

Abstract
CD80 and CD86 both costimulate T cell activation. Their individual effects in vivo are difficult to study as they are coordinately up-regulated on APCs. We have studied mice expressing rat insulin promoter (RIP)-CD80 and RIP-CD86 on the NOD and NOD.scid genetic background to generate in vivo models, using diabetes as a readout for cytotoxic T cell activation. Accelerated spontaneous diabetes onset was observed in NOD-RIP-CD80 mice and the transfer of diabetes from 6-wk-old NOD mice to NOD.scid-RIP-CD80 mice was greater compared with NOD-RIP-CD86 and NOD.scid-RIP-CD86 mice, respectively. However, the secondary in vivo response was maintained if T cells were activated through CD86 costimulation compared with CD80. This was demonstrated by greater ability to cause recurrent diabetes in NOD-RIP-CD86 diabetic mice transplanted with 6-wk-old NOD islets and adoptively transferred diabetes from diabetic NOD-RIP-CD86 mice to NOD.scid mice. In vitro, CD80 costimulation enhanced cytotoxicity, proliferation, and cytokine secretion in activated CD8 T cells compared with CD86 costimulation. We demonstrated increased CTLA-4 and programmed death-1 inhibitory molecule expression following costimulation by both CD80 and CD86 (CD80 > CD86). Furthermore, T cells stimulated by CD80 were more susceptible to inhibition by CD4+CD25+ T cells. Overall, while CD86 does not stimulate an initial response as strongly as CD80, there is greater sustained activity that is seen even in the absence of continued costimulation. These functions have implications for the engineered use of costimulatory molecules in altering immune responses in a therapeutic setting.
T cell activation is a multistep process that involves specific recognition of the MHC-peptide complex through the TCR (signal 1) as well as costimulatory influences through a variety of molecules (signal 2) (1). It is now clear that the group of molecules which include the B7 as well as the TNFR families are not only responsible for costimulation, but also have critical inhibitory and regulatory activity (2).
Costimulatory interactions are essential for T cells to undergo complete activation and occur when the constitutively expressed T cell costimulatory ligand CD28 binds either of the costimulatory molecules on APCs, CD80 (B7.1), or CD86 (B7.2). In the absence of signal 2 (and weak signal 3), T cells enter a state of anergy or apoptosis and hence CD28 is essential for T cell activation. However, in addition to the interaction of CD80 and CD86 molecules with CD28, CTLA-4, which is up-regulated after T cell activation (3) and reaches maximal cell surface expression 2-3 days after stimulation, acts as an inhibitory ligand (4). CTLA-4 may function either by direct competition with CD28 for CD80/CD86 binding, because CTLA-4 has a stronger affinity for both CD80 and CD86 compared with CD28 (5, 6), or through intracellular signaling. CTLA-4 becomes stabilized at the cell surface through phosphorylation and plays a role in initiating dephosphorylation events in the T cell synapse to down-regulate TCR signaling (reviewed in Ref. 7).
In recent years, other costimulatory molecules have been identified. ICOS and 4-1BB are expressed after initial T cell activation (8-11). ICOS is thought to be involved in effector phases of T cell function (12), while 4-1BB has been associated with proliferation and T cell survival (13). Programmed death-1 (PD-1)3 has been identified as another inhibitory costimulator, such that PD-1-deficient animals develop autoimmune disorders, such as lupus-like glomerulonephritis (14) and accelerated autoimmune diabetes (15).
CD80 and CD86 are primarily expressed on APCs with different kinetics, although there is expression on activated T cells as well (16, 17). CD86 is constitutively expressed on resting APCs, such as DCs and B cells, albeit at low levels, while CD80 is not detectable (18, 19). However, following activation, CD86 is up-regulated 6-h poststimulation, peaking between 18 and 24 h (17, 19). In contrast, CD80 expression on stimulated APCs is not evident for 24 h and demonstrates peak expression between 48 and 72 h (17, 19).
When studied individually, rather than on the same APCs, CD80 is a more potent costimulator of T cell activation compared with CD86. Tumors transfected with CD80 stimulated greater CTL activation compared with CD86 transfectants (20), while tumor-specific CTLs were only generated in mice immunized with irradiated CD80-transfected tumors and not CD86-transfected tumors (21). A greater number of CD4 T cells enter cell cycle with quicker kinetics when stimulated with CD80, compared with CD86 (22). These actions of CD80 have suggested that this molecule may be of use when expressed ectopically in tumors for enhancing immune responses. Much less is known of CD86 in this context.
The rat insulin promoter (RIP) has been used to direct expression of CD80 or CD86 costimulatory molecules to insulin-producing islet β cells, which do not normally express any costimulatory molecules. In conjunction with endogenous MHC class I molecules, islet β cells are effectively transformed into APCs for CD8 T cells. Model systems using RIP-CD80 and RIP-CD86 have been developed to test whether tissue expression of costimulators on islet β cells would break tolerance and cause diabetes. The experiments showed that, by itself, T cell activation with CD80 costimulation on a B6 or bm1 genetic background is not sufficient to cause disease (23-25). However, the presence of other factors, such as the coexpression of a viral protein or MHC class II, IE, could stimulate diabetes (26-28). Coexpression of cytokines TNF-α or IL-2 also precipitated diabetes (23, 28). Furthermore, the expression of CD80 on the NOD genetic background caused accelerated diabetes, as early as 3 wk of age (25, 29, 30). The effect of the MHC in this acceleration is particularly important as CD80 expression on the nonobese-resistant and congenic B6g7 mice also facilitates spontaneous diabetes in these diabetes-resistant strains (31). CD86 has been less studied in these models. When CD86 was expressed on the pancreatic islet β cells on the B6 genetic background, T cell tolerance to β cells was maintained and the mice did not develop diabetes (32). However, there was peri-islet infiltration with IL-4-producing cells in older mice. The effect of CD86 expression on the NOD genetic background was not previously reported.
In this study, we directly compared the effects of CD80 and CD86 costimulation on CD8 T cell activation using diabetes onset as an in vivo read out. We report that CD86 activates CD8 T cells with less potency during primary activation in vivo compared with CD80. In vitro studies, using CD80- or CD86-expressing islets as APCs, demonstrated less enhanced proliferation and cytotoxicity of CD86-activated CD8 T cells compared with CD80. However, CD80 costimulation also leads to greater increases of the inhibitory molecules CTLA-4 and PD-1 which could down-regulate CD8 T cell function. Furthermore, CD8 T cells activated by CD80 are more susceptible to inhibition by CD4+CD25+ T cells. These cells require continued costimulatory activity for maintenance of activation, whereas CD86 costimulation leads to lower but more sustained activity of CD8 T cells.
Materials and Methods
Mice
Two founder lines of C57BL/6-RIP-CD86 mice, lines 12 and 46, which expressed human CD86 on pancreatic β cells at medium and high levels, respectively (32), were backcrossed to NOD/caj mice (provided by the late C. Janeway, Yale School of Medicine, New Haven, CT) for at least 14 generations and were designated lines 12B and 46K. NOD-RIP-CD80 mice (29) were generated by backcrossing C57BL/6-RIP-CD80 mice to NOD/caj mice for at least three generations. NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 mice were generated by breeding NOD-RIP-CD80 or NOD-RIP-CD86 mice with NOD.scid mice. Transgene-positive F1 mice were backcrossed to NOD.scid mice and transgene-positive scid homozygous F2 mice were generated. These NOD.scid-RIP-CD80 mice were backcrossed nine generations to NOD.scid mice. NOD.scid-RIP-CD86 were derived from RIP-CD86 mice previously backcrossed 14 generations to NOD mice and therefore no further backcross was required.
Insulin-specific TCR-transgenic mice were generated by isolating TCR genomic DNA from G9C8 cloned T cells (33), which have previously been shown to have specific reactivity to amino acids 15-23 of the insulin B chain (34). TCR α (Vα18S1 Jα18 Cα) and β (Vβ6S1 Dβ1.1 Jβ2.3 Cα1) chain DNA was purified and cloned into pTαcass and pTβcass constructs, respectively, provided by D. Mathis (Joslin Diabetes Center, Harvard Medical School, Boston, MA) (35). The cloned constructs were injected directly into NOD OVA to generate independent TCR α and β founder lines, which were intercrossed to produce αβ TCR-transgenic mice (G9.NOD). G9Cα−/−.NOD mice were generated by crossing the αβ TCR-transgenic mice to NOD.Cα−/− mice (>20 generations backcross to NOD mice). The mice were specific pathogen free and housed in barrier rooms. All procedures were performed in accordance with U.K. Home Office-approved protocols.
Genotyping of mice for expression of CD80 or CD86 using PCR
Tail-tip DNA from NOD-RIP-CD80 and NOD-RIP-CD86 DNA was analyzed in a standard PCR. The samples were amplified for 35 cycles of 30 s denaturing at 95°C, 1 min annealing at 55°C, 1 min extension at 72°C and a final extension of 10 min. Primers B7-311 (TGA AGC CAT GGG CCA CAC) and B7-1192 (GAC ACT GTT ATA CAG GG) produced an 880-bp band for CD80 and primers B70 - 480 (CCC ACA GGA ATG ATT CGC ATC) and B70-1009 (TCA CTC TCT TCC CTC TCC ATT GTG) produced a 530-bp band for CD86 in transgene-positive mice. Primers were synthesized by Invitrogen Life Technologies.
