

Abstract
Objective
The following comprehensive review describes the evolution of stimulant drug formulations used in the treatment of attention-deficit/hyperactivity disorder (ADHD). Emphasis is placed on the basic and clinical pharmacology of the dl-methylphenidate (MPH) transdermal system (MTS).
Methods
The pharmacokinetic and pharmacodynamic literature pertaining to MPH and amphetamine enantiomers was reviewed in the context of ADHD therapy and MTS as a treatment option.
Results
MTS incorporates MPH into an adhesive monolithic matrix, using the free base form of the drug to facilitate transdermal absorption. MTS technology minimizes contact dermatitis by eliminating to need for percutaneous penetration enhancers. After a lag time of approximately 2 h, plasma concentrations of the therapeutic d-MPH isomer become detectable, then continuously rise over the course of the recommended 9 h wear time. Concentrations of l-MPH typically attain 40−50% that of d-MPH (vs. 1−2% following oral MPH). Unauthorized MTS removal poses some misuse liability and over 50% of MTS drug content remains in the discarded system.
Conclusions
While liquid or chewable MPH formulations overcome potential swallowing difficulties, as do sprinkled once-daily extended-release (ER) MPH products, only MTS addresses swallowing difficulties while also offering a flexible individualized MPH exposure time in a once-daily MPH regimen.
Keywords: methylphenidate, transdermal, pharmacokinetics, pharmacodynamics, enantiomer, amphetamine
INTRODUCTION
The central nervous system stimulant dl-methylphenidate (MPH) became available as a transdermal patch in 2006, using advanced proprietary technology from Noven Pharmaceuticals, Inc. (Miami, FL, USA). Regulatory approval has been obtained by Shire US Inc. (Wayne, PA, USA) for marketing this product under the trade name Daytrana®. The MPH transdermal delivery system (MTS) provides additional flexibility in the treatment options/regimens for attention-deficit/hyperactivity disorder (ADHD). The following literature review and commentary offers a perspective on MTS, addressing enantiospecific pharmacology, pharmaceutical technology, and potential therapeutic implications of MTS in the context of a broad consideration of pre-existing oral stimulant formulations used to treat ADHD.
HISTORICAL PERSPECTIVE ON STIMULANT DRUG DEVELOPMENT FOR ADHD
The use of psychomotor stimulants for the symptomatic control of ADHD was first reported by Bradley in 1937. In this seminal report, racemic amphetamine, that is, the 50:50 mixture of d- and l-amphetamine isomers (Figure 1; Benzedrine®), was shown to reduce the impulsivity, distractibility, and inattention characteristic of ADHD. Also in 1937, the pure isomer product d-amphetamine (Dexedrine®) became available as an alternative to the since discontinued racemic amphetamine product.
Figure 1.
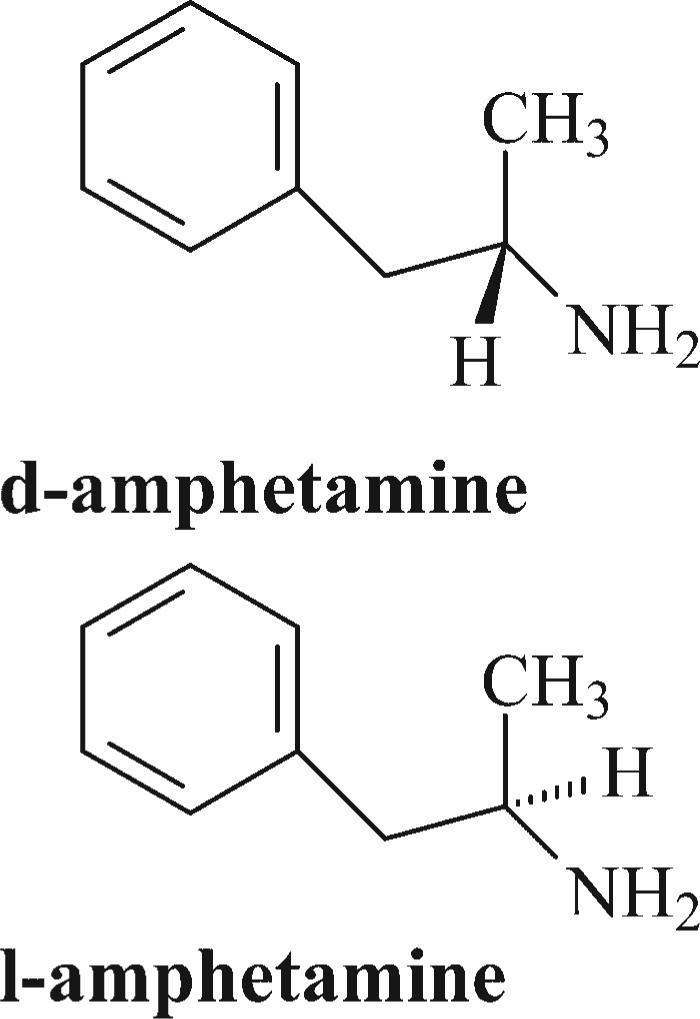
Stereostructures of amphetamine enantiomers
The optimal isomer content found in an amphetamine formulation for a specific ADHD patient may differ, as in accord with differing underlying etiologies of ADHD (Gozal and Molfeses, 2005). Bradley (1950) reported that dl-amphetamine provides conspicuously better therapeutic effects than d-amphetamine in children with compulsive traits. However, with the exception of anorexia, side effects were more prevalent with dl-amphetamine than with d-amphetamine. As an aside, Janowsky and Davis (1976) reported that in an experimental challenge with either isomer of amphetamine, l-amphetamine produced more irritability than d-amphetamine in a schizophrenic patient population.
The Snyder group (Arnold et al., 1972) found that l-amphetamine exhibited a longer latency of therapeutic onset in ADHD when compared to d-amphetamine. Further, each of the two amphetamine isomers were shown to be comparably efficacious in treating hyperactivity and aggressiveness, though d-amphetamine was found to be superior in maintaining attentiveness. Arnold et al. (1978) and McIntyre et al. (1981) also explored the use of either d-amphetamine or l-amphetamine to treat children with ADHD. Both studies found that while d-amphetamine provided the most beneficial effects for the majority of children, a sub-population responded most favorably to l-amphetamine. Of this latter group, some individuals exhibited behavioral deterioration when administered d-amphetamine. Indeed, the most widely prescribed amphetamine product used presently to treat ADHD (Adderall®; formerly marketed as Obetrol®) contains 75% d-amphetamine and 25% l-amphetamine formulated as sulfate, saccharate, and aspartate “mixed salts” (Popper, 1994). Note, in the context of amphetamine enantiospecific preclinical pharmacology, Zabik et al. (1978) found that while d-amphetamine increased locomotor activity in Swiss-Webster mice (4 mg/kg IP), l-amphetamine significantly decreased motor activity relative to controls.
Regardless of enantiomer considerations, a prodrug form of d-amphetamine became commercially available in 2007 as a treatment option for ADHD. This prodrug was synthesized by condensing the carboxylic acid of lysine with the amine of amphetamine to generate the amide lisdexamfetamine (Vyvanse®). As such, this derivative is devoid of pharmacological activity until metabolically activated by hydrolysis to d-amphetamine in the gut and/or liver. Though still a schedule II controlled substance, the parent prodrug exhibits greatly reduced intranasal or intravenous abuse liability because these routes circumvent presystemic activation (Blick and Keating, 2007). In the evolution of amphetamine products, a liquid d-amphetamine formulation (Liquadd®) was approved by the FDA in 2008 for the treatment of ADHD, designed specifically for patients who have swallowing difficulties when using solid amphetamine formulations.
Both MPH and amphetamine constitute first-line agents in the treatment of ADHD (Perrin et al., 1996; Rappley, 2005). The putative mode of action for both MPH and amphetamine primarily involves facilitation of dopaminergic transmission through interactions with the dopamine transporter. Amphetamine serves as a translocation substrate to promote subsequent release of extravesicular dopamine into the synapse (Kahlig and Galli, 2003). This specific mechanism may be kinetically potentiated by the presence of l-amphetamine, for example, the “mixed salts” (Glaser et al., 2005). In contrast, MPH binds to the dopamine transporter but does not trigger the conformational change required for transport into the presynaptic terminal. Thus, in a fashion analogous to cocaine (Froimowitz et al., 1995; Vastag, 2001), MPH transporter binding only impedes clearance of impulse-released dopamine from the synaptic cleft (Patrick et al., 1987; Hitri et al., 1994; Volkow et al., 2002b).
Several lines of evidence support a noradrenergic aspect of action in the therapeutic response to ADHD drugs, either in conjunction with dopaminergic influences (Solanto, 1998; Kuczenski and Segal, 2001; Berridge and Waterhouse, 2003; Oades et al., 2005), or largely independent of the dopamine synapse. In addition to acting on the dopamine transporter, d-MPH inhibits the norepinephrine transporter in a potent and enantioselective manner (Patrick et al., 1987; Markowitz et al., 2006; Williard et al., 2007; Markowitz and Patrick, 2008). It is noted that the ADHD approved drug atomoxetine (Strattera®) acts almost exclusively to inhibit the norepinephrine transporter (Glaze et al., 2002). This drug is neither a controlled substance nor a psychomotor stimulant (Heil et al., 2002; Bruno and Hess, 2006). Finally, emerging evidence indicates that receptor(s) for the monoamine serotonin can potentiate MPH behavioral effects in animals, pointing toward a novel direction in ADHD drug discovery (Borycz et al., 2008).
While amphetamine contains one chiral or stereogenic center, giving rise to two isomers as discussed above (Figure 1), MPH possesses two chiral centers which gives rise to four possible isomers. The earliest MPH formulation (Centedrin®; Rickter Works, Budapest, Hungary) contained all four isomers: 40% d-(R,S)-erthyro-, 40% l-(S,R)-erythro-, 10% d-(R,R)-threo-, and 10% l-(S,S)-threo-MPH. The dl-erythro-MPH components exacerbated cardiovascular toxicity (Szporny and Gorog, 1961) without significantly contributing to the desired central nervous system stimulant effects (Patrick et al., 1981). Accordingly, to improve the margin of safety for MPH products, an industrial method was developed to isolate only the dl-threo-MPH isomers (Figure 2; Rometsch, 1958). This MPH racemic mixture (e.g., Ritalin®) has been in clinical use in the USA since 1955 (Andreason, 2005).
Figure 2.
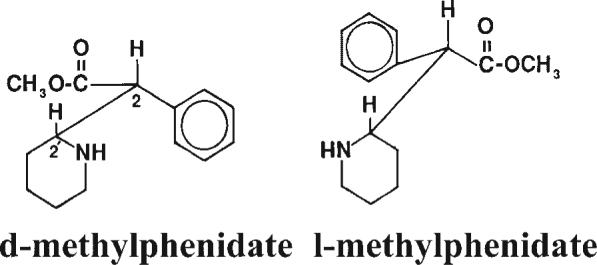
Stereostructures of MPH enantiomers as assigned by chemical correlation (Shafi'ee and Hite, 1969) and by X-ray crystallography (Froimowitz et al., 1995). Adapted from Patrick et al. (1987)
L-MPH AS PASSIVE “ISOMERIC BALLAST” OR ACTIVE COMPONENT OF DL-MPH
Therapeutic effects of oral dl-MPH appear to be limited to the d-MPH isomer (Srinivas et al., 1992; Aoyama et al., 1994; Ding et al., 2004; Markowitz and Patrick, 2008), as consistent with a pure d-MPH formulation (dexmethylphenidate; Focalin®) becoming available in 2002 (Arnold et al., 2004; Patrick, 2006a).
