoma diagnosis. Red line and shaded area denotes normal average and standard deviation in pineal gland size for children below 2 years of age (20). (B) Immunohistochemical staining for p53 in the tumor of the one patient developing pineal gland PNET.
Discussion
Our findings demonstrate that primary disruption of the Rb pathway by deregulated Cyclin D1 promoted a pre-malignant lesion in the mouse pineal gland. p53 prevented the progression to PNET by a strikingly robust process because transgenic mouse survival was not compromised without additional, germ-line genetic aberrations. This differs from many mouse models where loss of p53 accelerates tumorigenesis; here p53 loss was required for tumor development. In our model, p53 promoted the development of a state resembling cellular senescence; however, a well-accepted regulator and effector of p53 – p19Arf and p21Cip1 – were both dispensable. This highlights the tissue-specific and context-dependent importance of other components of the p53 pathway. We also found that a potent inhibitor of Cdk4(6), p18Ink4c, maintained cell cycle arrest and blocked progression to PNET. Ink4c has been increasingly implicated as a tumor suppressor in experimental brain tumor models and human tumor samples (35,40-43). While intuitively likely, its role in blocking Cyclin D1-driven neoplasia has not been previously established. Lastly, we demonstrate that subclinical pineal gland enlargement was frequent in children with heritable retinoblastoma (who are at high risk for developing pineal gland PNET), essentially mimicking the new model we have developed.
That p53 played a critical role to block PNET progression in the pineal gland was surprising because it is seemingly dispensable as a tumor suppressor in the retina of these mice (14). In the pineal gland, the largest measurable effect of p53 was to induce cell cycle arrest correlating with reversal of Cyclin D1-mediated Rb phosphorylation and the appearance of a senescence-like state with changes in heterochromatin bound proteins. Interestingly, this only occurred after a prolonged period of enhanced cell proliferation. Hence, the arrest was not simply a direct response to an oncogenic insult, a finding that is consistent with our observation that p19Arf was not required. While somewhat unexpected given the current model for Arf induction as an “oncogene-sensor” (44), it is not without precedent: Arf is not necessary for p53-dependent tumor suppression when SV40 T121 is expressed in the mouse choroid plexus (45). We can propose two alternative explanations for how p53 is engaged. First, Cyclin D1 might activate p53 in an oxidative stress- or DNA damage-dependent manner, bypassing Arf altogether (reviewed in ref. # 46). Alternatively, p53-dependent arrest might represent a developmental process intersecting with and blocking the oncogenic signals. Such an explanation would be consistent with the observation that genetically-susceptible children (like those with heritable retinoblastoma) are at highest risk of developing pineal gland PNET in the first several years of life (36,37).
Several recent papers have begun to address the role of Ink4c as a tumor suppressor in brain neoplasms. Zindy and colleagues showed that loss of one or both Ink4c alleles is needed to foster cerebellar PNET formation in p53 -/- mice (41); hence, a collaborative interaction exists. Members of the Roussel laboratory also established that Ink4c loss enhances cerebellar PNET driven by Sonic hedgehog (Shh) pathway activation (due to Ptc mutation) (35). However, Cyclin D1 is only one of several oncogenes induced when Shh is deregulated (35,47). Better definition of the relationship between Cyclin D1 and p18Ink4c stems from the finding that ectopic Cyclin D1 expression can drive tumor formation only in Ink4c-null mouse granule neuron precursors following orthotopic implantation (42). Our in vivo findings in a transgenic model complement this growing body of evidence by establishing that inactivation of either Ink4c or p53 was necessary and sufficient for PNET progression when Cyclin D1 represented the principal oncogenic signal. Parenthetically, tumor suppression by Ink4c did not apply to the retina as the Irbp-Cyclin D1 ocular phenotype was unaffected by Ink4c (or p21) loss (Supplemental Figure 7); hence, cellular context must be considered.
Our findings highlight the complex relationships between the Rb and p53 pathways. In the absence of p53, p18Ink4c expression increased but still was not sufficient to prevent PNET. While we have not evaluated p53 function in the Irbp-Cyclin D1, Ink4c -/- tumors, it remains intact in cerebellar PNETs occurring in Ptc +/-, Ink4c -/- mice (35). While both genes are needed for tumor suppression in our model, the basis for the interdependence is not clear from our studies. The two proteins did not seem to lie in a simple linear pathway because a greater fraction of pinealocytes proliferated at 2 months in the absence of p53 than in the absence of Ink4c (42% versus 1.5%, respectively). Also, the latency for tumor development was longer in Irbp-Cyclin D1, Ink4c -/- mice (~7 months) than Irbp-Cyclin D1, p53 -/- mice (3-4 months). Finally, one measure of p53 activity – p21Cip1 induction – was dampened at two months after pinealocytes arrest whereas p18Ink4c expression was high at P10 and even higher when the cells had arrested. The simplest explanation is that p53 and p18Ink4c represent parallel pathways that check pinealocyte proliferation by fundamentally different mechanisms. We can also speculate that the two genes might act sequentially such that p53 drives the Cyclin D1-expressing pinealocytes into an arrested state, and p18Ink4c maintains it. However, this concept needs to be evaluated experimentally.