Detection of CD80 and CD86 from islet cDNA
Pancreata from eight NOD-RIP-CD80 and eight NOD-RIP-CD86 mice at 6 wk of age were harvested and islets were isolated as described. RNA was extracted from the islet samples using TRIzol (Invitrogen Life Technologies). cDNA was generated from the RNA samples according to the manufacturer’s instructions. Concentrations of cDNA were measured using a spectrophotometer and 1/2 dilutions of cDNA (starting at 2 μg) were amplified using primers for CD80 and CD86 as described above. GAPDH was used as a control (GAPDH forward: GGT CAT CAT CTC CGC CCC TTC TGC, GAPDH reverse: GAG TGG GAG TTG CTG TTG AAG TCG; Invitrogen Life Technologies).
Histology
Pancreata, spleen, or kidney was harvested, fixed, and immunohistochemistry was performed as previously described (29, 30). Sections were incubated with specific biotinylated Abs (rat anti-murine CD4, CD8, B220 (Caltag Laboratories)), rat anti-human CD80 or CD86 (BD Pharmingen)) and detected with streptavidin-alkaline phosphatase (Vector Laboratories) and the AP substrate kit, VectorRed (Vector Laboratories). VectorRed working solution, made up in 100 mM Tris-HCl (pH 8.4) with the addition of levamisole (Vector Laboratories), was used in accordance with the manufacturer’s instructions. Sections were incubated in the dark for a preoptimized time and counterstained with hematoxylin (Vector Laboratories) before mounting under coverslips with Hydromount (National Diagnostics). Insulitis was measured in diabetic, 6- and 12-wk-old NOD, NOD-RIP-CD80, and NOD-RIP-CD86 mice. Sections were stained with H&E (Vector Laboratories) as described previously (29, 30) and 30-200 islets from four mice per group were scored for insulitis. Score 0, no infiltration; 1, peri-insulitis; 2, ≤50% infiltration; 3, 50-90% infiltration; and 4, ≥90% infiltration.
Diagnosis of diabetes
Groups of NOD-RIP-CD86 lines, NOD-RIP-CD80, and female NOD-RIP-CD86 transgene-negative mice were monitored weekly for spontaneous development of diabetes over a period of 25 wk. Diabetes was detected by initially testing for glycosuria (Bayer Diastix). Diagnosis was confirmed by blood glucose measurements >14 mM/L. Mice from adoptive transfer and transplant experiments were tested two to three times weekly for 112 days or until diabetic.
Adoptive transfer of diabetes
A total of 20 × 106 splenocytes at 108 cells/ml, isolated from diabetic NOD, NOD-RIP-CD80, and NOD-RIP-CD86 mice or 6-wk-old NOD mice or 107 purified CD8 T cells from G9Cα−/−.NOD mice were i.v. injected into 6- to 8-wk-old NOD.scid, NOD.scid-RIP-CD80, or NOD.scid-RIP-CD86-transgenic mice.
Murine islet isolation and transplantation
Perfusion of murine pancreas followed an adapted version based on a modification of the method established by Gotoh et al. (36). In brief, pancreata from NOD.scid, NOD.scid-RIP-CD80, NOD.scid-RIP-CD86, and diabetic transgenic mice were perfused with 2-3 ml of collagenase P (1 mg/ml; Boehringer Mannheim). Bile ducts were clamped off at the duodenal insertion to allow cannulation and specific perfusion to the pancreas. Perfused pancreata were digested for a batch-specific optimized time and collagenase activity was stopped with ice-cold HBSS, 10 mM HEPES, 5% FCS (Invitrogen Life Technologies). Digested pancreata were passed through a ∼100-μm strainer and centrifuged through layers of 25, 23, 20, and 11% Ficoll in HBSS-10 mM HEPES. Islets were collected at the interface between layers and for in vitro assays were handpicked under a dissecting microscope into RPMI complete medium (RPMI 1640-5% FCS, 50 μM 2-ME, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin; Invitrogen Life Technologies). When used for culture, the islets were irradiated with 4200 rad. For extraction of islet-infiltrating cells, the islets were trypsinized and filtered through a 70-μm cell strainer before flow cytometric analysis.
Flow cytometry
Single-cell suspensions of 0.1-1 × 106 cells were stained for various cell surface markers (rat anti-murine CD8-allophycocyanin; CD25, CD62L, CD95L, 4-1BB, ICOS, and PD-1-PE; CD107a and CD95-FITC). FcRs were blocked with anti-CD16 (2.4G2, provided by the late C. A. Janeway) and cells were stained for surface markers and costimulatory molecules. Intracellular staining was performed for CTLA-4 after cell surface staining following BD Pharmingen’s protocol. Intracellular staining for FoxP3 was performed using a FoxP3 staining kit (eBioscience/Insight Technology). The samples were acquired by flow cytometry using a FACSCalibur, results were analyzed using FlowJo software. All Abs had been previously titrated to determine optimal concentration and all were purchased from BD Pharmingen, except anti-murine CD8-allophycocyanin (Caltag Laboratories).
Analysis of cytokine secretion in T cell cultures
Supernatants from cell stimulation assays were harvested and analyzed for the presence of secreted cytokines using a multiplex fluorescent bead immunoassay from Bender Medsystems (Caltag Laboratories) (mouse Th1/Th2 cytokines: IFN-γ, IL-1α, IL-2, IL-4, IL-5, IL-6, IL-10, IL-17, TNF-α, and GM-CSF). Measurements were performed as described in the manufacturer’s instructions. Data were analyzed using BMS FlowCytomix Software, which was based in WinMDI and Microsoft Excel.
Proliferation, cytokine secretion, and cytotoxicity assays
Insulin-reactive CD8 T cells (3 × 105) from 5- to 8-wk-old G9Cα−/−.NOD mice were cocultured with 25 irradiated islets from NOD.scid, NOD.scid-RIP-CD80, or NOD.scid-RIP-CD86 mice in RPMI complete medium. CD8 T cells were purified from G9Cα−/−.NOD splenocytes using positive selection beads (Miltenyi Biotec) with >95% purities. Islets and CD8 T cells alone were used as controls. After 2 days of incubation, supernatants were taken for cytokine secretion analysis and cells were either pulsed with [3H]thymidine for 14 h to test proliferation or were used in cytotoxicity assays. Stimulated CD8 T cells were harvested and further cocultured with 104 51Cr-sodium chromate (Amersham) labeled P815 cells together with insulin peptide (B15-23) at an E:T ratio of 10:1 for 16 h. Specific lysis was calculated as ((cytotoxic release – min)/(max – min)) × 100%, where the minimal release (min) corresponds to the spontaneous lysis, and the maximal lysis corresponds to lysis induced by addition of hydrochloric acid (max).
CD4+CD25+ T cell depletion
CD25+ T cells were depleted using the CD25 depletion kit (Miltenyi Biotec) according to the manufacturer’s instructions. The depletion removed all the CD4+CD25high cells and <5% of the low-staining CD4+CD25+ cells remained after the procedure.
Statistical analysis
Statistical analysis was performed as stated using Graphpad Prism software. Analyses of Kaplan-Meier survival curves used a log-rank test similar to a Mantel-Haenszel test. Fisher’s exact tests were performed for insulitis comparisons. For both tests, Bonferroni corrections were made for multiple comparisons.
Results
Transgene expression is similar in NOD-RIP-CD86 and NOD-RIP-CD80 mice
A comparative study of spontaneous diabetes incidence was performed using NOD-RIP-CD80 and two founder lines of NOD-RIP-CD86-transgenic mice. Ab staining of pancreatic sections for the presence of transgenes was specific in the appropriate mice and demonstrated similar levels of staining in both NOD-RIP-CD80 and NOD-RIP-CD86 mice (Fig. 1A). A 2-fold limiting dilution PCR of NOD-RIP-CD80 and NOD-RIP-CD86 cDNA demonstrated comparatively similar levels of expression for the 880- and 530-bp products, respectively, related to the control products of the housekeeping gene GAPDH (Fig. 1, B and C).
FIGURE 1.