Any potential therapeutic benefit of removing the l-MPH isomer from an oral dl-MPH formulation is not obvious (Srinivas et al., 2001). Due to the very extensive oral pre-systemic metabolism of l-MPH, typically only low pg/ml concentrations of the l-isomer reach the systemic circulation (Modi et al., 2000; Patrick et al., 2005a; Patrick et al., 2007). This trace l-MPH plasma concentration represents only about 1% of the d-MPH plasma concentration. In effect, oral dl-MPH in humans is subject to “in vivo biocatalytic resolution” by carboxylesterase 1 (Zhu et al., 2008), analogous to the enzymatic approaches that have been used industrially to eliminate l-MPH from dl-MPH and yield enantiopure d-MPH (Prashad 2001).
In contrast, when dl-MPH is administered by the transdermal route, as in MTS, l-MPH circumvents both enteric and hepatic first-pass metabolism, allowing the l-MPH isomer to readily access the systemic circulation (see Sections “Methylphenidate transdermal system (MTS)” and “Pharmacokinetics of the MTS;” Pierce et al., 2008).
Most, but not all, pharmacological screening of l-MPH reveals that this isomer is devoid of activity, therapeutic (Srinivas et al., 1992) or otherwise. However, l-MPH may not necessarily represent merely a “passive” or inert component of MTS (see Heal and Pierce, 2006). In a pilot therapeutic drug monitoring study, distinguished from most other such studies by the use of enantiospecific gas chromatographic analytical methodology, poor responders to oral dl-MPH were reported to have significantly higher plasma concentrations of both d-MPH and l-MPH (Jonkman et al., 1998). Note that enantiospecific gas chromatographic methods may inherently lead to an overestimation of l-MPH plasma concentrations (see Patrick et al., 2005a).
In the absence of l-MPH, the duration of therapeutic action for d-MPH has been tentatively reported to extend beyond that of twice the mg dose of dl-MPH (Swanson et al., 2002; Wigal et al., 2004). While this possibility will require additional investigation, any potential differences in behavioral duration are unlikely to be based on pharmacokinetic influences of the l-isomer on that of d-MPH (Quinn et al., 2004).
l-MPH has been speculated to have abuse potential in its own right (Quinn, 2000) and using limited human testing to support patent claims, l-MPH has been reported to exhibit potentially therapeutic antidepressant activity and “energizing” effects (Midha et al., 2002; Rouhi, 2003). In addition, patents have stated that l-MPH offers anxiolytic and antipsychotic activity, serves as an antidote for stimulant overdose (Baldessarini and Cambell, 2001), and as an anticonvulsant (Davidson and Craig 2000), among other claims. However, caution must be exercised when interpreting assertions made in patents because their claims are not subject to peer review.
In vitro evaluations of l-MPH have revealed little or no activity (Patrick et al., 1987; Markowitz et al., 2006; Williard et al., 2007; Markowitz and Patrick, 2008). In vivo animal studies have found that l-MPH does not contribute to (1) locomotor activity (an index of dopaminergic agonism; Patrick et al., 1987; Williard et al., 2007), (2) pressor effects (Patrick et al., 1987), (3) nor to appetite suppression (Eckerman et al., 1991). The above reports notwithstanding, studies in rats using very high toxicological doses have found that l-MPH exhibits some limited behavioral effects, with females more sensitive than males (Teo et al., 2003a). Greater pupil dilation and vocalization in rats were reported for high doses of dl-MPH when compared to the pure d-enantiomer given at half the racemate mg/kg dose (Teo et al., 2002). Possible behavioral activity could be attributable to l-MPH when comparing high dose d-MPH administered at twice the dose of dl-MPH in pregnant animals (Teo et al., 2003b). In yet another high dose test comparing dl-MPH to d-MPH in rats, the racemate dosed at twice that of pure d-MPH resulted in potentiation of repetitive pawing, dilation of pupils, aggressive behavior, as well as of head-bobbing and hyperpnea in rabbits (Teo et al., 2003b).
In rodents, l-MPH has been reported to increase the behavioral response to cocaine (Ding et al., 2002), though these results were subsequently amended (Ding et al. 2004). It is noted that chromatographic evidence has tentatively supported the hypothesis that oral l-MPH, at least in baboons and rats, accumulates in the brain as the p-hydroxy-MPH metabolite (Ding et al., 2004; see Patrick et al., 1981) with unknown pharmacological implications.
Davids et al. (2002) assessed the activity of the separate MPH enantiomers in a 6-hydroxydopamine model of ADHD using rats. Challenges with d-MPH, l-MPH, racemic MPH, or saline in these chemically lesioned animals demonstrated that d-MPH was over three times more active in reducing motor activity than racemic MPH. A twofold reduction would be predicted were l-MPH inert. Finally, pre-treatment of these rats with l-MPH was reported to attenuate the motor activity response to d-MPH.
The presence of l-MPH in racemic products necessarily adds to the overall metabolic load, with the potential for drug interactions. For instance, following concomitant dl-MPH (0.3 mg/kg) and ethanol (0.6 gm/kg) in humans, l-MPH enantioselectively interacts with ethanol to yield the inactive (Patrick et al., 2005b; Williard et al., 2007) transesterification metabolite l-ethylphenidate. More importantly, this l-MPH—ethanol interaction was accompanied by a 40% mean elevation of d-MPH plasma Cmax and 25% increase in d-MPH exposure (AUC). Elevation in these parameters has been associated with an increase in abuse liability (see Patrick et al., 2007). The co-abuse of MPH and ethanol is of special health concern (Jaffe, 1991; Barrett and Pihl, 2002; DAWN, 2003; Barrett et al., 2005), and occurs very frequently during MPH polysubstance abuse (Darredeau et al., 2007; Novak et al., 2007). Further, as the diagnosis of ADHD in adults continues to increase (Okie, 2006), patient alcohol consumption emerges as an increasingly important aspect of drug individualization. The abuse implications of the above ethanol interaction with dl-MPH in part prompted a patent application for d-MPH devoid of the l-isomer (Khetani and Faleck, 2004).
The l-MPH isomer also warrants clinical consideration based on the recently reported instance of an MPH poor metabolizer who expressed a dysfunctional carboxylesterase 1, resulting in plasma concentrations of l-MPH over 100 times that of normal MPH metabolizers (Patrick et al., 2007). In addition, this subject had both elevated d-MPH plasma concentrations and elevated hemodynamic parameters (Zhu et al., 2008). Cardiovascular effects of stimulant use in ADHD may warrant further safety monitoring (Vetter et al., 2008). Whether this MPH poor metabolizer represents only a rare inborn error/defect of MPH metabolism, or a distinct pharmacogenetic polymorphism, that is, ≥1% of the population, is the subject of ongoing investigations using phenotyping (LeVasseur et al., 2008) and genotyping (Zhu et al., 2008) to advance ADHD personalized medicine (Stein and McGough, 2008).
Summarizing, existing studies of the single isomer l-MPH have reported some discernable pharmacological activity in animals, but only when administering extreme doses, or after invasive chemical lesioning of catecholaminergic structures in the central nervous system. Further, oral l-MPH exhibits virtually no absolute bioavailability due to its near complete pre-systemic hydrolysis. However, renewed clinical interest in l-MPH has resulted from: (1) the newly discovered interaction of l-MPH with ethanol; (2) the identification of an l-MPH poor metabolizer; and (3) the many-fold increase in l-MPH exposure when dosing dl-MPH transdermally as compared to the oral route.
EVOLUTION OF ORAL MPH FORMULATION TECHNOLOGY
The facile metabolic deesterification of MPH (Faraj et al., 1974) to the inactive (Patrick et al., 1981) amino acid ritalinic acid limits the elimination half-life to 2−3 h (Meyer et al., 2000). Because the desired behavioral effects of MPH usually require medicating the ADHD patient throughout the course of the school or work day, this short half-life of MPH generally dictates either a twice-daily regimen of immediate-release (IR) MPH, given at breakfast and again at lunch (Figure 3) or, depending on the patient's specific medication needs, a three-times-daily schedule with the third dose typically administered around 3−4 p.m. to promote, for example, homework concentration and/or family harmony. This three-times-daily MPH schedule carries with it a higher incidence of appetite suppression and insomnia (Stein et al., 1996).
Figure 3.
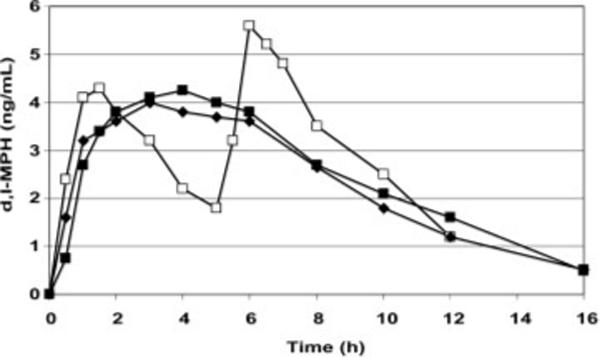
Mean plasma MPH concentration-times profiles (n = 18) comparing branded and generic 20 mg conventional ER-MPH formulations (◆; ■) versus 10 mg IR-MPH (□) dosed on the twice-daily schedule
These multiple dose IR-MPH schedules present concerns regarding compliance, peer ridicule during school dosing and medication storage at school/work to prevent diversion (Goyer et al., 1979; Dupont et al., 2007) of this Schedule II drug. To address these dosing issues, a once-daily extended-release (ER) MPH formulation was developed in the early 1980s using conventional max matrix technology (Figure 3; Ritalin-SR®). This ER product was followed by a generic form in 1990. ER formulations are classified by the USP as those that allow at least a twofold reduction in dosing frequency relative to IR (Abdou et al., 2000). The conventional ER dosage form of MPH provides a relatively constant blood concentration over the period between 2 and 6 h after dosing (Figure 3; Patrick et al., 1989).
There are possible differences in efficacy between multiple daily doses of IR-MPH relative to this conventional once-a-day ER-MPH formulation (Pelham et al., 1987; 1990) which have been theoretically attributed to tachyphylaxis, that is, the development of acute tolerance when there is little elevation in blood concentrations of MPH over time (Kollins et al., 1998; Swanson et al., 1999). Specifically, efficacy has been reported to correlate most robustly with the absorption phase of plasma MPH concentration-time profiles as opposed to periods of relatively constant plasma MPH concentrations (Swanson and Volkow, 2002; Volkow and Swanson, 2003). This hypothetical MPH pharmacokinetic–pharmacodynamic correlation, a clockwise hysteresis (Pleuvry, 2005), has been termed the “ramp effect” (Birmaher et al., 1989; Greenhill, 1992) or “gradient effect” (Patrick and Markowitz, 1997). Beneficial responses to amphetamine formulations in the treatment of ADHD have also been reported to correlate with the absorption phase of the pharmacokinetic profile (Brown et al., 1980). In a drug abuse context, very rapid elevation in blood and brain MPH concentrations accentuates euphoria (Spencer et al., 2006; Froimowitz et al., 2007; Parasrampuria et al., 2007a,b).
As it stands, therapeutic response to MPH may (Swanson et al., 1999) or may not (Schachar et al., 2008) be subject to acute MPH tolerance. Interestingly, in spite of this clinical focus on MPH tolerance, numerous preclinical MPH studies have demonstrated behavioral sensitization with chronic MPH dosing (e.g., McDougall et al., 1999; Yang et al., 2007).
