

Abstract
The reactive oxy-intermediate of the catalytic cycle of extradiol aromatic ring-cleaving dioxygenases is formed by binding the catecholic substrate and O2 in adjacent ligand positions of the active site metal (usually Fe(II)). This intermediate and the following Fe(II)-alkylperoxo intermediate resulting from oxygen attack on the substrate have been previously characterized in a crystal of homoprotocatechuate 2,3-dioxygenase (HPCD). Here a subsequent intermediate in which the O–O bond is broken to yield a gem diol species is structurally characterized. This new intermediate is stabilized in the crystal by using the alternative substrate, 4-sulfonylcatechol, and the Glu323Leu variant of HPCD, which alters the crystal packing.
Extradiol aromatic ring cleaving dioxygenases such as homoprotocatechuate 2,3-dioxygenase (HPCD) catalyze fissure of the O2 bond and incorporation of both atoms into catecholic substrates to give muconic semialdehyde products (1-3). These enzymes use an active site Fe(II) (and rarely Mn(II)) bound in a 2-His-1-carboxylate facial triad motif in the O2 activation process (4-6). X-ray crystal structures show that the substrate binds to the Fe(II) as an asymmetric chelate (6, 7). As substrate binds, two of the three solvents present on the accessible face of the iron in the resting enzyme are directly displaced and the third is either released or its bond to the iron is weakened. This prepares a site for O2 binding adjacent to the substrate.
We and others have speculated that O2 activation occurs as a result of electron transfer from the catecholic substrate to the bound O2 via the Fe(II) (1-3, 8-13). Radical recombination would yield an Fe(II)-bound alkylperoxo intermediate. O–O bond cleavage and insertion of one atom into the aromatic ring to form a 7-member lactone intermediate is proposed to follow. This step would be facilitated by an acid catalyst to protonate the oxygen atom proximal to the Fe(II). Hydrolysis of the lactone by the Fe(II)-bound oxygen, now at the level of hydroxide, would result in ring cleavage, and ultimately, product release.
Support for this mechanism has been gained using several experimental approaches. Neither the Fe(II) center nor the substrate, homoprotocatechuate (HPCA), has a convenient chromophore or EPR signal with which to monitor the reaction cycle. However, it was found that the chromophore of the alternative substrate 4-nitrocatechol (4NC) could be monitored, and it revealed the occurrence of at least 8 intermediates in the overall cycle (14, 15). Many of these had kinetic or spectroscopic properties consistent with intermediates predicted to occur in the cycle. Mutagenesis of His200, which is well positioned in the active site to serve as the catalytic acid, slowed the oxygen reaction steps (14). This allowed the detection of the first oxy-intermediate in the extradiol dioxygenase class based on its absorbance maximum at 610 nm.
Recently, we showed that HPCD in crystals retains catalytic activity (2, 16). When 4NC was used as the substrate and O2 was limited, the X-ray crystal structure of the homotetrameric enzyme showed that three of the four subunits in the asymmetric unit contained different reaction cycle intermediates. One subunit contained the predicted 4NC semiquinone- Fe(II) -superoxo intermediate, two more contained the Fe(II)-alkylperoxo intermediate, and the fourth contained the product complex. Thus, nearly the entire oxygen activation and insertion segment of the reaction cycle was represented in a single crystal. Differences in the identical subunits were presumably introduced by crystal packing forces.
The principal missing intermediate from the structural studies conducted thus far is the 7-membered lactone species that is postulated to be hydrolyzed to product. The solution transient kinetic studies showed that the substitution of the nitro group in place of the acetate group of HPCA resulted in at least a 200-fold decrease in the rate of decay of one of the oxy-intermediates prior to lactone formation (15). Consequently, it is possible that the lactone intermediate was not formed in sufficient quantity to be detected in the crystal. Here we show that use of an alternative substrate with a less electron-withdrawing substituent leads to the formation of a new intermediate that appears to occur between the Fe(II)-alkylperoxo and the product complexes in the reaction cycle.