Approximately 10% of children with heritable retinoblastoma develop a metachronous brain PNET, commonly pineoblastoma (36,37). That this only occurs in a subset and usually several years after the diagnosis of retinoblastoma implies that RB inactivation alone is insufficient for tumorigenesis in PNETs. Although our mouse model of deregulated Cyclin D1 expression may not totally equate to RB haploinsufficiency in children, we found parallel pineal gland enlargement in children with heritable retinoblastoma. In these children, progression from pineal gland enlargement to PNET might be halted in a p53- or Ink4c-dependent manner. Due to the aggressive nature of this tumor [currently the leading cause of death in these children in the first decade of life (36,37)], preventive therapy for children known to be at high risk is an attractive goal. Our finding of Ink4c-mediated tumor suppression in pineal PNET when RB function is compromised suggests that pharmacological blockade of Cdk4(6) activity may be a useful strategy to block progression of pre-malignant lesions.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. E. Y.-H. P. Lee, in whose laboratory the Irbp-Cyclin D1 mouse was originally generated using human Cyclin D1 cDNA from P. Hinds; donation of the Ink4c -/- knockout mice by Bristol-Myers Squibb through M. Barbacid and M. F. Roussel; helpful discussions with O. Ayrault, M. B. Kastan, E. Y.-H. P. Lee, K. Maclean, M. F. Roussel, C. J. Sherr, G. Zambetti, and F. Zindy, and with other members of the Kastan and Skapek laboratories; M. W. Wilson and B. G. Haik, who are integral members of the SJCRH retinoblastoma program; Jesse Jenkins for helpful discussions and assistance with pathology material, technical assistance by D. Bush (SJCRH Clinical Pathology Laboratory); and support from SJCRH shared resources: the Animal Resources Center, Biostatistics, and the Hartwell Center for Bioinformatics and Biotechnology.
This work was supported in part by NIH Cancer Center Support Core Grant CA21765 and the American Lebanese Syrian Associated Charities (ALSAC) of SJCRH.
Reference List
- 1.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24(17):2796–809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 2.Diehl JA. Cycling to cancer with Cyclin D1. Cancer Biol Ther. 2002;1:226–31. doi: 10.4161/cbt.72. [DOI] [PubMed] [Google Scholar]
- 3.Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol. 2005;23:4215–24. doi: 10.1200/JCO.2005.05.064. [DOI] [PubMed] [Google Scholar]
- 4.Lovec H, Grzeschiczek A, Kowalski M-B, Moroy T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma. EMBO J. 1994;13:3487–95. doi: 10.1002/j.1460-2075.1994.tb06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13:2124–30. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–71. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 7.Deane NG, Parker MA, Aramadla R, et al. Hepatocellular carcinoma results from chronic Cyclin D1 overexpression in transgenic mice. Cancer Res. 2001;61:5389–95. [PubMed] [Google Scholar]
- 8.Becker LE, Hinton D. Primitive neuroectodermal tumors of the central nervous system. Hum Pathol. 1983;14:538–50. doi: 10.1016/s0046-8177(83)80006-9. [DOI] [PubMed] [Google Scholar]
- 9.Neben K, Korshunov A, Benner A, et al. Microarray-based screening for molecular markers in medulloblastoma revealed STK15 as independent predictor for survival. Cancer Res. 2004;64:3103–11. doi: 10.1158/0008-5472.can-03-3968. [DOI] [PubMed] [Google Scholar]
- 10.Gilbertson RJ. Medulloblastoma: signaling a change in treatment. Lancet Oncol. 2004;5:209–18. doi: 10.1016/S1470-2045(04)01424-X. [DOI] [PubMed] [Google Scholar]
- 11.Oliver TG, Grasfeder LL, Carrol AL, et al. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci. 2003;100:7331–6. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Devel. 2006;133:3929–37. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liou GI, Geng L, Al-Ubaidi MR, et al. Tissue-specific expression in trangenic mice directed by the 5’-flanking sequences of the human gene encoding interphotoreceptor retinoid-binding protein. J Biol Chem. 1990;265:8373–6. [PubMed] [Google Scholar]
- 14.Skapek SX, Lin S-CJ, Jablonski MM, et al. Persistent expression of cyclin D1 disrupts normal photoreceptor differentiation and retina development. Oncogene. 2001;20:6742–51. doi: 10.1038/sj.onc.1204876. [DOI] [PubMed] [Google Scholar]