The pharmaceutical industry responded to the postulated issue of acute tolerance associated with the conventional monophasic release profile of the 1980s era ER-MPH by developing second generation ER-MPH formulations which provide biphasic or pulsed release of MPH. Each of these new products contains a percentage of the total MPH dose as IR-MPH but employ different technologies to obtain their own unique release characteristics. Biphasic MPH formulations not only facilitate once-daily dosing, but also could possibly reduce appetite suppression relative to the original ER-MPH product by providing MPH plasma concentration troughs (Ritalin-LA® and Focalin-XR®), or plateaus (Concerta®, Metadate CD®, Biphentin®, Medikinet®), during the typical lunchtime period.
The initial second generation ER-MPH product used the OROS® osmotic release technology developed by the Alza Corporation (Concerta®, 22% IR-MPH; Modi et al., 2000; Swanson et al., 2003). Beaded formulations followed (Metadate CD®, 30% IR-MPH; Gonzalez et al., 2002; Ritalin-LA®, 50% IR-MPH; Markowitz et al., 2003b; Biphentin®, 40% IR-MPH; Teicher et al., 2006; Reiz et al., 2008; Medikinet®, 50% IR-MPH; Haessler et al., 2008), and a biphasic-release formulation of pure d-MPH became available in 2005 (Focalin-XR®, 50% IR; Robinson and Keating, 2006). Of these several ER formulations, the mean plasma drug profiles for Ritalin-LA® and Focalin-XR® most closely resemble the time course of the twice-daily IR-MPH regimen (Figure 3; Tuerck et al., 2007). As might be anticipated, 50% IR-MPH biphasic products have been reported to provide better behavioral response at 2 h post-dosing than the 22% IR biphasic MPH dose, but then this disparity may be compensated by greater 12 h efficacy for the 22% IR formulation (Silva et al., 2008).
A multi-layered single composition beaded ERMPH formulation (Biphentin®, Canada) exhibits a less conspicuously pulsatile pharmacokinetic release profile. It was shown to provide a plasma MPH concentration time course approximating a 2−10 h plateau in ADHD children (n = 14; Quinn et al., 2007), or found to be only minimally biphasic in a normal adult subject study (n = 21; Reiz et al., 2008). Efficacy of this product has been compared against twice-daily schedules of IR-MPH (Weiss et al., 2007; Schachar et al., 2008) in children and adolescents, and versus placebo in adults (Jain et al., 2007).
Figure 4 depicts estimated shapes of mean d-MPH plasma profiles from select MPH formulations administered on a single day and serves to illustrate the various options available to modulate the MPH plasma concentration time course. Plasma Cmax values resulting from maintenance doses of ER-MPH formulations for most ADHD patients generally range from 10 to 20 ng/ml. For specific values, see Markowitz et al. (2003a), Patrick et al. (2005a), Quinn et al. (2007), Markowitz and Patrick (2008), Reiz et al. (2008).
Figure 4.
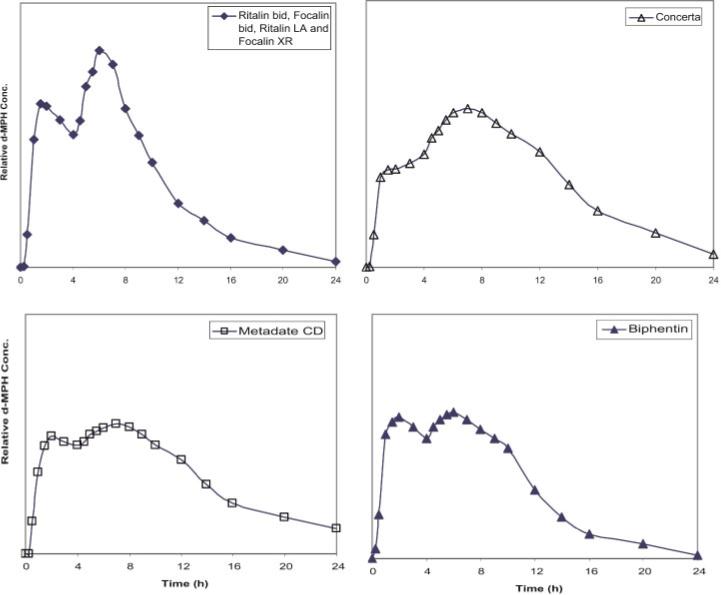
Estimate of relative mean plasma d-MPH concentrations from single doses of different biphasic/pulsatile (second generation) ER-MPH dosage forms when administered at doses that would provide approximately the same total exposure (AUC). In oral MPH pharmacokinetic analyses, virtually all detectable drug exists as the d-isomer due to the very extensive pre-systemic metabolism of l-MPH (See Historical perspective). Plasma Cmax values resulting from maintenance doses of ER-MPH formulations for most ADHD patients generally range from 10 to 20ng/ml. For specific values, see Markowitz et al. (2003a), Patrick et al. (2005a), Quinn et al. (2007), Markowitz and Patrick (2008), Reiz et al. (2008)
It is noted that the profiles for these formulations on a daily basis will result in no significant drug accumulation, that is, single-dose versus multiple-dose profiles will be essentially identical due to the short 2−3 h plasma half-life of MPH. When administered as equivalent doses of MPH, all these formulations have been demonstrated to result in the same total systemic exposure or area under the plasma concentration-time curve (AUC), but distinct differences in the time courses have the potential to elicit varying responses. However, there are no convincing clinical trials that distinguish any of these formulations are superior to dosage regimens of IR-MPH. All of these products have been shown superior to placebo treatment.
The choice of a specific MPH product or regimen for an individual patient should consider pharmacokinetic differences, yet from the standpoint of clinical efficacy, none should be eliminated, a priori, as a therapeutic option based on the shape of the mean plasma drug concentration profiles. It is emphasized that Figure 4 is only a composite picture of the relative d-MPH concentrations “estimated” by combining pharmacokinetic data from different studies (see Markowitz et al., 2003a, 2006; Markowitz and Patrick, 2008) and is only meant to represent the general shape of the profile expected. Individual patients will have the potential to vary widely from these ER-MPH profiles due to the many physiological variables associated with prolonged release drug products which course through various regions of the gut (McConnell et al., 2008; Kagan and Hoffman, 2008). Accordingly, clinicians are advised to use Figures 3 and 4 only as a general guide in pharmacokinetically distinguishing these oral dosage forms. Unfortunately, MPH product selection for optimal efficacy in treating ADHD largely relies on empiricism, a treatment area where therapeutic drug monitoring has not been of established value.
METHYLPHENIDATE TRANSDERMAL SYSTEM (MTS)
MTS became available in 2006 as four strengths (patches with different surface areas). The transdermal route of MPH administration offers another alternative to MPH sprinkles (Pentikis et al., 2002; Lee et al., 2003) in addressing the drug needs of patients with oral tablet/capsule swallowing difficulties, while also providing for the once-daily dosing regimen now characterizing most ADHD drug prescribing practices (Scheffler et al., 2007). Transdermal administration of metabolically labile dl-MPH also overcomes both enteric and hepatic pre-systemic deesterification—the first-pass effect—which otherwise limits the oral bioavailability of d-MPH to approximately 25% (Chan et al., 1983) and l-MPH to approximately 1% (Modi et al., 2000; Patrick et al., 2007). Accordingly, after dosing with MTS, both d-MPH and l-MPH are readily absorbed into the systemic circulation (Figure 5). Prior to development of MTS, the only significant amounts of l-MPH to reach the bloodstream occurred following intravenous dl-MPH when the drug had been indicated for intractable hiccups (Vasiloff et al., 1965); or when assessing absolute bioavailability (Chan et al., 1983); or during intravenous (Levine et al., 1986; Shlomi et al., 2008)/intranasal (Llana and Crismon, 1999) MPH abuse; and finally, in the instance of a dl-MPH poor metabolizer who expresses a dysfunctional carboxylesterase 1 enzyme which otherwise is primarily responsible for l-MPH metabolic clearance (see Section “l-MPH as passive “isomeric ballast” or active component of dl-MPH;” Patrick et al., 2007; LeVasseur et al., 2008; Zhu et al., 2008).
Figure 5.
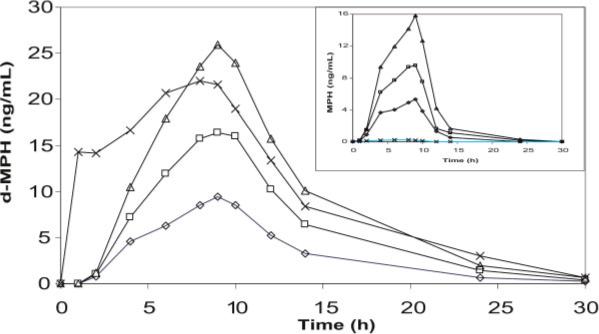
Plasma d-MPH and l-MPH (inset) concentration-time profiles for 12.5 (◇), 25 (□), and 37.5 cm2 (Δ) MTS compared to 54 mg Concerta® (X) (Andreason, 2005)
While MTS does avoid oral first-pass hydrolytic metabolism, pre-systemic percutaneous metabolism may still differentially influence d-MPH versus l-MPH bioavailability. For instance, enantioselective deesterification of a racemic ester prodrug has been reported in the course of transdermal delivery (Ahmed et al., 1997).
MTS PRODUCT DEVELOPMENT
Novel transdermal and adhesive technology, when applied to MPH delivery, has resulted in MTS. The technology used for MTS is a patented process which minimizes the surface area of the transdermal system required in the delivery of once-daily MPH. MTS relies on a blend of drug, acrylic polymers and silicone adhesives containing a high drug load. Such a MPH load establishes a large concentration gradient which facilitates drug diffusion into the skin and avoids the need to incorporate transdermal permeability enhancers which otherwise have been found to promote localized contact dermatitis (Pappinen and Urtti, 2006).
While oral MPH products use the hydrochloride salt form of the drug, MTS uses the skin permeable free base form of MPH in the multipolymeric adhesive. The drug-adhesive blend is coated onto a backing film which is a polyester/ethylene vinyl acetate laminate serving as an occlusive barrier in addition to holding the drug reservoir. The adhesive is protected with a release liner which the caregiver or patient removes prior to applying MTS to skin. When MTS is applied to a patient, the backing film remains visible and displays the product labeling to allow a clinician, parent, or teacher to know that MPH is being administered.
MTS overcomes the negative social issues associated with multiple daily oral dosing, allowing controlled drug release over an extended period of time, and with the added flexibility of allowing termination of drug delivery at any time simply by removing the transdermal product (Wilens et al., 2008b). The range of MTS patch sizes also provides for different delivery rates and facilitates dose titration and optimization for individual patients.
Since Shire acquired the marketing rights for MTS from Noven, they have conducted pediatric studies in which evidence of efficacy and safety was demonstrated in 6−12 years old patients with ADHD. In April 2006, the FDA approval of MTS was announced. The product was approved for pediatric patients with ADHD ≥ 6 years old, though the target patient population most likely will also include adults with ADHD. Although initially this will be an off label use, the product is capable of achieving and maintaining effective plasma MPH levels in healthy adults. Duration of drug release beyond 9 h may be particularly desirable for adults with ADHD; although, the typical dosing duration for MPH rarely exceeds 12 h. Any longer duration of stimulant drug delivery has been accompanied by an unacceptable incidence of anorexia and insomnia (Noven/Shire, 2005a, 2005b).