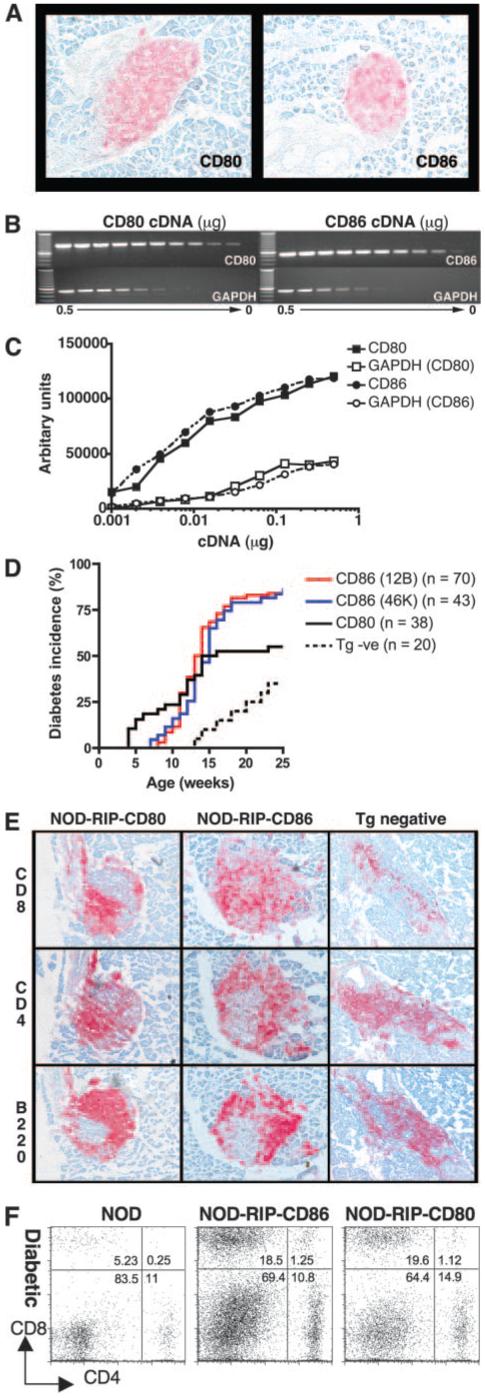
Accelerated diabetes in NOD-RIP-CD80 and NOD-RIP-CD86 mice. A, Expression of CD80 or CD86 was confirmed at 6-wk old by immunohistochemistry on frozen sections. Sections shown are representative of three individual mice per strain. B, Comparative expression of CD80 and CD86 levels was tested using PCR amplification of 2-fold dilutions of cDNA (0.5-9.8 × 10−4 μg) extracted from islets isolated from eight mice per strain. GAPDH was used as a housekeeping control. C, Densitometry confirmed similar expression of CD80 and CD86 expression. D, Male and female NOD-RIP-CD80, NOD-RIP-CD86 (12B and 46K), and transgene-negative mice were monitored weekly for diabetes onset by testing for glycosuria and confirmed by testing blood glucose levels (>14 mM/L). Log-rank tests showed no statistically significant difference between 12B and 46K mice (p = 0.28) but significant differences were found between NOD-RIP-CD80 and NOD-RIP-CD86 (12B and 46K) mice (p = 0.002). E, Islet infiltrates from diabetic NOD-RIP-CD80, NOD-RIP-CD86, and transgene (Tg) negative mice were tested for the presence of CD8, CD4, and B cells by immunohistochemistry on frozen sections. F, Islet-infiltrating cells isolated from diabetic NOD, NOD-RIP-CD80, and NOD-RIP-CD86 islets were stained for CD8 and CD4. Cells were extracted from islets pooled from three mice per group.
NOD-RIP-CD86 mice develop accelerated diabetes but less rapidly than NOD-RIP-CD80 mice
A previous report demonstrated no difference in the onset of autoimmune diabetes in two founder lines of NOD-RIP-CD80-transgenic mice with differing levels of CD80 expression or a difference between genders (29). Similar to NOD-RIP-CD80 mice, no gender difference in the onset of diabetes was observed between the medium (12B) and high (46K) expressing founder lines of NOD-RIP-CD86-transgenic mice (data not shown). Combined male and female diabetes incidence curves of NOD-RIP-CD86 12B and 46K-transgenic mice showed comparable results with an onset of diabetes from 7 to 8 wk of age, 50% diabetes incidence between 13 and 14 wk and >80% diabetes incidence by 20 wk of age. There was no statistically significant difference in the diabetes incidence between the two lines of NOD-RIP-CD86 (Fig. 1D). In contrast, NOD-RIP-CD80 mice developed diabetes from an earlier age of 4 wk, 50% were diabetic by 13 wk and at 20 wk of age diabetes incidence was 58% (Fig. 1D), which was similar to previously published work (29, 30). In addition, at the point of diabetes onset in NOD-RIP-CD86-transgenic mice, 15% of NOD-RIP-CD80-transgenic mice had become diabetic (Fig. 1D). Log-rank tests comparing NOD-RIP-CD80 and combined 46K and 12B NOD-RIP-CD86 incidence curves showed that diabetes incidence was significantly different (p = 0.002). Both NOD-RIP-CD80 and NOD-RIP-CD86 mice had accelerated onset of diabetes compared with 13-wk-old transgene-negative mice.
Expression of RIP-CD80 and RIP-CD86 and acceleration of autoimmune diabetes was associated with greater levels of islet infiltration (Table I) and an increased ratio of CD8 T cells within the islets (Fig. 1, E and F). At 6 wk of age, islet infiltration was most pronounced in NOD-RIP-CD80 mice, where ∼50% of islets had high levels of insulitis (scores 2-4), compared with 10% in NOD-RIP-CD86 mice and only low levels (scores 0-1) of infiltration were observed in transgene-negative mice (Table I). By 12 wk of age, the degree of insulitis in NOD-RIP-CD80 and NOD-RIP-CD86 islets was comparable but more insulitis was observed compared with transgene-negative mice. Islet infiltration was similar in all strains after diabetes development (Table I). Islet infiltrates in both diabetic NOD-RIP-CD80 and NOD-RIP-CD86 mice comprised a high proportion of CD8 T cells compared with transgene-negative mice, while the proportion of CD4 T cells and B cells was comparable (Fig. 1E). This was confirmed by flow cytometry showing that isolated islet infiltrates from diabetic mice demonstrated a 3-fold increase in the proportion of CD8 T cells from NOD-RIP-CD80 and NOD-RIP-CD86 mice compared with transgene-negative mice (Fig. 1F).
Table I.
Lymphocytic infiltration of NOD-RIP-CD80, NOD-RIP-CD86, and transgene-negative micea
| 6 wk |
12 wk |
Diabetic |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Score | CD80 | CD86 | Tg negative | CD80 | CD86 | Tg negative | CD80 | CD86 | Tg negative |
| 0 | 20.0 | 73.3 | 98.1 | 20.4 | 25.0 | 32.4 | 0.0 | 0.0 | 0.0 |
| 1 | 30.4 | 17.2 | 1.9 | 12.2 | 8.0 | 18.3 | 0.6 | 0.0 | 2.9 |
| 2 | 34.8 | 7.8 | 0.0 | 41.5 | 23.5 | 27.5 | 9.7 | 20.2 | 26.5 |
| 3 | 12.2 | 1.7 | 0.0 | 17.7 | 29.5 | 16.9 | 39.2 | 33.7 | 38.2 |
| 4 | 2.6 | 0.0 | 0.0 | 8.2 | 14.0 | 4.9 | 50.6 | 46.1 | 32.3 |
Insulitis in the islets of the different strains was examined at 6 wk of age where scores were grouped into low (scores 0 and 1) and high (scores 2-4) levels of insulitis and compared by Fisher’s exact test. One hundred percent of islets from transgene-negative mice had low levels of infiltration compared to 90 and 50% in NOD-RIP-CD86 and NOD-RIP-CD80 islets, respectively. Values of p ≤ 0.0009 for all three comparisons. The difference in NOD-RIP-CD80 and NOD-RIP-CD86 islet infiltration compared to transgene negative mice was statistically different by Fisher’s exact test (p = 0.002 and 0.0012, respectively). Similar degrees of infiltration between NOD-RIP-CD80 and NOD-RIP-CD86 mice at 12 wk corresponded with the convergence of diabetes incidence in these mice at 12 wk of age. In diabetic mice, there was no difference in degree of insulitis between all strains, where >97% of all islets had high levels of infiltration (p > 0.3 for all comparisons).
Recurrent diabetes occurs at a faster rate in diabetic NOD-RIP-CD86 mice compared with diabetic NOD-RIP-CD80 mice transplanted with healthy NOD islets
We tested secondary responses of cells from diabetic NOD, NOD-RIP-CD86, and NOD-RIP-CD80 mice in islet transplant experiments. Islets from 6-wk-old NOD mice were transplanted into diabetic NOD, NOD-RIP-CD86, and NOD-RIP-CD80 recipient mice under the kidney capsule. The recipients of the transplants became euglycemic by 48 h. Infiltration and destruction of the transplanted islets recurred with a reversed pattern of onset compared with the natural history experiments (Fig. 2A). Diabetes recurred in NOD mice between 7 and 12 days post-islet transplantation and in NOD-RIP-CD86 mice between 17 and 18 days post-islet transplantation. In contrast, diabetes recurred in NOD-RIP-CD80 mice from 25 days posttransplantation and one recipient even survived for 45 days posttransplantation (Fig. 2A). This suggested that the cells had been “tuned” such that cells from RIP-CD80 mice were less effective than those from RIP-CD86 mice, which in turn were less effective than cells from the NOD mice. Curiously, when transplanted islets were examined for the presence of CD8, CD4, and B cells, a marked reduction in the CD8:CD4 T cell ratio was observed in NOD-RIP-CD80-transplanted mice (Fig. 2B). The relative number of infiltrating CD4 T cells was at least double compared with infiltrating CD8 T cells in the NOD-RIP-CD80-transplanted mice. In contrast, both NOD and NOD-RIP-CD86 diabetic mice had similar proportions of infiltrating CD8 and CD4 T cells although the NOD-RIP-CD86 islets had a greater total infiltrate. B cells followed a similar pattern of infiltration into islet transplants of diabetic NOD and NOD-RIP-CD86 mice.
FIGURE 2.