- 15.Silva RL, Thornton JD, Martin AC, et al. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005;24:2803–14. doi: 10.1038/sj.emboj.7600751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latres E, Malumbres M, Sotillo R, et al. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan PKS, Chan DPC, To K-F, Cheung JLK, Cheng AF. Evaluation of extraction methods from paraffin wax embedded tissues for PCR amplification of human and viral DNA. Journal of Clinical Pathology. 2001;54:401–3. doi: 10.1136/jcp.54.5.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fung K-M, Trojanowski JQ. Animal models of medulloblastomas and related primitive neuroectodermal tumor. A review. J Neuropath Exp Neurol. 1995;54:285–96. doi: 10.1097/00005072-199505000-00001. [DOI] [PubMed] [Google Scholar]
- 19.McKeller RN, Fowler JL, Cunningham JJ, et al. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc Nat Acad Sci. 2002;99:3848–53. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumida M, Barkovich AJ, Newton TH. Development of the pineal gland: measurement with MR. Am J Neuroradiol. 1996;17:233–6. [PMC free article] [PubMed] [Google Scholar]
- 21.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 22.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Devel. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 23.Kitagawa M, Higashi H, Jung HK, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15:7060–9. [PMC free article] [PubMed] [Google Scholar]
- 24.Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997:287–301. doi: 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends in Cell Biology. 2001;11:S27–S31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 26.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;2004:2919–33. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 27.Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–46. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 28.Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 29.Ferbeyre G, de SE, Lin AW, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 31.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 32.Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994;7:480–4. doi: 10.1038/ng0894-480. [DOI] [PubMed] [Google Scholar]
- 33.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–22. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Thornton JD, Silva RLA, Martin AC, Skapek SX. The Arf tumor suppressor regulates Platelet-derived growth factor receptor β signaling: A new view through the eyes of Arf -/- mice. Cell Cycle. 2005;4:1316–9. doi: 10.4161/cc.4.10.2109. [DOI] [PubMed] [Google Scholar]
- 35.Uziel T, Zindy F, Xie S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Devel. 2005;19:2656–67. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blach LE, McCormick B, Abramson DH, Ellsworth RM. Trilateral retinoblastoma - incidence and outcome: a decade of experience. Int J Radiation Oncol Biol Phys. 1994;29:729–33. doi: 10.1016/0360-3016(94)90560-6. [DOI] [PubMed] [Google Scholar]
- 37.Kivela T. Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17:1829–37. doi: 10.1200/JCO.1999.17.6.1829. [DOI] [PubMed] [Google Scholar]
- 38.Cordon-Cardo C, Latres E, Drobnjak M, Oliva MR, Brennan MF, Levine AJ. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res. 1994;54:794–9. [PubMed] [Google Scholar]
- 39.Soussi T, Beroud C. Assessing TP53 status in human tumours to evaluate clinical outcome. Nature Rev Cancer. 2001;1:233–40. doi: 10.1038/35106009. [DOI] [PubMed] [Google Scholar]
- 40.Ramsey MR, Krishnamurthy J, Pei X-H, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res. 2007;67:4732–41. doi: 10.1158/0008-5472.CAN-06-3437. [DOI] [PubMed] [Google Scholar]
- 41.Zindy F, Nilsson LM, Nguyen L, et al. Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res. 2003;63:5420–7. [PubMed] [Google Scholar]
- 42.Zindy F, Uziel T, Ayrault O, et al. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67:2676–84. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 43.Solomon DA, Kim J-S, Jenkins S, et al. Identification of p18INK4c as a tumor suppressor gene in glioblastoma multiforme. Cancer Res. 2008;68:2564–9. doi: 10.1158/0008-5472.CAN-07-6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherr CJ. Principles of tumor suppression. Cell. 2004;116:235–46. doi: 10.1016/s0092-8674(03)01075-4. [DOI] [PubMed] [Google Scholar]
- 45.Tolbert D, Lu X, Yin C, Tantama M, Van Dyke T. p19Arf is dispensable for oncogenic stress-induced p53-mediated apoptosis and tumor suppression in vivo. Mol Cell Biol. 2002;22:370–7. doi: 10.1128/MCB.22.1.370-377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 47.Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–67. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.