4-Sulfonylcatechol (4SC) was selected as an alternative substrate in which the substituent electronegativity is midway between those of 4–NO2 and the normal 4–CH2COO- substituents. HPCD catalyzes conversion of 4SC to a yellow product with spectral features and pKa values similar to those of the extradiol product from HPCA cleavage between the 2 and 3 positions. The turnover number is 10% of that observed when using HPCA (Table S1). Diffusion of 4SC into a crystal of HPCD at either atmospheric or very low O2 concentrations (as used successfully in previous studies) resulted in mixtures of intermediates in the subunit active sites such that structural assignments were not possible. During ongoing studies of the influence of crystal packing on intermediate stability, we have noted that surface modifications affect the subunit-specific stability of intermediates in the active sites. Consequently, the effects of one such mutation on the stability of intermediates formed during in crystallo 4SC turnover were tested. Residue Glu323 occurs in a region of the surface of HPCD where significant unfavorable crystal contacts occur in several locations (Figure S2). This residue also marks the beginning of a 43-residue C-terminal segment of the enzyme that that forms a flap over the active site (6, 16). The E323L mutant of HPCD was constructed and found not to significantly alter the steady state kinetic parameters of HPCA or 4SC turnover (Table S1). E323L crystallized in the same space group as the native enzyme (P21212), but the unit cell was found to be 10 Å longer in one dimension and 2 Å longer in a second, indicating that crystal packing had been altered. The X-ray crystal structure of mutant enzyme was solved at 1.65 Å resolution (Tables S2, S3). Superposition of the structures of the mutant and WT enzymes shows that the overall structure of each subunit as well as the overall tetrameric assembly of the enzyme is unchanged by the mutation (rmsd 0.35-0.45 Å over all atoms), but substantial changes occur in the crystal packing interfaces (Figures S2B, S3).
In crystallo reaction was initiated by transfer of E323L crystal into the crystallization solution containing 4SC in a low-O2 (∼5-10 ppm) glovebox, followed by flash freezing (see Supporting Information for Experimental Procedures). The resulting X-ray structure of the complex, solved at 1.60 Å resolution (Tables S2, S3), showed an apparent single intermediate species in subunits C (occupancy 85 %) and D (occupancy 65 %) of the enzyme molecule in the asymmetric unit. Neither 4SC nor any intermediate was detected in subunits A and B.
The structure shown in Figure 1A was obtained by fitting the electron density in subunit C with a model built by breaking the O–O bond of a putative 4SC-alkylperoxo intermediate, resulting in a C2-gem diol adduct of 4SC bound to the iron. The observed electron density and bond length for the new atom bonded to C2 are both consistent with its assignment as oxygen. Several other potential ligands were tested, but did not account for the observed density (see Supporting Information). The electron density of a complete substrate ring is observed, showing that the ring-open product has not yet formed. However, the ring is puckered, consistent with a sp3 hybridized carbon 2.
FIGURE 1.
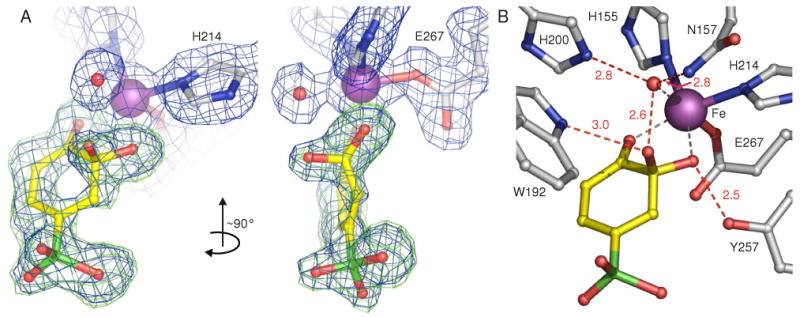
Structure of a species trapped in the active sites of E323L mutant of 2,3-HPCD during in crystallo reaction with 4SC and O2 (PDB code 3ECK). (A) Gem diol adduct of 4SC. The blue 2Fobs–Fcalc maps are contoured at 1.0 σ. The green Fobs–Fcalc ligand-omit maps were computed by removing ligands from the final model and are contoured at +3 σ. (B) Hydrogen-bonding interactions of the intermediate with active site residues. Atom color code: gray, carbon (enzyme residues); yellow, carbon (ligand); blue, nitrogen; red, oxygen; green, sulfur; purple, iron. Red dashed lines show hydrogen-bonds (Å). Gray dashed lines indicate bonds or potential bonds to iron; distances and angles are given in Table S3; additional views and comparison of active site structures in subunits C and D are given in Figure S1.