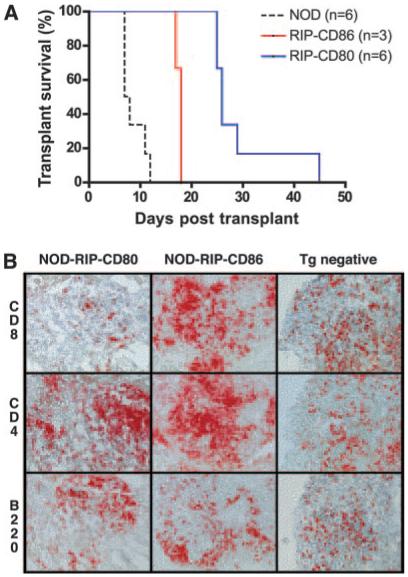
Diabetes recurred more slowly in diabetic NOD-RIP-CD80 mice. A, Transplant survival was monitored in diabetic NOD, NOD-RIP-CD80, and NOD-RIP-CD86 mice in which islets from 6-wk-old NOD mice were transplanted under the kidney capsule and rendered euglycemic after 48 h. The difference in time of development of recurrent diabetes was statistically significant (p < 0.05). B, Cellular phenotypes infiltrating transplanted islets in diabetic NOD (Tg negative (−ve)), NOD-RIP-CD80, and NOD-RIP-CD86 were examined by immunohistochemistry on frozen sections.
CD8 T cells from diabetic NOD-RIP-CD86 mice have a greater ability to transfer diabetes compared with CD8 T cells from diabetic NOD-RIP-CD80 mice
Previous studies have shown that splenocytes from diabetic NOD-RIP-CD80 mice have a reduced capacity to transfer diabetes to NOD.scid recipients (F. S. Wong, unpublished data and Ref. 25). In this study, we compared the ability of splenocytes from diabetic NOD-RIP-CD86 and NOD-RIP-CD80 mice to transfer diabetes to NOD.scid recipients. Eighty-nine percent of recipient mice developed diabetes after transfer of NOD-RIP-CD86 diabetic splenocytes. In striking contrast, only 24% of NOD.scid recipient mice transferred with splenocytes from diabetic NOD-RIP-CD80 mice developed diabetes within the 16-wk (112 day) experimental period (Fig. 3A). As expected, 100% of NOD.scid mice transferred with splenocytes from diabetic NOD mice developed diabetes. Additionally, a marked difference in the onset of diabetes was observed, with recipients of NOD diabetic splenocytes developing diabetes 20 days posttransfer, while onset occurred at 43 and 56 days following transfer with NOD-RIP-CD86 and NOD-RIP-CD80 diabetic splenocytes, respectively (Fig. 3A). There was no correlation between the age at which the mice became diabetic and their ability to transfer diabetes, although in general, the diabetic NOD-RIP-CD80 mice were younger (4-10 wk) than the diabetic NOD-RIP-CD86 mice (8-12 wk), because they developed earlier diabetes.
FIGURE 3.
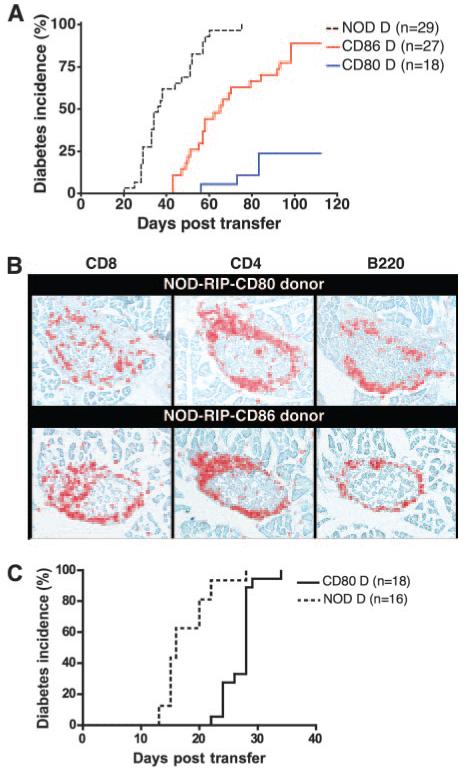
Cells from diabetic NOD-RIP-CD80 mice adoptively transfer diabetes with reduced efficiency to NOD.scid mice, which can be overcome by local expression of costimulatory molecules. A, Diabetes incidence in NOD.scid recipients following adoptive transfer of 20 × 106 splenocytes from diabetic NOD (Tg negative (−ve)), NOD-RIP-CD80, or NOD-RIP-CD86 mice. Mice were tested for diabetes every 2 days. The difference in incidence was statistically significant (p < 0.0001). B, Islet infiltration in nondiabetic NOD.scid mice 16-wk postadoptive transfer of splenocytes from diabetic NOD-RIP-CD80 (top panel) or NOD-RIP-CD86 (bottom panel) mice. The sections were stained for CD4 and CD8 T cells and for B cells (B220). C, Diabetes incidence in NOD.scid-RIP-CD80 recipients following adoptive transfer of 20 × 106 splenocytes from diabetic NOD-RIP-CD80 mice. Mice were tested every 2 days.
The reduced ability of NOD-RIP-CD80 diabetic splenocytes to adoptively transfer diabetes was not due to an inability to migrate to pancreatic islets. Peri-insulitis consisting of CD8 T cells, CD4 T cells, and B cells was observed in the islets of nondiabetic recipient NOD.scid mice at 112 days posttransfer with NOD-RIP-CD80 or NOD-RIP-CD86 diabetic splenocytes (Fig. 3B). Clearly, transferred splenocytes from diabetic NOD-RIP-CD80 mice were capable of reaching the islets and when further costimulation was provided by transfer into NOD.scid-RIP-CD80 recipients, these cells were capable of adoptively transferring diabetes in 100% of recipients (Fig. 3C). Thus, cells costimulated by CD80 were CD80 dependent whereas those stimulated by CD86 were not.
T cell development does not play a role in the difference in T cell activation by CD86 or CD80
To test that the different outcomes following adoptive transfer were not related to differences in T cell development in the thymus, we used adoptive transfer of splenocytes from 6-wk-old NOD mice into NOD.scid-RIP-CD86 and NOD.scid-RIP-CD80 mice and tested for their ability to develop diabetes (Fig. 4A). A similar pattern of development of diabetes compared with the natural history of diabetes in NOD-RIP-CD86 and NOD-RIP-CD80 mice was observed. One hundred percent of NOD.scid-RIP-CD80 recipients had developed diabetes by 40 days posttransfer, while the earliest time of diabetes onset occurred 45 and 54 days in NOD.scid-RIP-CD86 and NOD.scid mice, respectively. By 70 days after transfer, 90% of NOD.scid-RIP-CD86 recipients had developed diabetes whereas only 45% of NOD.scid recipients had developed disease at this point (Fig. 4A). Thus, this experiment removed the possibility that any differences seen in diabetes onset in the NOD-RIP-CD86 or NOD-RIP-CD80 mice related to expression of the transgenes in the thymus and hence to development of diabetogenic T cells.
FIGURE 4.
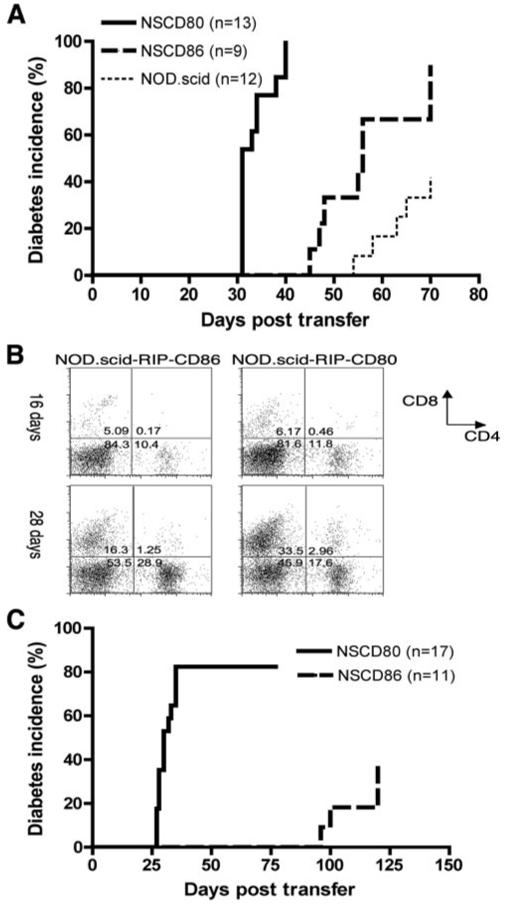
Accelerated diabetes in RIP-CD80 and RIP-CD86 mice is not due to differences in thymic T cell development. A, Adoptive transfer of diabetes into NOD.scid, NOD.scid-RIP-CD80, and NOD.scid-RIP-CD86 mice showed a similar pattern of diabetes development to the natural history of diabetes in NOD, NOD-RIP-CD80, and NOD-RIP-CD86 mice. A total of 20 × 106 splenocytes from 6-wk-old mice were transferred into NOD.scid, NOD.scid-RIP-CD80 (NSCD80), and NOD.scid-RIP-CD86 (NSCD86) mice and monitored weekly for diabetes development. The difference in the time taken to develop diabetes and the final incidence of diabetes was statistically significant (p < 0.0001). B, Islet expression of CD80 promotes increased islet infiltration of CD8 T cells in NOD.scid-RIP-CD80 recipients over time. Islet-infiltrating cells isolated from NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 islets after transfer with splenocytes from 6-wk-old mice were stained for CD8 and CD4 T cells. Top panels, Dot plots for cells from 16 days posttransfer; bottom panels, 28 days posttransfer. Islets from three mice per group were used. C, Purified insulin-specific CD8 T cells cause increased incidence of diabetes in NOD.scid-RIP-CD80 recipients compared with NOD.scid-RIP-CD86 recipients. Purified insulin-specific CD8 T cells (107) (>95% pure) were transferred into NOD.scid-RIP-CD80 (NSCD80) recipients compared with NOD.scid-RIP-CD86 (NSCD86) recipients. The difference in the time taken to develop diabetes and the final incidence of diabetes was statistically significant (p < 0.0001). No diabetes is seen when these cells are transferred to NOD.scid recipients (n = 10, data not shown).