MTS is available in four patch sizes: 12.5, 18.75, 25, and 37.5 cm2. These correspond to the active surface area from which MPH can be absorbed. As with some other transdermal drugs, the total amount of MPH delivered increases proportionately with patch size (Noven/Shire, 2005a, 2005b). The different sizes offer a nominal delivery of 10, 15, 20, and 30 mg/9 h, respectively, based on pediatric pharmacokinetic studies where MTS was applied on the hip area of these children for 9 h (Noven/Shire, 2005a, 2005b). If MTS is applied for shorter periods of time, the dose delivered will be proportionately less. Therefore, it is necessary to view dosing as the amount delivered per hour. For this reason, the labeling for this product also includes hourly dose rates of 1.1, 1.6, 2.2, and 3.3 mg/h, corresponding to the four MTS sizes. Wearing MTS for longer periods than the recommended 9 h will result in correspondingly larger doses of MPH, for example, 3.3 mg/h × 12 h will result in an average dose of 39.6 mg from the MTS with a nominal dose of 30 mg. It is emphasized that these labeled doses were obtained as average doses in groups of pediatric patients. Individual titration and dose optimization should always be implemented (Wilens et al., 2008b).
Both patients and health care providers need awareness of special considerations when using transdermal drug delivery systems. On average, only 36% of the MPH contained in an MTS is absorbed during a 9 h application. Therefore, a substantial amount of drug remains in the system after removal from a patient. This residual content represents a potential source of MPH diversion or accidental poisoning. Accordingly, MTS Prescribing Information (Shire, 2006) contains the following statement: “Fold the used Daytrana® patch in half and press firmly so that the sticky side sticks to itself. Flush the used patch down the toilet or dispose of it in a lidded container right away.”
To prevent MPH absorption through the fingers, whoever applies the MTS should avoid contact with the adhesive surface and with the inside of the protective pouch. Hands should be washed with soap immediately after application of the MTS. After removal of the MTS from a patient, any adhesive residue visible on the skin should be removed since this residue has the potential to continue delivering drug for systemic absorption. Plasma concentrations will decline after removal of an MTS (Figure 5) but, as with most transdermal formulations, the drug concentration does not initially decline as rapidly as from, for example, an IV dose (Chan et al., 1983), consistent with absorption persisting from residual drug on or in the skin.
Heat, including fever, generally increases the absorption rate of drugs during transdermal delivery systems (Hull, 2002), and in the case of MPH from MTS this rate can increase up to twice the normal delivery rate (Noven/Shire, 2005a, 2005b). For example, a heating pad, heated water bed or electric blanket is never to be used while wearing MTS. As a warning precedent, heat sources have led to fentanyl transdermal patch overdoses (Boswell, 1992; Frolich et al., 2001; Gutstein and Akil, 2006).
In addition, MTS should be applied only to healthy undamaged skin since a healthy stratum corneum is the principle barrier to drug absorption. MTS should be applied on a clean, dry area of the hip, alternating the hip daily. When properly applied, bathing and swimming should not interfere with MTS adherence.
NEW DRUG APPLICATION (NDA) STUDIES
Sixteen studies in humans were used to support the MTS NDA. Clinical development involved studies in pediatric patients in the age range of 6−12 years old, as well as studies in healthy male and female adult volunteers. There were four Phase I studies, five in Phase II, three Phase III studies of short duration, and four long-term Phase III studies (Noven/Shire, 2005a, 2005b). The first MTS Phase I/II study in 12 ADHD pediatric patients demonstrated that the product had similar efficacy to three-times-daily oral MPH. This study also suggested the need for reformulation of MTS to improve adhesion, though the early formulation served to characterize the pharmacokinetics of transdermal MPH and its metabolism.
Transdermal absorption has previously been described as the Apparent Dose (Noonan and González, 1989) and can be calculated by the following relationship:
where the Initial Potency is the amount of drug contained in an MTS prior to dosing and Final Potency is the quantity determined by analyzing the MTS after removal from the patient.
Several pharmacokinetic and tolerability/dose ranging studies followed, initially using a wear time of 12 h. However, the FDA noted too many adverse events (Mays and Findling, 2005) with a 12 h dosing regimen (insomnia, anorexia, and weight loss). In addition to these concerns, the potential for skin sensitization and overexposure were also outlined in an FDA action letter. These issues required addressing prior to obtaining approval. The FDA suggested decreasing the wear time. Accordingly, safety and efficacy studies were redesigned to shorten the wear time to reduce total MPH exposure.
A 25 cm2 patch worn for 8 h resulted in lower overall exposure to MPH than dosing with 36 mg Concerta®, while administration of MTS for 10 h yielded greater exposure than Concerta®. The Tmax for Concerta® typically occurs typically within 6−8 h, and the plasma MPH profile has a fairly broad peak (Gonzalez et al., 2002; Figure 4). A 9 h wear time for MTS was thus chosen for further dose ranging and dose proportionality studies. This reduced MTS wear time brought tolerability in line with oral MPH formulations (Noven/Shire, 2005a, 2005b).
In a pediatric Phase III study using 9 h dosing of MTS, efficacy was compared to that of Concerta® (after optimization of the oral dose) and placebo. Low intra-subject variability, but a high inter-subject variability, was reported. These results emphasize the need for careful titration to optimize the maintenance dose of MTS.
Other specialized studies included evaluating buccal absorption of MPH from MTS as a potential route for drug abuse (see Section “Abuse potential of transdermal MPH”), as well as defining the pharmacokinetics of MPH in subjects with irritated or inflamed skin. With both buccal mucosa and irritated skin, MPH absorption was more rapid. A major safety study was also conducted to address an FDA request to investigate the potential for irritation and/or sensitization after a non-clinically relevant 24 h MTS wear (Noven/Shire, 2005a, 2005b).
PHARMACOKINETICS OF THE MTS
From Figure 5, it can be seen that there is generally a proportional increase in d-MPH maximum plasma concentration (Cmax) with increases in nominal dose using a 9 h wear time. In data submitted to the FDA, wear times longer or shorter than 9 h resulted in correspondingly higher or lower maximum concentrations (Section “New drug application (NDA) studies”). The extent of exposure to d-MPH, as determined from the area under the concentration-time profiles (AUC), also indicates dose proportionality with increases or decreases in exposure depending on wear time (Pierce et al., 2008). Although the mean time of d-MPH maximum concentration occurs at the time of patch removal, individual subjects did exhibit somewhat shorter or longer times to maximum plasma concentration (Tmax) than the actual wear time. Unlike the oral dosage forms of MPH, which begin reaching the systemic circulation soon after dosing, MTS exhibits a lag times ranging from 1 h to as long as 4 h. However, unlike the biphasic ER-MPH dosage forms, once the concentrations of MPH are detectable in the blood, there is essentially a constant increase up to the 9 h recommended wear time. Upon removal of MTS there is a biexponential decline of d-MPH (Shire, 2006; Kowalik et al., 2007), with a terminal half-life in the range seen with orally administered IR-MPH products (2−4 h). This relatively short half-life resulted in clinically inconsequential concentrations at the time of the following morning new patch application, and explains the negligible drug accumulation seen with multiple daily dosing.
When the 30 mg/9 h MTS was compared to a 54 mg oral dose of the ER formulation Concerta®, the mean Cmax and AUC of d-MPH were not significantly different. However, the l-MPH concentrations for Concerta® were mostly below the limit of assay quantification, while the concentrations of l-MPH for the MTS were approximately half as high as that of d-MPH (Figure 5; Pierce et al., 2005; 2008).
The recommended initiation dose starts at 10 mg (per 9 h), followed by weekly upward titration to establish the optimal individualized maintenance dose. The original NDA studies followed a 5-week optimization period with tapering of the dose one time if needed.
LITERATURE EFFICACY TRIALS OF MTS
Efficacy of MTS in children diagnosed with ADHD has been evaluated in several clinical trials where MTS has demonstrated efficacy above that of placebo. Trials have been conducted comparing MTS and Concerta to placebo, but were not designed to compare the efficacy between the two active treatments. The following trials have been limited to those using the presently recommended 9 h MTS wear times.
McGough et al. (2006) conducted a randomized, double-blind, placebo-controlled laboratory classroom assessment of MTS in ADHD children. Efficacy was measured using standardized and validated rating instruments (Penberthy et al., 2005), such as the SKAMP and ADHD-RS-IV, to assess efficacy. The primary measurement was that of deportment. This multi-center, crossover trial studied children between 6 and 12 years of age. Measures, taken in laboratory classroom days at 2 and 9 h post-dose, demonstrated that MTS participants scored significantly better than with a Placebo Transdermal System (PTS). McGough also reported that “the mean number of math problems attempted and math problems correct were significantly higher for participants treated with MTS compared to participants treated with PTS.” Those participants receiving MTS (79.8%) were more likely to be deemed improved by clinicians compared with PTS treatment (11.6%; p < .0001). Similarly, significant differences were observed by parents: 71.1% of MTS patients and 15.8% of PTS participants were rated as improved.
Other studies (Findling and Lopez 2005; Findling et al., 2008) have investigated MTS with the comparator Concerta® in treating ADHD using a double-blind, placebo-controlled, multi-center, dose optimized study of children 6−12 years of age. Parents, clinicians, and teachers assessed scores commencing at the beginning visit and at each study site visit thereafter. Participants were randomized in receiving the MTS, Concerta®, or PTS. Results demonstrated significant improvements in participants treated with MTS over PTS. Although the investigators stated that the study design was not intended to compare efficacy of Concerta® to that of MTS, the results did not show a significant difference between the two regimens. At the study endpoint, approximately 70% of subjects showed improvement compared to 25% for the placebo group.
Following a preliminary dose-ranging study (Pelham et al., 2005a), the efficacy of MTS was further investigated through pairing MTS with behavioral modification (Pelham et al., 2005b). The study was a 6-week, dose-ranging, single-center trial in 27 children. The team sought to further the findings of previous studies by increasing the dose by 50% and extending the trial from 6 to 8 weeks. Behavior modification was crossed with medication. The results revealed that MTS was efficacious in all doses tested and that MTS was well tolerated.
ADVANTAGES OF MTS
MTS, like the oral ER-MPH formulations, was in part designed to overcome the need for supplemental ADHD medication after the morning dose. Patent literature (Mantelle and Dixon, 2001) proposes a further therapeutic advantage for the MTS, that of eliminating the fluctuating blood levels associated not only with twice-daily and three-times-daily IR-MPH schedules, but also with once-daily biphasic ER-MPH formulations (Figures 4 and 5). It should be noted that avoiding blood MPH concentration fluctuations, in and of itself, may not optimize MPH therapy. For instance, the original ER-MPH formulation provides a near plateau blood MPH concentration between 2 and 6 h following oral administration, yet some clinicians have been less than satisfied with patient response to ER-MPH (Pelham et al., 1987; Dulcan, 1990; Greenhill, 1992; Perrin et al., 1996). However, the constantly rising MPH blood concentrations provided by the recommended 9 h wear time for the MTS may be optimal for a given patient.
Children often report that they do not like to, or refuse to, take solid dosage forms and/or have difficulty swallowing them (Hurwitz, 2001). MTS represents an alternative to ER-MPH sprinkles (Pentikis et al., 2002; Lee et al., 2003) in children with tablet or capsule swallowing difficulties. MTS could be especially beneficial if such an individual requires a high MPH dose which can involve administration of oral MPH formulations with larger physical dimensions. As an example of the potential seriousness of swallowing problems, Wagner et al. (2001) reported a case involving an ER-MPH formulation lodging in the throat of an adolescent. Treatment required general anesthesia and laryngoscopic removal of the tablet. The emergency nature of this case was compounded by the non-absorbable composition of the formulation shell which precluded any prospect of eventual tablet dissolution. The patient had previously chewed another brand of MPH tablet. While a parental guide for minimizing typical swallowing difficulties in children is available (Hurwitz, 2001), the use of MTS obviates this rare (Bass et al., 2002), but serious problem.