The position of the Fe(II) ligation that is occupied by the proximal O of the alkylperoxo intermediate, O2, and small molecules in other ligand complex structures of HPCD appears to be occupied by a single heavy atom species such as one oxygen derived from O2 or solvent. Attempts to fit this density with a diatomic molecule were unsuccessful.
The second sphere residues of the active site are positioned very similarly to those in the structures of intermediates previously reported (2, 16). In particular, the proposed active site acid catalyst, His200, is positioned 2.8 Å from, and hydrogen bonded to, the putative oxygen iron ligand as shown in Figure 1B. Tyr257 forms a strong hydrogen bond to the original hydroxyl group of the 4SC C2, stabilizing the sp3 hybridization of this carbon, as it does in the Fe(II)-alkylperoxo intermediate.
It is clear from the structure of the new intermediate that it does not represent the 7-membered lactone species postulated in the mechanism. We originally proposed (2, 8) that this species arises from a Criegee rearrangement, which is traditionally considered to be a concerted process that would not produce a discrete gem diol intermediate.
Recently, computational studies of the mechanism have been reported based on the X-ray crystal structures for the enzyme-substrate complex (10, 11). As illustrated in Figure 2, these studies predict that a lactone will be formed as an intermediate, but in each case, the lactone forms from an intermediate epoxide. The epoxide, in turn, forms from a transient species in which the O–O bond is broken such that one oxygen is added to the substrate C2 while the second is retained on the iron. This substrate intermediate would formally be a gem diol, however, the computational studies find that the intermediate would be short-lived and likely to have radical character.
FIGURE 2.

Proposed mechanism of formation of the 7-membered lactone intermediate. Adapted from ref (10, 11)
The remarkable stability of the new intermediate versus that predicted in computations may have several origins. First, midway through the catalytic cycle the enzyme active site may provide a somewhat different environment for the substrate intermediate than that predicted by calculations, which were based on the enzyme-substrate complex structure. Second, 4SC has a different distribution of ring electron density than that of the HPCA used in these calculations. Third, the current results suggest that crystal packing forces have an important role in stabilizing the new intermediate, but they are not brought into computations.
It is interesting to note that the active sites of E323L and native HPCD show no significant structural differences despite the difference in their abilities to stabilize the new intermediate. This might be expected if the underlying cause is not in the structure, per se, but rather in the active site and subunit dynamics that contribute to progress along the reaction coordinate.
The O-O bond cleaved intermediate that we propose here has not been previously recognized for any ring cleaving dioxygenase. It strongly suggests that the oxygen insertion reaction is not concerted, and thus, under the right conditions, an epoxide or lactone intermediate may also be trapped. In this regard, the approach of using a crystal to both trap and structurally characterize enzyme intermediates appears to hold great promise (17).
Supplementary Material
Experimental procedures, a table of kinetic parameters, a table of statistics for the X-ray crystal structures, a table of interatomic distances and angles, a figure comparing the intermediates in subunits C and D, a figure showing the location of residue E323, and a figure comparing the surface contacts for the E323 (wild-type) and the L323 (E323L mutant) residues. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
X-ray data were collected at Advanced Photon Source, Argonne National Laboratory and Kahlert Structural Biology Laboratory, University of Minnesota. Data analysis was carried out at the Basic Sciences Computing Laboratory at the University of Minnesota supported by Minnesota Supercomputing Institute. We thank Frank Rotella, Caroline Wilmot, Stephanie Groce and Michael Mbughuni for assistance and discussions.
Footnotes
This research was supported by nih grant GM24689 and a Minnesota Partnership for Biotechnology Grant (SPAP-05-0013-P-FY06).
The coordinates for structures reported here have been deposited in the Protein Data Bank as 3ECJ and 3ECK.