Additionally, greater expansion (or recruitment) of CD8 T cells in islet infiltrates was observed in NOD.scid-RIP-CD80 recipients transferred with spleen cells from 6-wk-old NOD mice (Fig. 4B). Islet infiltration was similar at 16 days post-transfer, where ∼5-6% CD8 and 10-11% CD4 T cells were observed (Fig. 4B). By 28 days, the proportion of CD8 T cells in NOD-scid-RIP-CD80 recipients had risen to >30%, double that compared with NOD.scid.RIP-CD86 recipients. Conversely, the level of CD4 T cells was increased in the NOD.scid-RIP-CD86 recipient mice (Fig. 4B). Early diabetes onset, increased islet infiltration, and greater numbers of CD8 T cells all suggest that islet expression of CD80 and CD86 enables specific activation of CD8 T cells. The fact that these observations occur at an earlier time point in NOD-RIP-CD80 mice strongly indicates that activation of CD8 T cells through CD80 generates enhanced responses, compared with CD86.
To demonstrate that the expression of CD80 or CD86 could differentially stimulate CD8 T cells directly in our model system, rather than mainly through an effect on CD4 T cells, we adoptively transferred purified insulin-specific CD8 T cells directly ex vivo from the splenocytes of G9Cα−/−. NOD-transgenic mice into NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 recipients. We showed that the naive CD8 insulin-reactive T cells transfer diabetes more rapidly and with a higher final incidence to NOD.scid-RIP-CD80 recipients compared with NOD.scid.RIP-CD86 recipients shown in Fig. 4C.
In vitro stimulation of lymphocytes with islets from NOD.scid-RIP-CD80 mice induces greater proliferation and cytokine production than islets from NOD.scid-RIP-CD86 mice and nontransgenic mice
To test the islet reactivity of T cells of known specificity, CD8 T cells were isolated from insulin-specific CD8 TCR (G9.Cα−/−) transgenic mice and in vitro assays were performed using isolated islets as both a source of CD8 T cell-specific APCs as well as insulin peptides. Proliferation assays clearly demonstrated that stimulation of insulin-specific CD8 T cells using islets from NOD.scid-RIP-CD80 mice generated heightened activity, ∼4-fold greater compared with proliferation to NOD.scid islets (Fig. 5A). NOD.scid-RIP-CD86 islets also demonstrated enhanced stimulation, albeit to a lesser extent, where proliferation was ∼3-fold greater than the proliferation to NOD.scid islets (Fig. 5A). Additionally, it was observed that stimulation of insulin-reactive CD8 T cells with CD80 expressing islets produced increasing levels of IL-2 and IFN-γ secretion over time, while CD86 costimulation, under these conditions, generated very low levels of IL-2 and negligible amounts of IFN-γ (Fig. 5B). In addition, the production of cytokines IL-1α, IL-4, IL-5, IL-6, IL-10, IL-17, TNF-α, and GM-CSF was also tested but these were not detected (data not shown). Higher levels of CD25 and reduced expression of CD62L on CD8 T cells activated by NOD.scid-RIP-CD80 islets compared with NOD.scid-RIP-CD86 islets reflected enhanced T cell activation through CD80 (Fig. 5C).
FIGURE 5.
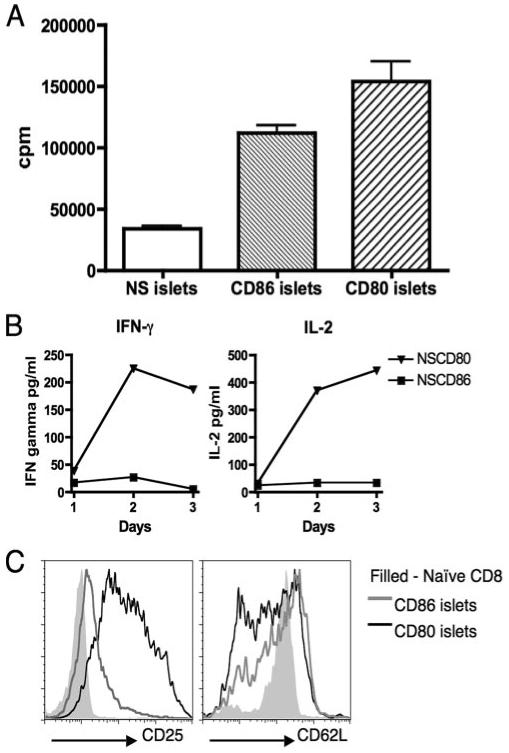
In vitro activation of insulin-reactive CD8 T cells by CD80 results in enhanced proliferation, cytokine production, and change in expression of activation markers compared with CD86 stimulation. A, Naive insulin-reactive CD8 T cells proliferated to islets from all strains, but CD80 costimulation induced the highest level of proliferation in a 3-day culture, followed by CD86 stimulation. B, Levels of IL-2 and IFN-γ secretion were measured in supernatants taken from proliferation assays of insulin-reactive CD8 T cells cultured with NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 islets. C, Cell surface expression of activation markers CD25 and CD62L were measured 2 days after insulin-reactive CD8 T cells were activated by both NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 islets. Filled histograms represent naive ex vivo insulin-reactive CD8 T cells. Results are representative of three similar assays.
In vitro stimulation of lymphocytes with islets from NOD.scid-RIP-CD80 mice alters the threshold of activation of cytotoxic responses compared with islets from NOD.scid-RIP-CD86 mice and nontransgenic mice
The cytotoxicity of insulin-reactive CD8 T cells after activation with islets from the different transgenic strains showed that CD80 costimulation induced greater levels of effector function with specific target cell lysis reaching a maximum of 68%, compared with 34 and 31% for CD86 and NOD.scid stimulated cells, respectively (Fig. 6A). Cells stimulated by both CD80 and CD86 respond to lower peptide concentrations, but to a much greater degree in CD80-costimulated CD8 T cells (Fig. 6A), indicating lowering of activation threshold.
FIGURE 6.
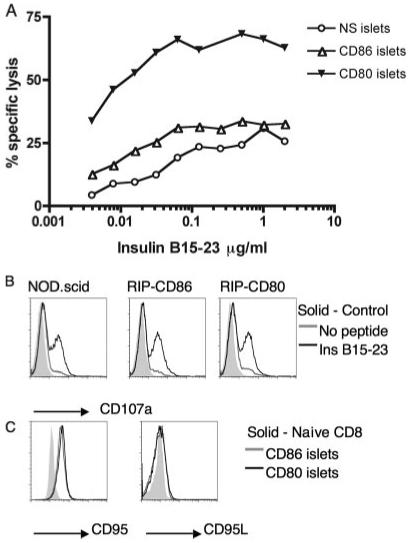
Stimulation of insulin-reactive CD8 T cells by CD80 produces cells with greater cytotoxic effects compared with stimulation through CD86. A, Cytotoxicity of insulin-reactive CD8 T cells prestimulated with NOD.scid (NS), NOD.scid-RIP-CD80, or NOD.scid-RIP-CD86 islets was measured as specific lysis of insulin B15-23-pulsed 51Cr-labeled P815 cells. B, Levels of cell surface CD107a was measured on prestimulated insulin-reactive CD8 T cells in response to P815 cells in the presence or absence of insulin B15-23. C, Expression levels of death receptor molecules CD95 and CD95L were monitored after activation of insulin-reactive CD8 T cells with islets from NOD.scid-RIP-CD80 or NOD.scid-RIP-CD86 mice. Results are representative of two similar assays.
With insulin peptide at 3.9 × 10−3μg/ml, CD8 T cells prestimulated with CD80 islets caused ∼34% specific lysis, at which point specific lysis was only 12 and 4% by CD86 and NOD.scid prestimulated CD8 T cells, respectively. For CD86 prestimulated CD8 T cells to cause the same level of cytotoxic killing, 0.5 μg/ml peptide was required (Fig. 6A). Thus, >100 times more peptide was required for CD86-stimulated cells to cause the same level of cytotoxicity as CD80-stimulated cells.
Furthermore, the results demonstrate that specific cell lysis could be perforin/granzyme or death receptor mediated as increased levels of CD107a were found on CD80-prestimulated CD8 T cells following coculture with insulin peptide-coated P815 cells (Fig. 6B) as well as elevated expression of CD95 on islet-activated CD8 T cells, although the overall level of CD95L was low (Fig. 6C).