Since the MTS drug reservoir remains outside of the gastrointestinal tract, this offers additional flexibility in the duration of exposure when compared to oral MPH formulations, that is, the MTS may be removed at any time to tailor the individual's optimal daily interval of medication treatment as demonstrated by Wilens et al. (2008b) when testing 4 and 6 h wear times. Removal of MTS earlier than the typical 9 h wear time should minimize the incidence of appetite/growth suppression (Faraone and Giefer, 2007)) and insomnia, as consistent with the side effect reduction when comparing a twice-daily IR-MPH regimen to that of three-times-daily (Stein et al., 1996; MTA, 2004). If serious adverse events were to emerge using MTS, only the transdermal dose also allows for PRN removal, as in the cases of three small children exhibiting MTS induced buccal-lingual movements (Pelham et al., 2005a).
Accordingly, a single morning MTS application offers a more flexible alternative than oral once-daily ER-MPH formulations and can help address, but not eliminate, compliance, diversion and stigmatizing issues attendant with using multiple dose IR-MPH regimens. MTS is both discrete and nearly colorless, while still allowing parents and teachers to have visual evidence of compliance. It is noted that the oral administration of drugs to children has anecdotally been associated with “cheeking,” then discarding the oral dose in some obstinate patients. This is not possible with transdermal dosing, since the patch applied to the skin will be readily visible to a parent or teacher. If the patch should be removed, reapplication will result in poor adhesion.
The MTS studies used for FDA approval (Noven/Shire, 2005a, 2005b) point to an advantage over oral MPH formulations by allowing lower doses through the circumvention of enteric and hepatic pre-systemic metabolism. By avoiding oral first pass metabolism, MTS pharmacokinetics should not be significantly affected by food intake. While food intake has repeatedly been shown to have little influence on the extent of oral MPH absorption, food has been shown to delay the Tmax (slow the rate of absorption; see Patrick et al., 2005a) or differentially influence the biphasic nature of ER-MPH products (Haessler et al., 2008). However, while no enteric or hepatic first-pass metabolism can occur using MTS, dermal metabolic enzyme activity has been shown to substantially influence skin bioavailability of drugs (Block, 2000), including drugs subject to deesterification (Ahmed et al., 1997; Section “Methylphenidate transdermal system (MTS)”). No evidence of any dermal metabolism has been reported for MTS though further studies in this area may be warranted.
The Mantelle and Dixon (2001) patent proposes that MTS affords the capability of providing a “sleep window” for certain patients with ADHD, depending on the wear time. Some skin drug loading is characteristic of transdermal drug delivery: the skin depoteffect(Cefali et al., 1993). Thus, the patent claims that after patch removal, MTS can provide adequate blood concentrations of MPH for the first 1−1.5 h past bedtime to suppress a potential MPH rebound effect (exacerbation of ADHD symptoms upon drug offset). This hypothetical situation, however, would apply only if MTS were worn for 12 h or longer, where tolerability has been problematic.
The pharmacokinetics of MTS has not been reported to be influenced by gender (Shire, 2006). In view of the recently reported greater oral bioavailability found in men versus women dosed with ER-MPH (Markowitz et al., 2003b), the transdermal route of MPH administration may eliminate gender differences in MPH pharmacokinetic, especially if this dimorphism is based on differential levels of enteric/hepatic pre-systemic metabolism.
CONCERNS/LIMITATIONS OF MTS
Although MTS was designed to facilitate compliance with once-daily dosing, the patch is also subject to unscheduled/unauthorized removal with potential for diversion. This may be of special concern with oppositional children. As discussed above, the absence of the patch should be obvious to a parent, teacher, or health-care giver.
Minimal to moderate localized contact dermatitis (erythema) was frequently reported during Phase III efficacy studies of MTS, typically observed as slight and transient pinkness at application site. As with other transdermal drug products, some incidence of contact dermatitis should be expected to occur and may be seen even with placebo patches. The patented technology used in MTS eliminates the need for skin permeability enhancers, minimizes dermatitis especially when application sites are alternated as directed (Section “MTS product development”). This generally clears overnight and, in part, is the result of the mechanical removal of stratum corneum by the adhesive of a transdermal system. The limited dermatitis appears to be more of a reaction to the physical properties of the pharmaceutical excipients/adhesives in MTS than to the drug itself (Pelham et al., 2005a, 2005b). However, in a controlled study designed to explore the possibility of significant skin sensitization, 18 of 133 subjects were confirmed to have become sensitized after intentionally applying MTS to the same skin site for 3 weeks. This 13.5% sensitization rate underscores the importance of alternating the skin application sites. The possibility may even exist that some MTS sensitized individuals may thereafter be unable to receive MPH by any route of administration (Anderson and Scott, 2006; Shire, 2006). Cutaneous adverse events with MPH are not limited to administration by the transdermal route and successful desensitization to oral MPH induced rash has been reported (Confino-Cohen and Goldberg, 2005).
On occasion, patches have come off prematurely such as when swimming or changing clothes. Adjunctive taping along the sides of the patch can remedy this (Shire, 2006). Some individuals have reported discomfort at the time of MTS removal. Again, alternating the patch application site minimizes this effect.
MTS should be applied approximately 2 h before therapeutic effects are desired in order to allow for the greater skin transit of MPH than when dosed orally (Anonymous, 2005). Parents have reported an apparent long offset, with effects continuing into the evening even when the MTS was removed at 3:30 p.m. (Pelham et al., 2005a). This time course is consistent with a skin depot of MPH at the application site.
The substantial drug reservoir contained in the MTS poses some accidental poisoning liability. Considering that the 37.5 cm patch contains 82.5 mg of MPH (95 mg of MPH when calculated as the hydrochloride salt as in oral formulations), a single patch chewed and/or swallowed by a small child may result in a medical emergency and the entire content of the patch should be considered in such an event rather than only the labeled dose (Scarman et al., 2007). Even a used MTS poses a poisoning risk since over 60% of the MPH content of a used MTS generally remains in the discarded system. In this context, it is noted that a child suffered a seizure after chewing an antitussive transdermal patch, Triaminic Vapor Patche®, and this misadventure resulted in a product recall (McDermott, 2006).
Development of a pure active isomer of a drug which formerly contained an inactive isomer offers the prospect of retaining efficacy while reducing overall metabolic burden (Aboul-Enein and Wainer, 1997). Consequently, the potential for drug–drug interactions decreases, for instance when the active isomer and the inactive isomer compete for a common metabolic pathway/enzyme. In the case of IR-dl-MPH, the l-isomer component of oral dl-MPH appears to interact with ethanol through the catalytic action of carboxylesterase 1 to yield l-ethylphenidate, and importantly, with a concurrent elevation of d-MPH plasma concentrations (see Section “l-MPH as passive “isomeric ballast” or active component of dl-MPH;” Patrick et al., 2005b, 2007). d-MPH is also a substrate for carboxylesterase 1. Use of MTS results in 20−50 times higher plasma concentrations of the non-therapeutic l-MPH isomer than when using oral dosing (Section “l-MPH as passive “isomeric ballast” or active component of dl-MPH;” Figure 5; Modi et al., 2000; Patrick et al., 2007; Pierce et al., 2008). No ethanol interaction has been noted in the MTS product literature (Shire, 2006) though an ethanol interaction has been indicated in the product literature of an oral ER-MPH product (Concerta®, 2006). Possibly ethanol only interacts with l-MPH during the enteric and hepatic pre-systemic metabolism associated with oral dl-MPH dosing. However, due to the ion trapping effect, which generally drives accumulation of basic drugs such as MPH from the systemic circulation into the stomach (Shore et al., 1957; Levine et al., 1986), even MPH absorbed from MTS may still interact with ethanol in the gut.
The MTS monthly regimen can be expected to cost more than preexisting product line MPH therapies in 2008, for example: $161 for 15 mg or 20 mg/30 d versus $134 for Concerta (36 mg) versus $34 generic (2 × 10 mg).
The influence of geographical differences in ambient temperature on the rate and extent of MPH delivery has potential clinical implications, though such studies have not been reported.
Finally, long-term use of MTS has not been systematically studied (Shire, 2006) and the long-term effects of increased exposure to l-MPH are unknown (Silva, 2006). A recent long-term report on growth of children with ADHD did find that MTS can lead to a growth reduction (Faraone and Giefer, 2007), but this side effect is well known to be associated with any chronic stimulant therapy in growing children (Lisska and Rivkees, 2003).
ABUSE POTENTIAL OF TRANSDERMAL MPH
As consistent with its Schedule II categorization, considerable abuse potential for MPH exists (Klein-Schwartz, 2002; Parasrampuria et al., 2007a). The level of abuse liability appears to be influenced by the specific oral formulation (Kollins et al., 1998; Parasrampuria et al., 2007b). Formulating MPH as MTS requires conversion of the USP MPH hydro-chloride salt into the water insoluble free base form to impart the lipophilicity required for percutaneous penetration. MPH free base can be expected to extract from MTS using any number of commercially available non-polar organic solvents, for example, lighter fluid, cook and lantern fuel, or cold weather engine starter (diethyl ether). However, as the free base, MPH can no longer be solubilized in water, thus eliminating the potential for intravenous abuse (Levine et al., 1986). As with the free base form of cocaine, MPH free base should also prevent intranasal abuse (Barrett et al., 2005) due to its inability to be dissolved in the moist mucosal sinuses. This insolubility characteristic not withstanding, the possibility exists that MPH free base could be extracted from MTS and smoked, as in the abuse of the free base forms of cocaine “crack” (Lakoski et al., 1992) and meth-amphetamine “ice” (Karch, 1996). Note that as the oral hydrochloride salt, MPH (and certainly cocaine) is expected to primarily pyrolyze rather than volatilize under flame which limits, though not eliminates, the abuse potential of MPH by smoking (Darredeau et al., 2007).
In studies of the abuse potential of MTS, Shire, with prompting from the FDA, gauged the subjective effects in adult volunteers with a history of stimulant abuse. Upon application of MTS (3 or 6 × 25 cm2 MTS), mild euphoria was reported which was found to be similar to the comparator stimulant oral phentermine (a Schedule IV drug). All subjects experienced euphoria with the three MTS applications. However, 42% of subjects reported dysphoria when administered the simultaneous six MTS doses. Further, when MTS was applied to the buccal mucosa, over 50% of the dose was delivered within 2 h (vs. 36% over 9 h for torso placement, see Section “MTS product development”). This rapid absorption appears to be facilitated by the lack of a stratum corneum in the buccal mucosa and the high vascularity in this region. The addition of an astringent or distasteful ingredient to the patch matrix formulation may be worth considering to reduce oral cavity MTS abuse liability (Noven/Shire, 2005a, 2005b), though the existing MTS can be expected to be very distasteful in its own right.
As with other ADHD stimulant formulations (Wilens et al., 2008a), used MTS patches are potential candidates for drug diversion. For instance, a 25 cm2 patch may contain approximately 35 mg of MPH free base left in the patch after the recommended 9 h wear. This patch originally contains 55 mg which delivers MPH at 2.2 mg/h = 20 mg/9 h. When the same patch is worn a second time, appreciable drug absorption continues, although at a much reduced rate than from the fresh product. Quantifying the delivery of MPH after repeated applications, Shire found that following a 16 h wear, when the same patch was again applied for another 16 h, that is, a 32 h total, 27.4 mg of the 55 mg MPH content was delivered, of which 60% was delivered in the first 16 h and 40% during the second 16 h period (Andreason, 2005).