References
- 1.Lipscomb JD, Orville AM. Mechanistic aspects of dihydroxybenzoate dioxygenases. Metal Ions Biol Syst. 1992;28:243–298. [Google Scholar]
- 2.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit Rev Biochem Mol Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- 4.Han S, Eltis LD, Timmis KN, Muchmore SW, Bolin JT. Crystal structure of the biphenyl-cleaving extradiol dioxygenase from a PCB-degrading pseudomonad. Science. 1995;270:976–980. doi: 10.1126/science.270.5238.976. [DOI] [PubMed] [Google Scholar]
- 5.Sugiyama K, Senda T, Narita H, Yamamoto T, Kimbara K, Fukuda M, Yano K, Mitsui Y. Three-dimensional structure of 2,3-dihydroxybiphenyl dioxygenase (BphC enzyme) from Pseudomonas sp strain KKS102 having polychlorinated biphenyl (PCB)-degrading activity. Proceedings of the Japan Academy, Series B: Physical and Biological Sciences. 1995;71:32–35. [Google Scholar]
- 6.Vetting MW, Wackett LP, Que L, Jr, Lipscomb JD, Ohlendorf DH. Crystallographic comparison of manganese- and iron-dependent homoprotocatechuate 2,3-dioxygenases. J Bacteriol. 2004;186:1945–1958. doi: 10.1128/JB.186.7.1945-1958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato N, Uragami Y, Nishizaki T, Takahashi Y, Sazaki G, Sugimoto K, Nonaka T, Masai E, Fukuda M, Senda T. Crystal structures of the reaction intermediate and its homologue of an extradiol-cleaving catecholic dioxygenase. J Mol Biol. 2002;321:621–636. doi: 10.1016/s0022-2836(02)00673-3. [DOI] [PubMed] [Google Scholar]
- 8.Arciero DM, Lipscomb JD. Binding of 17O-labeled substrate and inhibitors to protocatechuate 4,5-dioxygenase-nitrosyl complex Evidence for direct substrate binding to the active site Fe2+ of extradiol dioxygenases. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 9.Bugg TDH. Dioxygenase enzymes: catalytic mechanisms and chemical models. Tetrahedron. 2003;59:7075–7101. [Google Scholar]
- 10.Deeth RJ, Bugg TDH. A density functional investigation of the extradiol cleavage mechanism in non-heme iron catechol dioxygenases. J Biol Inorg Chem. 2003;8:409–418. doi: 10.1007/s00775-002-0430-7. [DOI] [PubMed] [Google Scholar]
- 11.Siegbahn PEM, Haeffner F. Mechanism for catechol ring-cleavage by non-heme iron extradiol dioxygenases. J Am Chem Soc. 2004;126:8919–8932. doi: 10.1021/ja0493805. [DOI] [PubMed] [Google Scholar]
- 12.Kovaleva EG, Lipscomb JD. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emerson JP, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L., Jr Swapping metals in Fe- and Mn-dependent dioxygenases: evidence for oxygen activation without a change in metal redox state. Proc Natl Acad Sci U S A. 2008;105:7347–7352. doi: 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groce SL, Lipscomb JD. Aromatic ring cleavage by homoprotocatechuate 2,3-dioxygenase: Role of His200 in the kinetics of interconversion of reaction cycle intermediates. Biochemistry. 2005;44:7175–7188. doi: 10.1021/bi050180v. [DOI] [PubMed] [Google Scholar]
- 15.Groce SL, Miller-Rodeberg MA, Lipscomb JD. Single-turnover kinetics of homoprotocatechuate 2,3-dioxygenase. Biochemistry. 2004;43:15141–15153. doi: 10.1021/bi048690x. [DOI] [PubMed] [Google Scholar]
- 16.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hajdu J, Neutze R, Sjogren T, Edman K, Szoke A, Wilmouth R, Wilmot CM. Analyzing protein functions in four dimensions. Nat Struct Biol. 2000;7:1006–1012. doi: 10.1038/80911. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, a table of kinetic parameters, a table of statistics for the X-ray crystal structures, a table of interatomic distances and angles, a figure comparing the intermediates in subunits C and D, a figure showing the location of residue E323, and a figure comparing the surface contacts for the E323 (wild-type) and the L323 (E323L mutant) residues. This material is available free of charge via the Internet at http://pubs.acs.org.