CD86 and CD80 both stimulate up-regulation of CTLA-4 and PD-1
Although CD86 did not stimulate CD8 T cells to the same extent as CD80 in terms of increased proliferation, cytotoxicity, and increased diabetogenicity, we also did not observe the inhibition seen upon secondary stimulation. We therefore tested expression of inhibitory costimulatory ligands. We examined the expression of CTLA-4 in cells infiltrating islets from adoptively transferred diabetic NOD.scid-RIP-CD86 mice and found that the inhibitory coreceptor CTLA-4 was increased but less than with CD80 stimulation (Fig. 7A). Additionally, PD-1 was also expressed in islet infiltrates from NOD.scid-RIP-CD86 and NOD.scid-RIP-CD80 mice transferred with splenocytes from 6-wk-old NOD mice (Fig. 7B). Sixteen days posttransfer, CD8 T cells isolated from NOD.scid-RIP-CD80 mice expressed higher levels of PD-1 but by 28 days expression was similar in CD8 T cells isolated from both strains (Fig. 7B). Other costimulatory ligands that include 4-1BB and ICOS were studied but they were not detected (data not shown). We tested the effect of blocking PD-1 and CTLA-4 on stimulation of in vitro cytotoxicity using CD107a assays. However, we did not demonstrate any differences in generation of in vitro cytotoxicity in these assays.
FIGURE 7.
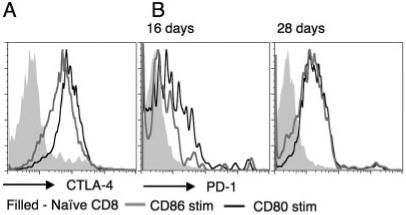
CD80 and CD86 stimulation of CD8 T cells results in upregulation of inhibitory coreceptors CTLA-4 and PD-1 but CD80 stimulation causes earlier and greater expression levels. A, Intracellular CTLA-4 expression was examined following in vitro activation of insulin-reactive CD8 T cells by NOD.scid-RIP-CD80 or NOD.scid-RIP-CD86 islets. B, Cell surface expression of PD-1 was studied on islet infiltrate CD8 T cells following transfer of splenocytes from 6-wk-old NOD mice into NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 mice 16 and 28 days posttransfer. Filled histograms represent naive ex vivo CD8 T cells (A and B). Results are representative of two similar assays.
Depletion of CD4+CD25+ cells facilitates adoptive transfer of diabetes by splenocytes from diabetic NOD.scid-RIP-CD80 mice
We had shown earlier (Fig. 3) a reduced ability of splenocytes from diabetic RIP-CD80 mice to transfer diabetes to secondary NOD.scid recipients. To examine further possible inhibitory mechanisms for this reduced ability to transfer disease, we performed adoptive transfer of spleen cells from 6-wk-old NOD mice into NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 mice, similar to the experiment shown in Fig. 4A. When the mice developed diabetes, the spleen cells were harvested and the expression of CD4+CD25+FoxP3 was examined by flow cytometry in an aliquot of cells. We found that both sets of spleen cells expressed a similar number of CD4+CD25+FoxP3+ cells in the spleen shown in Fig. 8A.
FIGURE 8.
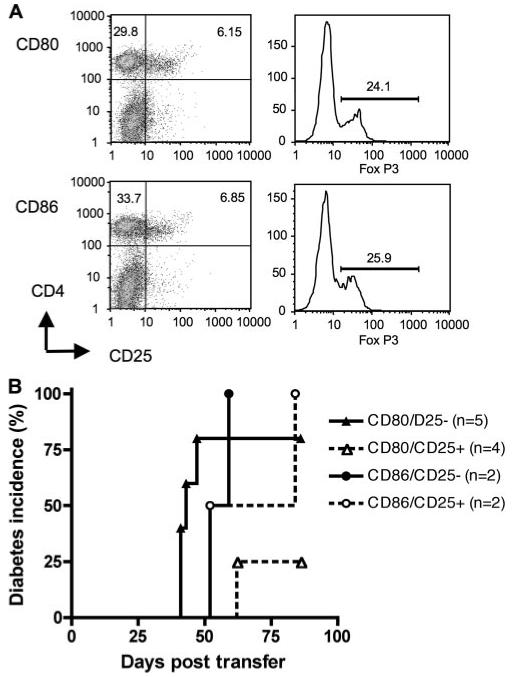
CD4+CD25+ depletion of CD80-stimulated splenocytes restores the ability to cause diabetes. A, Splenocytes from 6-wk-old NOD mice were adoptively transferred into NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 mice. When the mice became diabetic, the splenocytes were harvested and stained for CD4+CD25+ and FoxP3+ cells. Top panels, Representative cells from NOD.scid-RIP-CD80 mice; lower panels, cells from NOD.scid-RIP-CD86 mice. B, The splenocytes were depleted of CD4+CD25+ T cells (>80% depleted) and then adoptively transferred to NOD.scid mice which were observed for diabetes. These were compared with cells that had not been depleted of CD4+CD25+ cells. The results shown for the transfer from diabetic NOD.scid-RIP-CD80 mice are combined from two separate experiments.
From these spleen cells in diabetic mice, CD4+CD25+ T cells were depleted immediately, in a proportion of the splenocytes, before secondary adoptive transfer to NOD.scid mice. The depletion process removed all the CD4+CD25high T cells. Spleen cells that were not depleted of CD4+CD25+ cells were used as controls. We found that after depletion of the CD4+CD25+ T cells, splenocytes from diabetic NOD.scid-RIP-CD80 were able to transfer diabetes at a similar rate to cells obtained from diabetic NOD.scid-RIP-CD86 mice (Fig. 8B).
Discussion
Costimulation is essential for the full activation of CD8 T cells. In this study, we have aimed first to study the effect of CD80 (B7.1) and CD86 (B7.2) individually, a feature not possible in natural APCs as both are coexpressed and regulation is interdependent. Second, we are studying cells that respond to an important autoantigen, insulin, that is presented endogenously in the islets. Third, the strength of signal 1 is also an important factor. Using this model, we are able to use the ability of the islet to naturally present the range of Ag concentrations that the cells would encounter in vivo in the islet. The ability of CD8 T cells to cause diabetes is a complex process and our model allows us to test effects on cell expansion, cytokine production, cytotoxicity in vitro and in vivo as well as homing in vivo. We are using diabetes as an in vivo readout to study the individual effects of costimulatory molecules and not the development of natural diabetes, as islet β cells do not normally express costimulatory molecules (37).
In this study, we have demonstrated a clear difference in the activation of CD8 T cells after CD80 or CD86 costimulation. In vivo, we observed accelerated diabetes in RIP-CD80 mice (4 wk) as noted in previous studies (25, 29, 30) as well as in RIP-CD86 mice (7 wk) compared with transgene-negative mice (13 wk). The greater acceleration of diabetes incidence in RIP-CD80 mice was not likely to be due to a difference in expression levels of CD80 compared with CD86. Diabetes development in the medium (12B) and high (46K) expressing NOD-RIP-CD86 mice was almost identical, while previous studies showed a similar result with different expression levels in RIP-CD80 mice (29). Additionally, Ab staining of pancreatic sections from NOD-RIP-CD80 and NOD-RIP-CD86 (12B) had similar protein levels, and PCR products from cDNA isolated from islets of these transgenic mice demonstrated comparable levels of expression. Hence, we infer that accelerated diabetes was due to enhanced stimulation of T cells by CD80 compared with CD86.
The proportion of CD8 T cells in islet infiltrates was increased in diabetic NOD-RIP-CD80 and NOD-RIP-CD86 mice compared with diabetic transgene-negative littermates. However, proportions of B and CD4 T cells were similar in all the diabetic mice examined, which suggested that costimulation and MHC class I on islet β cells specifically activated CD8 T cells. We further demonstrated that CD80 enhanced the proportion of CD8 T cells in islet infiltrates more than CD86 costimulation. NOD.scid-RIP-CD80 and NOD.scid-RIP-CD86 mice i.v. transferred with splenocytes from 6-wk-old NOD mice showed similar proportions of CD8 and CD4 T cells in islet infiltrates after 16 days but after 28 days, twice the CD8 T cells were present in NOD.scid-RIP-CD80 islets compared with NOD.scid-RIP-CD86. This suggested that CD8 T cells were being further activated in situ and hence proliferating in the islets themselves.
Further evidence that the expression of CD80 and CD86 expressed on the islets directly stimulate CD8 T cells was shown by experiments using purified, naive low-avidity CD8 T cells recognizing insulin peptide B15-23. These cells are unable to transfer diabetes to NOD.scid mice unless they have been preactivated (L. K. Siew, F. S. Wong, unpublished observations). However, they cause diabetes in NOD.scid-RIP-CD80 mice with faster kinetics than NOD.scid-RIP-CD86 mice. No CD4 T cells were transferred in these experiments indicating that the expression of the costimulatory molecules on the islets has a direct effect on CD8 T cells.
When the cells from the diabetic NOD-RIP-CD80 and NOD-RIP-CD86-transgenic mice were subjected to a secondary stimulation, CD86-stimulated cells were more effective in causing recurrent diabetes in both NOD islet-transplanted mice and in NOD.scid recipients receiving cells from diabetic NOD-RIP-CD86-transgenic mice. Although the cells from diabetic NOD-RIP-CD80-transgenic mice caused delayed diabetes in NOD.scid recipients or did not transfer diabetes, when the cells from diabetic NOD-RIP-CD80-transgenic mice were retransferred into NOD.scid-RIP-CD80-transgenic mice, diabetes occurred rapidly and in all mice. This indicated that the threshold of activation of the CD80-stimulated cells was altered such that they could respond in the presence of CD80 but only poorly in the absence of the costimulatory molecule.