In response to the above abuse concerns, a comprehensive risk management program has been instituted, with support from the FDA, to address the proper control of the used MTS. This program includes a chart system with the administrator's initials, wear times, and the method of disposal to minimize the risk of diversion. Proper disposal entails folding the adhesive surface over on itself whereby the patch cannot be reopened without destruction (Noven/Shire, 2005a, 2005b).
CONCLUSIONS
While oral solutions and chewable tablets of MPH have become available within the last several years to overcome potential swallowing difficulties, only MTS or sprinkled beaded MPH formulations also provide for single daily dosing. In addition, MTS offers the unique flexibility of individualizing MPH exposure by allowing patient-specific wear times of this transdermal patch. Detectable plasma concentrations following MTS application typically exhibit a longer lag time than after oral MPH dosing. Then, unique to MTS product, the drug plasma concentration ascends at a nearly constant rate until the end of the recommended 9 h patch removal. This novel MPH absorption profile characterizing MTS offers yet another treatment option in the drug individualization of ADHD patients. Finally, it has not been established whether there is any clinical significance to the much greater patient exposure (AUC) to l-MPH when dl-MPH is dosed transdermally as compared to oral dosing.
ACKNOWLEDGEMENTS
This review was supported in part by the NIH/NIDA (RO1 DA-15797) and NIAAA (RO1 AA016707).
Footnotes
FINANCIAL DISCLOSURES
K.S. Patrick serves as a consultant to Johnson & Johnson, Celgene, Janssen-Ortho, UCB, Lilly and has consulted with Novartis and Clariant.
A.B. Straughn serves as a consultant to Novartis and Purdue Pharma.
M.A. González has consulted with Noven, Shire, and Johnson & Johnson.
Ethics
Only ethical practices were engaged in the development of this review manuscript.
REFERENCES
- Abdou HM, Hanna S, Muhammad N. Dissolution. In: Gennado AR, editor. Remington: The Science and Practice of Pharmacy. 20th edn Lippincott Williams & Wilkins; Baltimore: 2000. pp. 654–666. [Google Scholar]
- Aboul-Enein HY, Wainer IW. The Impact of Stereochemisrty on Drug Development and Use. John Wiley & Sons; New York: 1997. [Google Scholar]
- Ahmed S, Imai T, Yoshigae Y, Otagiri M. Stereospecific activity and nature of metabolizing esterases for propranolol in hairless mouse skin, liver and plasma. Life Sci. 1997;61:1879–1887. doi: 10.1016/s0024-3205(97)00827-8. [DOI] [PubMed] [Google Scholar]
- Anderson VR, Scott LJ. Methylphenidate transdermal system. Drugs. 2006;66:1117–1126. doi: 10.2165/00003495-200666080-00007. [DOI] [PubMed] [Google Scholar]
- Andreason PJ. Methylphenidate transdermal patch. 2005 www.fda.gov/ohrms/dockets/ac/05/slides/2005-4195 S1 01 FDA-Andreason.ppt.
- Anonymous Transdermal methylphenidate (Daytrana) for ADHD. Med Lett. 2006;48(1237):49–51. [PubMed] [Google Scholar]
- Aoyama T, Sasaki T, Kotaki J. Pharmacokinetics and pharmacodynamics of (+)-threo-methylphenidate enantiomer in patients with hypersomnia. Clin Pharmacol Ther. 1994;55:270–276. doi: 10.1038/clpt.1994.27. [DOI] [PubMed] [Google Scholar]
- Arnold LE, Huestis RD, Wemmer D, Smeltzer DJ. Differential effect of amphetamine optical isomers on Bender Gestalt performance of the minimally brain dysfunctional. J Learn Disabil. 1978;11:14–19. doi: 10.1177/002221947801100303. [DOI] [PubMed] [Google Scholar]
- Arnold LE, Lindsey RL, Conners CK, et al. A double-blind, placebo-controlled withdrawal trial of dexmethylphenidate hydrochloride in children with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol. 2004;14:542–554. doi: 10.1089/cap.2004.14.542. [DOI] [PubMed] [Google Scholar]
- Arnold LE, Wender PH, McCloskey K, Snyder PH. Levoamphetamine and dextroamphetamine: comparative efficacy in the hyperkinetic syndrome. Arch Gen Psychiatry. 1972;27:816–822. doi: 10.1001/archpsyc.1972.01750300078015. [DOI] [PubMed] [Google Scholar]
- Baldessarini B, Cambell A. Method of dopamine inhibition using l-methylphenidate. 2001. US Patent 6,221,883.
- Barrett SP, Darredeau C, Bordy LE, Pihl RO. Charateristics of methylphenidate misuse in a university student sample. Can J Psychiatry. 2005;50:457–461. doi: 10.1177/070674370505000805. [DOI] [PubMed] [Google Scholar]
- Barrett SP, Pihl RO. Oral methylphenidate-alcohol co-abuse. J Clin Psychopharmacol. 2002;22:633–634. doi: 10.1097/00004714-200212000-00020. [DOI] [PubMed] [Google Scholar]
- Bass DM, Prevo M, Waxman DS. Gastrointestinal safety of an extended-release, nondeformable, oral dosage form (OROS): a retrospective study. Drug Safety. 2002;25:1021–1024. doi: 10.2165/00002018-200225140-00004. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Birmaher B, Greenhill LL, Cooper TB, Fried J, Maminski B. Sustained release methylphenidate: pharmacokinetic studies in ADDH males. J Am Acad Child Adolesc Psychiatry. 1989;28:768–772. doi: 10.1097/00004583-198909000-00020. [DOI] [PubMed] [Google Scholar]
- Blick SKA, Keating GM. Lisdexamfetamine. Pediatr Drugs. 2007;9:129–135. doi: 10.2165/00148581-200709020-00007. [DOI] [PubMed] [Google Scholar]
- Block LH. Medicated topicals. In: Gennaro AR, editor. Remington: The Practice and Science of Pharmacy. 20th edn Lippincott Williams & Wilkins; Baltimore MD: 2000. pp. 836–857. [Google Scholar]
- Borycz J, Zapata A, Quiroz C, Volkow ND, Ferre S. 5-HT1B receptor-mediated serotonergic modulation of methylphenidate-induced lcomotor activation in rats. Neuropsychopharmacology. 2008;33:619–626. doi: 10.1038/sj.npp.1301445. [DOI] [PubMed] [Google Scholar]
- Boswell MV. Fentanyl transdermal system overdose secondary to cutaneous hyperthemlia. Anesth Analg. 1992;77:390–391. doi: 10.1213/00000539-199308000-00029. [DOI] [PubMed] [Google Scholar]
- Bradley C. Behavior of children receiving benzedrine. Am J Psychiatry. 1937;94:577–585. [Google Scholar]
- Bradley C. Benzedrine and Dexedrine in the treatment of children's behavior disorders. Pediatrics. 1950;5:24–36. [PubMed] [Google Scholar]
- Brown GL, Ebert MH, Mikkelsen EJ, Hunt RD. Behavioral and motor activity response in hyperactive children and plasma amphetamine levels following a sustained release preparation. J Am Acad Child Psychiatry. 1980;19:225–239. doi: 10.1016/s0002-7138(09)60699-3. [DOI] [PubMed] [Google Scholar]
- Bruno KJ, Hess EJ. The α2c-adrenergic receptor mediates hyperactivity of coloboma mice, a model of attention deficit hyperactivity disorder. Neurobiol Dis. 2006;23:679–688. doi: 10.1016/j.nbd.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Cefali EA, Banfield CR, González MA, Wagner JG. In vivo determination of zero-order absorption from a transdermal glycerol trinitrate system. Eur J Pharm Biopharm. 1993;39:140–143. [Google Scholar]
- Chan YP, Swanson JM, Soldi SS, Thiessen JJ, Macleod SM, Logan W. Methylphenidate hydrochloride given with or before breakfast: II. Effect on plasma concentrations of methylphenidate and ritalinic acid. Pediatrics. 1983;72:56–59. [PubMed] [Google Scholar]
- Concerta Janssen-Ortho. Product monograph. 2006. 25 July; No: 106348.
- Confino-Cohen R, Goldberg A. Sussessful desensitization of methylphenidate-induced rash. J Child Adolesc Psychopharmacol. 2005;15:703–705. doi: 10.1089/cap.2005.15.703. [DOI] [PubMed] [Google Scholar]
- Darredeau C, Barrett SF, Jardin B, Pihl RO. Patterns and predictors of medication compliance, diversion, and misuse in adult prescribed methylphenidate users. Hum Psychophaharmacol. 2007;22:529–536. doi: 10.1002/hup.883. [DOI] [PubMed] [Google Scholar]
- Davidson EJ, Craig F. Treatment of convulsive states. 2000. US Patent 7384959.
- Davids E, Zhang K, Tarazi FI, Baldessarini RJ. Stereoselective effects of methylphenidate on motor hyperactivity in juvenile rats induced by neonatal 6-hydroxydopamine lesioning. Psychopharmacology. 2002;160:92–98. doi: 10.1007/s00213-001-0962-5. [DOI] [PubMed] [Google Scholar]
- DAWN: Drug Abuse Warning Network Annual emergency department data. DHHS. Substance Abuse and Mental Health Services Administration. 2003 [PubMed] [Google Scholar]
- Ding YS, Fowler JS, Volkow ND. Is the l-threo enantiomer of methylphenidate (Ritalin) inactive in the brain when given orally? [abstr]. Am Col Neuropsychopharmacol. 2002;8–12:255. [Google Scholar]
- Ding YS, Gatley SJ, Thanos PK, et al. Brain kinetics of methylphenidate (Ritalin) enantiomers after oral administration. Synapse. 2004;53:168–175. doi: 10.1002/syn.20046. [DOI] [PubMed] [Google Scholar]
- Dulcan MK. Using psychostimulants to treat behavioral disorders of children and adolescents. J Child Adoles Psychopharmcol. 1990;1:7–20. doi: 10.1089/cap.1990.1.7. [DOI] [PubMed] [Google Scholar]
- DuPont RL, Buched RH, Wilford BB, Coleman JJ. School-based administration of ADHD drugs decline, along with diversion, theft, and misuse. J Sch Nurs. 2007;23:349–352. doi: 10.1177/10598405070230060801. [DOI] [PubMed] [Google Scholar]
- Eckerman DA, Moy SS, Perkins AN, Patrick KS, Breese GR. Enantioselective behavioral effects of methylphenidate in rats. Pharmacol Biochem Behav. 1991;40:875–880. doi: 10.1016/0091-3057(91)90100-g. [DOI] [PubMed] [Google Scholar]
- Faraj BA, Israili ZH, Perel JM, et al. Metabolism and disposition of methylphenidate-14C in man and animals. J Exper Pharmacol Ther. 1974;191:535–547. [PubMed] [Google Scholar]
- Faraone SV, Giefer EE. Long-term effects of methylphenidate transdermal delivery system treatment of ADHD on growth. J Am Acad Adolesc Psychiatry. 2007;46:1138–1147. doi: 10.1097/chi.0b013e31806ad1d7. [DOI] [PubMed] [Google Scholar]
- Findling R, Lopez FA. The effects of transdermal methylphenidate with reference to Concerta in ADHD. Sci Proc Am Acad Child Adolesc Psychiatry. 2005;32:123. [Google Scholar]
- Findling RL, Bukstein OG, Melmed RD, et al. A randomized, double-blind, plascebo-controlled, parallel-group study of methylphenidate transdermal system in pediatric patientswith attention-deficit/hyperactivity disorder. J Clin Psychiatre. 2008;69:149–159. doi: 10.4088/jcp.v69n0120. [DOI] [PubMed] [Google Scholar]
- Froimowitz M, Gu Y, Dakin LA, et al. Slow-onset, long-duration, alkyl analogues of methylphenidate with selectivity for the dopamine transporter. J Med Chem. 2007;50:219–232. doi: 10.1021/jm0608614. [DOI] [PubMed] [Google Scholar]
- Froimowitz M, Patrick K, Cody V. Conformational analysis of methylphenidate and its structural relationship to other dopamine reuptake blockers such as CFT. Pharm Res. 1995;12:1430–1434. doi: 10.1023/a:1016262815984. [DOI] [PubMed] [Google Scholar]
- Frolich M, Giannotti A, Modell JH. Opioid overdose in a patient using a fentanyl patch during treatment with a warming blanket. Anesth Analg. 2001;93:647–648. doi: 10.1097/00000539-200109000-00023. [DOI] [PubMed] [Google Scholar]
- Glaser PEA, Thomas TC, Joyce BM, Castellanos FX, Gerhardt GA. Differential effects of amphetamine isomers on dopamine release in the rat striatum and nucleus accumbens. Psychopharmacology. 2005;178:250–258. doi: 10.1007/s00213-004-2012-6. [DOI] [PubMed] [Google Scholar]
- Glaze SA, Robertson DW, Wise LD. Attention deficit-hyperactivity disorder: pathophysiology and design of new treatments. Annu Rep Med Chem. 2002;37:11–20. [Google Scholar]
- Gonzalez MA, Pentikis HS, Anden N, et al. Methylphenidate bioavailability from two extended-release formulations. Inter J Clin Pharmacol Ther. 2002;40:175–184. doi: 10.5414/cpp40175. [DOI] [PubMed] [Google Scholar]
- Goyer PF, Davies GC, Rapoport JL. Abuse of prescription stimulant medication by a 13-year-old hyperactive boy. J Am Acad Child Psychiatry. 1979;18:170–175. doi: 10.1016/s0002-7138(09)60486-6. [DOI] [PubMed] [Google Scholar]
- Gozal D, Molfeses DL. Attention Deficit Hyperactivity Disorder: From Genes to Patients. Humana Press; Totowa, NJ: 2005. [Google Scholar]
- Greenhill LL. Pharmacologic treatment of attention deficit hyperactivity disorder. Psychiatr Clin North Am. 1992;15:1–27. [PubMed] [Google Scholar]
- Gutstein HB, Akil H. Opioid analgesics. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's the Pharmacological Basis of Therpeutics. 11th edn McGraw-Hill; New York: 2006. pp. 547–590. [Google Scholar]
- Haessler F, Tracik F, Stammer H, Klatt J. A pharmacokinetic study of two modified-release methylphenidate formulations under different food conditions in healthy volunteers. Int J Clin Pharmacol Ther. 2008;46:466–476. doi: 10.5414/cpp46466. [DOI] [PubMed] [Google Scholar]
- Heal DJ, Pierce DM. Methylphenidate and its isomers: their role in the treatment of attention-deficit hyperactivity disorder using transdermal delivery system. CNS Drugs. 2006;20:713–734. doi: 10.2165/00023210-200620090-00002. [DOI] [PubMed] [Google Scholar]
- Heil SH, Holmes HW, Bickel WK, et al. Comparison of the subjective, physiological, and psychotor effects of atomexetine and methylphenidate in light drug abusers. Drug Alcohol Depend. 2002;67:149–156. doi: 10.1016/s0376-8716(02)00053-4. [DOI] [PubMed] [Google Scholar]
- Hitri A, Hurd Y, Wyatt RJ, Deutsch SI. Molecular, functional and biochemical characteristics of the dopamine transporter: regional differences and clinical relevance. Clin Neuropharmacol. 1994;17:1–22. doi: 10.1097/00002826-199402000-00001. [DOI] [PubMed] [Google Scholar]
- Hull W. Heat-enhanced drug delivery: a survey paper. J Appl Res Clin Exp Ther. 2002;2:69–76. [Google Scholar]
- Hurwitz J. Swallowing tablets and capsules: a guide for parents, care givers, and children 4 years and up. Duke Pediatric Clinical Research Program. 2001. Room 1080, Duke South Hospital, Durham, NC 27710.
- Jaffe SL. Intranasal abuse of prescribed methylphenidate by an alcohol and drug abusing adolescent with ADHD. J Am Acad Child Adolesc Psychiatry. 1991;30:773–775. [PubMed] [Google Scholar]
- Jain U, Hechtman L, Weiss M, et al. Efficacy of a novel biphasic controlled-release methylphenidate formula in adults with attention-deficit/hyperactivity disorder: results of a double-blind, placebo-controlled crossover study. J Clin Psychiatry. 2007;68:268–277. doi: 10.4088/jcp.v68n0213. [DOI] [PubMed] [Google Scholar]
- Janowsky DS, Davis JM. Methylphenidate, dextroamphetamine and levamfetamine. Arch Gen Psychiatry. 1976;33:304–308. doi: 10.1001/archpsyc.1976.01770030024003. [DOI] [PubMed] [Google Scholar]
- Jonkman LM, Verbaten MN, Deboer D, et al. Differences in plasma concentrations of the D- and L-threo methylphenidate enantiomers in responding and non-responding children with attention-deficit hyperactivity disorder. Psychiatry Res. 1998;78:115–118. doi: 10.1016/s0165-1781(97)00138-8. [DOI] [PubMed] [Google Scholar]
- Kagan L, Hoffman A. Systems for region selective drug delivery in the gastrointestinal tract: biopharmaceutical considerations. Expert Opin Drug Deliv. 2008;5:681–692. doi: 10.1517/17425247.5.6.681. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Galli A. Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine. Eur J Pharmacol. 2003;479:153–158. doi: 10.1016/j.ejphar.2003.08.065. [DOI] [PubMed] [Google Scholar]
- Karch SB. The Pathology of Drug Abuse. 2nd edn. CRC Press; Boca Raton: 1996. pp. 199–240. [Google Scholar]
- Khetani V, Faleck H. Method of diminishing co-abuse potential. 2004. US Patent App. 20050239830.
- Klein-Schwartz W. Abuse and toxicity of methylphenidate. Curr Opin Pediatr. 2002;14:219–223. doi: 10.1097/00008480-200204000-00013. [DOI] [PubMed] [Google Scholar]
- Kollins SH, Rush CR, Pazzaglia PJ, Ali JA. Comparison of acute behavioral effects of sustained-release and immediate-release methylphenidate. Exp Clin Psychopharmacol. 1998;6:367–374. doi: 10.1037//1064-1297.6.4.367. [DOI] [PubMed] [Google Scholar]
- Kowalik S, Minami H, Silva RR. Critical assessment of the methylphenidate transdermal system. Drugs Today. 2007;43:515–527. doi: 10.1358/dot.2007.43.8.1062674. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Locomotor effects of acute and repeated threshold doses of amphephetamine and methylphenidate: Relative roles of dopamine and norepinephrine. J Pharmacol Exper Ther. 2001;296:876–883. [PubMed] [Google Scholar]
- Lakoski JM, Galloway MP, White FJ. Cocaine Pharmacology, Physiology and Clinical Strategies. CRC Press; Boca Raton: 1992. [Google Scholar]
- Lee L, Kepple J, Wang Y, et al. Bioavailability of modified-release methylphenidate: influence of high-fat breakfast when administered intact and when capsule content sprinkled on applesauce. Biopharm Drug Dispos. 2003;24:233–243. doi: 10.1002/bdd.358. [DOI] [PubMed] [Google Scholar]
- LeVasseur NL, Zhu H-J, Markowitz JS, DeVane CL, Patrick KS. Enantiospecific gas chromatographic-mass spectrometric analysis of urinary methylphenidate: implications for phenotyping. J Chromatogr B. 2008;862:140–149. doi: 10.1016/j.jchromb.2007.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Caplan YH, Kauffman G. Fatality resulting from methylphenidate overdose. J Anal Toxicol. 1986;10:209–210. doi: 10.1093/jat/10.5.209. [DOI] [PubMed] [Google Scholar]
- Lisska M, Rivkees SA. Dailey methylphenidate use slows the growth of children: a community based study. J Pediatr Endocrinol Metab. 2003;16:711–718. doi: 10.1515/jpem.2003.16.5.711. [DOI] [PubMed] [Google Scholar]
- Llana ME, Crismon ML. Methylphenidate: increased abuse or appropriate use? J Am Pharm Assoc. 1999;39:526–530. doi: 10.1016/s1086-5802(16)30473-9. [DOI] [PubMed] [Google Scholar]
- Mantelle J, Dixon TA. Compositions and methods for treatment of attention deficit disorder and attention deficit/hyperactivity disorder with methylphenidate. 2001. US Patent 6,210,705 B1.
- Markowitz JS, DeVane CL, Pestreich LK, Patrick KS, Muniz R. Comprehensive in vitro screening of d-, l- and dl-methylphenidate: an exploratory study. J Child Adolesc Psychopharmacol. 2006;16:687–698. doi: 10.1089/cap.2006.16.687. [DOI] [PubMed] [Google Scholar]
- Markowitz JS, Patrick KS. Differential pharmacokinetics and pharmacodynamics of methylphenidate enantiomers: does chirality matter? J Clin Psychopharmacol. 2008;28:S54–S61. doi: 10.1097/JCP.0b013e3181733560. [DOI] [PubMed] [Google Scholar]
- Markowitz JS, Straughn AB, Patrick KS. Advances in the pharmacotherapy of attention—deficit hyperactivity disorder. Pharmacotherapy. 2003a;23:1281–1299. doi: 10.1592/phco.23.12.1281.32697. [DOI] [PubMed] [Google Scholar]
- Markowitz JS, Straughn AB, Patrick KS, et al. Pharmacokinetics of two modified-release methylphenidate formulations in normal volunteers: Ritalin LA versus Concerta. Clin Pharmacokinet. 2003b;42:393–401. doi: 10.2165/00003088-200342040-00007. [DOI] [PubMed] [Google Scholar]
- Mays DA, Findling R. Methylphenidate transdermal system. J Am Acad Child Adolesc Psychiatry. 2005;44:1223. doi: 10.1097/01.chi.0000183461.18907.bc. [DOI] [PubMed] [Google Scholar]
- McConnell EL, Fadda HM, Basit AW. Gut instincts: explorations in intestinal physiology and drug delivery. Int J Pharm. 2008 doi: 10.1016/j.ijpharm.2008.05.012. in press. Available online 20 May 2008, DOI: 10.1016/j.ipharm.2008.05.012. [DOI] [PubMed] [Google Scholar]
- McDermott C. FDA warns consumers not to use Triaminic vapor patch. FDA News. 2006 June 19;:P06–P82. [Google Scholar]
- McDougall SA, Collins RL, Karper PE. Effects of repeated methylphenidate treatment in the young rat: sensitization of both locomotor activity and stereotyped sniffing. Exp Clin Psychopharmacol. 1999;7:208–218. doi: 10.1037//1064-1297.7.3.208. [DOI] [PubMed] [Google Scholar]
- McGough JJ, Wigal SB, Abikoff H, Turnbow JM, Posner K, Moon E. A randomized, double-blind, placebo-controlled, laboratory classroom assessment of methylphenidate transdermal system in children with ADHD. J Attent Disord. 2006;9:476–485. doi: 10.1177/1087054705284089. [DOI] [PubMed] [Google Scholar]
- McIntyre MB, Firemark HM, Cho AK, Cho AK, Bodner L, Gomez M. Computer analyzed EEG in amphetamine-response hyperactive children. Psychiatry Res. 1981;4:189–198. doi: 10.1016/0165-1781(81)90022-6. [DOI] [PubMed] [Google Scholar]
- Meyer MC, Straughn AB, Jarvi EJ, et al. Bioequivalence of methylphenidate immediate-release tablets using a replicated study design. Pharmaceut Res. 2000;17:381–384. doi: 10.1023/a:1007560500301. [DOI] [PubMed] [Google Scholar]
- Midha KK, Teicher M, Kumar V. Method of treating depression using l-threo-methylphenidate. 2002. US Patent 6,395,752.