To examine the effect of the stimulation by islet Ag with either CD80 or 86, we performed in vitro studies with T cells of a defined specificity that are able to react to the islet Ag, insulin. Islets isolated from both NOD.scid-RIP-CD86 and NOD.scid-RIP-CD80 mice induced higher levels of proliferation in naive insulin-reactive CD8 T cells compared with NOD.scid islets but greater proliferation was seen with RIP-CD80 islets. CD8 T cells secreted limited IL-2 and IFN-γ upon stimulation with RIP-CD86 islets, while RIP-CD80 islets induced production of high levels of both cytokines. Other cytokines were not detected and therefore, we infer that the results seen were not due to production of some protective cytokines. The insulin-reactive CD8 T cells stimulated in vitro with RIP-CD80 islets also demonstrated higher levels of cytotoxicity, compared with CD86 stimulation, which in turn was higher than cells stimulated with NOD.scid islets. Additionally, responses were more sensitive in CD8 T cells stimulated with CD80 where ∼50% specific lysis occurred with peptide concentrations at 0.01 μg/ml. In contrast, ∼20 and 10% lysis was observed in CD86-stimulated and unstimulated CD8 T cells, respectively. Our studies also demonstrated increased levels of CD107a and CD95 on the surface of activated insulin-reactive CD8 T cells after stimulation with all sets of islets, suggesting that the cytotoxic effects could be manifested through both the death receptor and perforin/granzyme mechanisms. Even though the levels of CD107a were similar throughout, the cytotoxicity shown by CD80-stimulated cells toward peptide coated targets was much higher. It is conceivable that an increased quantity of perforin and the various granzyme molecules are responsible for activating the caspase pathway within the cytotoxic vesicles of CD80-stimulated cells, although we have no direct evidence for this.
We have clearly shown here that stimulation of naive CD8 T cells in vivo and in vitro by both CD80 and CD86 results in activated cells with a greater potential to proliferate and kill through cytotoxic mechanisms. Previously it has been demonstrated that cells from diabetic NOD-RIP-CD80 mice have an impaired ability to transfer diabetes to NOD.scid mice (Ref. 25 and F. S. Wong, unpublished observations) whereas splenocytes from diabetic nontransgenic NOD mice transfer diabetes to 100% of NOD.scid recipients. We confirm these results in respect of cells derived from diabetic NOD-RIP-CD80 mice. In addition, however, we show here although the primary stimulation in the presence of CD86 is not as great as with CD80, the CD86-stimulated cells retain the ability to kill targets and cause diabetes in the absence of continued costimulation, and nearly 90% of recipients become diabetic following adoptive transfer of splenocytes from diabetic NOD-RIP-CD86 mice. Thus, although activation through CD80 results in enhanced proliferation and cytotoxicity in a primary response, the secondary responses are markedly reduced. This was further demonstrated in diabetic NOD, NOD-RIP-CD80, and NOD-RIP-CD86 mice transplanted with islets from 6-wk-old mice, in which grafts transplanted into diabetic NOD-RIP-CD86 mice were destroyed faster compared with the longest survival in NOD-RIP-CD80 mice.
The reduced efficacy of CD8 T cells previously stimulated with CD80 compared with CD86 makes CD86 overall a more effective costimulator. It is likely that more than one mechanism is involved in the reduced secondary stimulation of CD8 T cells by CD80 in this in vivo model. These could include:
Cells previously stimulated by CD80 are not able to migrate to the relevant site (healthy islets), although this is unlikely because CD8, CD4, and B cells were all present in peri-islet infiltrates in nondiabetic transfer recipients at 10 wk. Similarly, in the transplant experiments, lymphocytes were present as infiltrates in the islet grafts. However, in the transplants, the proportion of CD8 T cells was greatly reduced in NOD-RIP-CD80 recipients. The fact that the cells were able to migrate to the islets but not penetrate and destroy the islet cells suggests that they were not sufficiently activated by the islet cells that did not express the costimulatory molecules.
Stronger stimulation of CD8 T cells by CD80 induces islet reactive cells to die by activation-induced cell death. However, after in vitro stimulation with islets expressing CD80 or CD86, survival of the cells is similar as shown by similar cell numbers and annexin V staining (data not shown). Furthermore, following in vitro stimulation with islets and then a short maintenance period over 3 days in IL-2, a greater proportion of CD80 prestimulated CD8 T cells remained viable (data not shown).
A possible explanation behind the reduced efficacy of CD80-stimulated CD8 T cells is increased and sustained expression of inhibitory coreceptors, such as CTLA-4 and PD-1, although CD86 also up-regulated these inhibitory receptors. It is well known that CTLA-4 is up-regulated on activation and interacts with CD80 and cells are inhibited as a result of this interaction. However, PD-1 up-regulation has not previously been linked to cells activated by CD80. In this study, we have demonstrated that in vivo, PD-1 is up-regulated on the cell surface of CD8 T cells stimulated by CD80 and CD86 but higher expression is evident at an earlier time in CD80-stimulated CD8 T cells. Additionally, intracellular expression of CTLA-4 is apparent in insulin-reactive CD8 T cells at a greater level after activation through CD80 stimulation compared with CD86 stimulation. We propose that after the initial aggressive activation of CD8 T cells by CD80, the T cells up-regulate inhibitory coreceptors to a greater degree and potentially for a longer period of time, compared with CD86 stimulation. This would enable T cells to be “turned off” and, depending on the duration of inhibitory coreceptor expression, prevent or reduce the possibility of a secondary stimulation. This would most likely be the case when peptides of interest are displayed at endogenous concentrations, such as in the studies presented here. However, by providing increased levels of costimulation, this inhibition can be overcome. We performed in vitro blocking experiments using CD8 cells stimulated by RIP-CD80 and RIP-CD86 islets with Abs to block CTLA-4 or PD-1 and tested in vitro cytotoxicity measuring CD107a but did not observe consistent alteration in the in vitro responses (data not shown). Clearly, it would be interesting to monitor the kinetics of inhibitory receptor expression before, during, and after primary and secondary activation of CD8 T cells through CD80 or CD86 stimulation in vivo. Most recently, it has been shown that in addition to binding to the known inhibitory ligand, CTLA-4 on T cells, B7-1 is also able to directly bind to PD-L1 and deliver an inhibitory signal (38). This interaction could also explain the inhibition observed after the initial activation. It would certainly be of importance also to test for the up-regulation of PD-L1 and the effect of blocking this interaction in this system.
A further possibility for the reduced ability of CD80-stimulated cells to cause diabetes in secondary adoptive transfer could be an increase in regulatory T cell inhibition of cells stimulated with CD80 compared with CD86. We measured the percentage of CD4+CD25+ cells in the splenocytes of mice that had become diabetic after the primary transfer of 6-wk-old NOD splenocytes into NOD.scid-RIP-CD86 and NOD.scid-RIP-CD80 mice. The percentage of these cells in the splenocytes was not different between the two types of mice. However, following depletion of the CD4+CD25+ T cells, the cells from the diabetic NOD.scid-RIP-CD80 mice were able to transfer diabetes with similar kinetics to cells from diabetic NOD.scid-RIP-CD86 mice. Thus, this suggests that either an increase in the levels of CD4+CD25+ T cells in the diabetic RIP-CD80 splenocytes were inhibiting the CD80-stimulated CD8 T cells (our evidence did not support this), or the CD8 cells were more susceptible to inhibition by these regulatory cells. Removing the CD4+CD25+ T cells facilitated the secondary adoptive transfer of diabetes by the CD80-stimulated splenocytes.
Finally, the use of ectopic expression of costimulatory molecules to boost immune responses is a strategy that has been used for modulating immune responses to tumors. The fact that CD80 stimulates stronger activating responses also has an impact in vivo. The increased stimulation of cells with more rapid responses and expansion of CD8 T cells that is not maintained if the costimulatory molecule is removed may also suggest that more cells are initially recruited by islets expressing CD80 as previously suggested by Allison et al. (25). Diabetes in the NOD mouse is caused by a heterogeneous group of cells and the ability of CD80 to lower the threshold of activation of lower avidity cells could allow for recruitment of these lower avidity cells to the islet and in situ expansion of cells. However, the low-avidity cells, although stimulated to full activation in response to CD80 stimulation, are not able to maintain the level of activation important for cytotoxicity in the absence of costimulatory molecules. Thus, on encounter of low levels of Ag in the absence of the costimulatory molecule, such as in the case of transplantation with islets that do not express CD80, or in secondary transfer when the recipients do not express CD80, the low-avidity CD8 T cells are unable to further contribute to the disease process. Our results also suggest that these CD80-stimulated T cells are more susceptible to inhibition by CD4+CD25+ regulatory T cells. This may further imply that the islet destructive repertoire is different in the RIP-CD80 mice compared with the RIP-CD86 mice and nontransgenic NOD mice.
In conclusion, our studies have shown that when CD80 and CD86 stimulation is separated and tested in vivo, both CD80 and CD86 can stimulate activating and inhibitory responses but CD80 induces, overall, stronger responses. Thus, CD80 targeted locally may be useful in the context where CD80 remains expressed and continues to be active (39) to stimulate lower avidity T cells and overcome inhibition by CD4+CD25+ T cells. Although once activated, effector cells may not require continued costimulation under all circumstances, this depends on continued lower threshold of activation and is likely also to be dependent on the level of Ag expression. Overall, while CD86 does not stimulate an initial response as strongly as CD80, there is greater sustained activity that is seen, even in the absence of continued costimulation. These functions have implications for the engineered use of costimulatory molecules in altering immune responses in a therapeutic setting.
Acknowledgments
We thank Andrew Herman for assistance in flow cytometry, Irene Visintin for advice on immunohistochemistry, Stephen Chapman for excellent technical assistance, and Gwen Scott for assistance at the end stages of the experiments. The original RIP-CD80, and RIP-CD86 mice were generated by the Yale Diabetes Endocrinology Research Center Transgenic Core DK45735.
Footnotes
I.J.T. was supported by a Wellcome Trust Prize Studentship, R.A.F. is an investigator of the Howard Hughes Medical Institute, and F.S.W. was a Wellcome Trust Senior Fellow in Clinical Science.
- PD-1
- programmed death-1
- RIP
- rat insulin promoter
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu. Rev. Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 2.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 3.Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, Damle NK. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J. Exp. Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 5.Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, Davis SJ. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 6.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 7.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat. Rev. Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 8.Beier KC, Hutloff A, Dittrich AM, Heuck C, Rauch A, Buchner K, Ludewig B, Ochs HD, Mages HW, Kroczek RA. Induction, binding specificity and function of human ICOS. Eur. J. Immunol. 2000;30:3707–3717. doi: 10.1002/1521-4141(200012)30:12<3707::AID-IMMU3707>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, Nguyen T, Burwell T, Schneider H, Gonzalo JA, et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity. 2000;13:95–105. doi: 10.1016/s1074-7613(00)00011-x. [DOI] [PubMed] [Google Scholar]
- 10.Futagawa T, Akiba H, Kodama T, Takeda K, Hosoda Y, Yagita H, Okumura K. Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int. Immunol. 2002;14:275–286. doi: 10.1093/intimm/14.3.275. [DOI] [PubMed] [Google Scholar]
- 11.McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, Chernova T, Malenkovich N, Jabs C, Kuchroo VK, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 2000;165:5035–5040. doi: 10.4049/jimmunol.165.9.5035. [DOI] [PubMed] [Google Scholar]
- 12.Rottman JB, Smith T, Tonra JR, Ganley K, Bloom T, Silva R, Pierce B, Gutierrez-Ramos JC, Ozkaynak E, Coyle AJ. The costimulatory molecule ICOS plays an important role in the immunopathogenesis of EAE. Nat. Immunol. 2001;2:605–611. doi: 10.1038/89750. [DOI] [PubMed] [Google Scholar]
- 13.Hurtado JC, Kim YJ, Kwon BS. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J. Immunol. 1997;158:2600–2609. [PubMed] [Google Scholar]
- 14.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 15.Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003;198:63–69. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, Somoza C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature. 1993;366:76–79. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 17.Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7-1 and B7-2 costimulatory ligands: expression and function. J. Exp. Med. 1994;180:631–640. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman GJ, Gray GS, Gimmi CD, Lombard DB, Zhou LJ, White M, Fingeroth JD, Gribben JG, Nadler LM. Structure, expression, and T cell costimulatory activity of the murine homologue of the human B lymphocyte activation antigen B7. J. Exp. Med. 1991;174:625–631. doi: 10.1084/jem.174.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenschow DJ, Su GH, Zuckerman LA, Nabavi N, Jellis CL, Gray GS, Miller J, Bluestone JA. Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA. 1993;90:11054–11058. doi: 10.1073/pnas.90.23.11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fields PE, Finch RJ, Gray GS, Zollner R, Thomas JL, Sturmhoefel K, Lee K, Wolf S, Gajewski TF, Fitch FW. B7.1 is a quantitatively stronger costimulus than B7.2 in the activation of naive CD8+ TCR-transgenic T cells. J. Immunol. 1998;161:5268–5275. [PubMed] [Google Scholar]
- 21.Gajewski TF, Fallarino F, Uyttenhove C, Boon T. Tumor rejection requires a CTLA4 ligand provided by the host or expressed on the tumor: superiority of B7-1 over B7-2 for active tumor immunization. J. Immunol. 1996;156:2909–2917. [PubMed] [Google Scholar]
- 22.Manzotti CN, Liu MK, Burke F, Dussably L, Zheng Y, Sansom DM. Integration of CD28 and CTLA-4 function results in differential responses of T cells to CD80 and CD86. Eur. J. Immunol. 2006;36:1413–1422. doi: 10.1002/eji.200535170. [DOI] [PubMed] [Google Scholar]
- 23.Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor α leads to autoimmunity in transgenic mice. Proc. Natl. Acad. Sci. USA. 1994;91:5138–5142. doi: 10.1073/pnas.91.11.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herrera PL, Harlan DM, Fossati L, Izui S, Huarte J, Orci L, Vassalli JD, Vassalli P. A CD8+ T-lymphocyte-mediated and CD4+ T-lymphocyte-independent autoimmune diabetes of early onset in transgenic mice. Diabetologia. 1994;37:1277–1279. doi: 10.1007/BF00399802. [DOI] [PubMed] [Google Scholar]
- 25.Allison J, Stephens LA, Kay TW, Kurts C, Heath WR, Miller JF, Krummel MF. The threshold for autoimmune T cell killing is influenced by B7-1. Eur. J. Immunol. 1998;28:949–960. doi: 10.1002/(SICI)1521-4141(199803)28:03<949::AID-IMMU949>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Harlan DM, Hengartner H, Huang ML, Kang YH, Abe R, Moreadith RW, Pircher H, Gray GS, Ohashi PS, Freeman GJ, et al. Mice expressing both B7-1 and viral glycoprotein on pancreatic β cells along with glycoprotein-specific transgenic T cells develop diabetes due to a breakdown of T-lymphocyte unresponsiveness. Proc. Natl. Acad. Sci. USA. 1994;91:3137–3141. doi: 10.1073/pnas.91.8.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Herrath MG, Guerder S, Lewicki H, Flavell RA, Oldstone MB. Coexpression of B7-1 and viral (“self”) transgenes in pancreatic β cells can break peripheral ignorance and lead to spontaneous autoimmune diabetes. Immunity. 1995;3:727–738. doi: 10.1016/1074-7613(95)90062-4. [DOI] [PubMed] [Google Scholar]
- 28.Guerder S, Meyerhoff J, Flavell R. The role of the T cell costimulator B7-1 in autoimmunity and the induction and maintenance of tolerance to peripheral antigen. Immunity. 1994;1:155–166. doi: 10.1016/1074-7613(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 29.Wong S, Guerder S, Visintin I, Reich EP, Swenson KE, Flavell RA, Janeway CA., Jr Expression of the co-stimulator molecule B7-1 in pancreatic β-cells accelerates diabetes in the NOD mouse. Diabetes. 1995;44:326–329. doi: 10.2337/diab.44.3.326. [DOI] [PubMed] [Google Scholar]
- 30.Wong FS, Visintin I, Wen L, Granata J, Flavell R, Janeway CA. The role of lymphocyte subsets in accelerated diabetes in nonobese diabetic-rat insulin promoter-B7-1 (NOD-RIP-B7-1) mice. J. Exp. Med. 1998;187:1985–1993. doi: 10.1084/jem.187.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong FS, Du W, Thomas IJ, Wen L. The influence of the major histocompatibility complex on development of autoimmune diabetes in RIP-B7.1 mice. Diabetes. 2005;54:2032–2040. doi: 10.2337/diabetes.54.7.2032. [DOI] [PubMed] [Google Scholar]
- 32.Guerder S, Eynon EE, Flavell RA. Autoimmunity without diabetes in transgenic mice expressing β cell-specific CD86, but not CD80: parameters that trigger progression to diabetes. J. Immunol. 1998;161:2128–2140. [PubMed] [Google Scholar]
- 33.Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA., Jr CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J. Exp. Med. 1996;183:67–76. doi: 10.1084/jem.183.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong FS, Karttunen J, Dumont C, Wen L, Visintin I, Pilip IM, Shastri N, Pamer EG, Janeway CA., Jr Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat. Med. 1999;5:1026–1031. doi: 10.1038/12465. [DOI] [PubMed] [Google Scholar]
- 35.Kouskoff V, Signorelli K, Benoist C, Mathis D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J. Immunol. Methods. 1995;180:273–280. doi: 10.1016/0022-1759(95)00002-r. [DOI] [PubMed] [Google Scholar]
- 36.Gotoh M, Maki T, Satomi S, Porter J, Monaco AP. Immunological characteristics of purified pancreatic islet grafts. Transplantation. 1986;42:387–390. doi: 10.1097/00007890-198610000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Stephens LA, Kay TW. Pancreatic expression of B7 co-stimulatory molecules in the non-obese diabetic mouse. Int. Immunol. 1995;7:1885–1895. doi: 10.1093/intimm/7.12.1885. [DOI] [PubMed] [Google Scholar]
- 38.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 Ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaufman HL, Deraffele G, Mitcham J, Moroziewicz D, Cohen SM, Hurst-Wicker KS, Cheung K, Lee DS, Divito J, Voulo M, et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J. Clin. Invest. 2005;115:1903–1912. doi: 10.1172/JCI24624. [DOI] [PMC free article] [PubMed] [Google Scholar]