- Modi NB, Lindemulder G, Gupta SK. Single-and multiple-dose pharmacokinetics of oral once-a-day osmotic controlled-release OROS™ (methylphenidate HCl) formulation. J Clin Pharmacol. 2000;40:379–388. doi: 10.1177/00912700022009080. [DOI] [PubMed] [Google Scholar]
- MTA Cooperative Group National Institute of Mental Health Multimodal Treatment Study of ADHD follow-up: changes in effectiveness and growth after the end of treatment. Pediatrics. 2004;113:762–769. doi: 10.1542/peds.113.4.762. [DOI] [PubMed] [Google Scholar]
- Noonan PK, González MA. Pharmacokinetics and the variability of percutaneous absorption. J Toxicol Cutaneous Ocul Toxicol. 1989;8:511–516. [Google Scholar]
- Novak SP, Kroutil LA, Williams RL, Van Brunt DL. The nonmedical use of prescription ADHD medications: results from a national Internet panel. Subst Abuse Treat Prevent Policy. 2007;2:1–17. doi: 10.1186/1747-597X-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noven/Shire [3 May 2007];2005a Pharmacokinetics/pharmacodynamics summary NDA 21− 514. 24 October: 1–43. http://fda.gov/ohrms/dockets/ac/o5/briefing/2005-4195 B1 01 02-Noven-Appendix-1.pdf.
- Noven/Shire Methylphenidate transdermal system NDA 21−514. FDA Psychopharmacologic Drugs Advisory Committee Briefing Document. 2005b December 2;:1–30. [Google Scholar]
- Oades RD, Sadile AG, Sagvolden T, et al. The control of responsiveness in ADHD by catecholamines: evidence for dopaminergic, noradrenergic and interactive roles. Dev Sci. 2005;8:122–131. doi: 10.1111/j.1467-7687.2005.00399.x. [DOI] [PubMed] [Google Scholar]
- Okie S. ADHD in adults. N Engl J Med. 2006;354:2637–2641. doi: 10.1056/NEJMp068113. [DOI] [PubMed] [Google Scholar]
- Pappinen S, Urtti A. Microemulsions in topical drug delivery. In: Smith EW, Mailbach HI, editors. Percutaneous Penetration Enhancers. 2nd edn CRC; New York: 2006. pp. 109–116. [Google Scholar]
- Parasrampuria DA, Schoedel KA, Gu J, Ciccone P, Silber SA, Sellers EM. Assessment of pharmacokinetics and pharmacodynamic effects related to abuse potential of a unique oral osmotic-controlled extended-release methylphenidate formulation in humans. J Clin Pharamcol. 2007a;47:1476–1488. doi: 10.1177/0091270007308615. [DOI] [PubMed] [Google Scholar]
- Parasrampuria DA, Schoedel KA, Schuller R, et al. Do formulation differences alter abuse liability of methylphenidate?: a placebo-controlled, randomized, double-blind, crossover study in recreational drug users. J Clin Psychopharmacol. 2007b;27:459–467. doi: 10.1097/jcp.0b013e3181515205. [DOI] [PubMed] [Google Scholar]
- Patrick KS. Dexmethylphenidate extended-release; guest commentary. Drugs. 2006a;66:669–670. [Google Scholar]
- Patrick KS, Caldwell RW, Ferris RM, Breese GR. Pharmacology of the enantiomers of threo-methylphenidate. J Pharmacol Exp Ther. 1987;241:152–158. [PubMed] [Google Scholar]
- Patrick KS, Kilts CD, Breese GR. Synthesis and pharmacology of hydroxylated metabolites of methylphenidate. J Med Chem. 1981;29:1237–1240. doi: 10.1021/jm00142a021. [DOI] [PubMed] [Google Scholar]
- Patrick KS, Gonzalez MA, Straughn AB, Markowitz JS. New methylphenidate formulations for the treatment of attentiondeficit/hyperactivitydisorder. Exp Opin Drug Deliv. 2005a;2:121–143. doi: 10.1517/17425247.2.1.121. [DOI] [PubMed] [Google Scholar]
- Patrick KS, Markowitz JS. Methylphenidate, amphetamine enantiomers and pemoline in the treatment of attention-deficit hyperactivity disorder. Human Psychopharmacol. 1997;12:527–546. [Google Scholar]
- Patrick KS, Straughn AB, Jarvi EJ, Meyer MC. The absorption of sustained-release methylphenidate formulations compared to an immediate-release formulation. Biopharm Drug Dispos. 1989;10:165–171. doi: 10.1002/bdd.2510100206. [DOI] [PubMed] [Google Scholar]
- Patrick KS, Straughn AB, Minhinnett RR, et al. Influence of ethanol and gender on methylphenidate pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2007;81:346–353. doi: 10.1038/sj.clpt.6100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick KS, Williard RL, VanWert AL, Dowd JJ, Oatis JE, Jr, Middaugh LD. Synthesis and pharmacology of ethylphenidate enantiomers: the human transesterification metabolite of methylphenidate and ethanol. J Med Chem. 2005b;48:2876–2881. doi: 10.1021/jm0490989. [DOI] [PubMed] [Google Scholar]
- Pelham WE, Burrows-MacLean L, Gnagy EM, et al. Transdermal methylphenidate, behavioral, and combined treatment for children with ADH. Exper Clin Psychopharmacol. 2005a;13:111–126. doi: 10.1037/1064-1297.13.2.111. [DOI] [PubMed] [Google Scholar]
- Pelham WE, Greenslade KE, Vodde-Hamilton M, et al. Relative efficacy of long-acting stimulants on children with attention deficithyperactivity disorder: a comparison of standard methylphenidate, sustained-release methylphenidate, sustained-release dextroamphetamine, and pemoline. Pediatrics. 1990;86:226–237. [PubMed] [Google Scholar]
- Pelham WE, Manos MJ, Essell CE, et al. A dose-ranging study of a methylphenidate transdermal system in children with ADHD. J Am Acad Child Adolesc Psychiatry. 2005b;44:522–529. doi: 10.1097/01.chi.0000157548.48960.95. [DOI] [PubMed] [Google Scholar]
- Pelham WE, Stuges J, Hoza J, et al. Sustained release and standard methylphenidate effects on cognitive and social behavior in children with attention deficit disorder. Pediatrics. 1987;80:491–501. [PubMed] [Google Scholar]
- Penberthy JK, Cox D, Breton M, et al. Calibration of ADHD assessments across studies: a meta-analysis tool. Appl Psychophysiol Biofeedback. 2005;30:31–51. doi: 10.1007/s10484-005-2172-0. [DOI] [PubMed] [Google Scholar]
- Pentikis HS, Simmons RD, Benedict MF, Hatch SJ. Methylphenidate bioavailability in adults when an extended-release mutiparticulate formulation is administered on food or as an intact capsule. J Am Acad Child Adolesc Psychiatry. 2002;41:443–449. doi: 10.1097/00004583-200204000-00017. [DOI] [PubMed] [Google Scholar]
- Perrin J, Erenberg G, LaCamera R, et al. Medication for children with attentional disorders. Pediatrics. 1996;98:301–304. [PubMed] [Google Scholar]
- Pierce DM, Dixon C, Wigal S, McGough J. Pharmacokinetics of methylphenidate transdermal system in children with ADHD. 52nd Sci Proc Am Acad Child Adolesc Psychiatry. 2005;131:B43. [Google Scholar]
- Pierce D, Dixon CM, Wigal SB, McGough JJ. Pharmacokinetics of methylphenidate transdermal system (MTS): Results from a laboratory classroom study. J Child Adolesc Psychopharmacol. 2008;18:355–364. doi: 10.1089/cap.2007.0148. [DOI] [PubMed] [Google Scholar]
- Pleuvry BJ. Hystertesis in drug response. Anesth Inten Care Med. 2005;6:286–287. [Google Scholar]
- Popper CW. The story of four salts. J Child Adolesc Psychiatry. 1994;4:217–223. [Google Scholar]
- Prashad M. Approaches to the preparation of enantiomerically pure (2R,2’R)-(+)-threo-methylphenidate hydrochloride. Adv Synth Catal. 2001;343:379–392. [Google Scholar]
- Quinn D, Bode T, Reiz JL, Donnelly GAE, Darke AC. Single-dose pharmacokinetics of multilayer-release methylphenidate and immediate-release methylphenidate in children with attention-deficit/hyperactivity disorder. J Clin Pharmacol. 2007;47:760–766. doi: 10.1177/0091270007299759. [DOI] [PubMed] [Google Scholar]
- Quinn DMP. Ritalin: Theory and Practice. 2nd edn Liebert Pub; New York: 2000. Methylphenidate: the role of the d-isomer. pp. 369–374. [Google Scholar]
- Quinn D, Wigal S, Swanson J, et al. Comparative pharmacodynamics and plasma concentrations of d-threo-methylphenidate hydrochloride after single doses of d-threo-methylphenidate and dl-threo-methylphenidate hydrochloride in a double-blind, placebo-controlled, crossover laboratory school study in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2004;43:1422–1429. doi: 10.1097/01.chi.0000140455.96946.2b. [DOI] [PubMed] [Google Scholar]
- Rappley MD. Attention deficit-hyperactivity disorder. N Engl J Med. 2005;352:165–173. doi: 10.1056/NEJMcp032387. [DOI] [PubMed] [Google Scholar]
- Reiz JL, Donnelly GAE, Michalko K. Comparative bioavailability of single-dose methylphenidate from a multilayer-release bead formulation and an osomotic system: a two-way crossover study in health young adults. Clin Ther. 2008;30:59–69. doi: 10.1016/j.clinthera.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Robinson DM, Keating GM. Dexmethylphenidate extended-release. Drugs. 2006;66:661–668. doi: 10.2165/00003495-200666050-00006. [DOI] [PubMed] [Google Scholar]
- Rometsch R. Process for conversion of streoisomers. 1958. US Patent 2,838,519.
- Rouhi AM. Chirality at work. Chem Eng News May. 2003;5:56–61. [Google Scholar]
- Scarman EJ, Erdman AR, Cobaugh DJ, et al. Methylphenidate poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol. 2007;45:737–752. doi: 10.1080/15563650701665175. [DOI] [PubMed] [Google Scholar]
- Schachar R, Ickowitz A, Crosbie J, et al. Cognitive and behavioral effects of multilayer-release methylphenidate in the treatment of children with attention-deficit/hyperactivity disorder. J Child Adoles Psychopharmacol. 2008;18:11–24. doi: 10.1089/cap.2007.0039. [DOI] [PubMed] [Google Scholar]
- Scheffler RM, Hinshaw SP, Modrek S, Levine P. The global market for ADHD medications. Health Trends. 2007;26:450–457. doi: 10.1377/hlthaff.26.2.450. [DOI] [PubMed] [Google Scholar]
- Shafi'ee A, Hite G. Stereochemical studies on medicinal agents. IV. Conformational analysis of ephedrine isomers and related compounds. J Med Chem. 1969;10:1057–1063. doi: 10.1021/jm00318a015. [DOI] [PubMed] [Google Scholar]
- Shire Pharmaceuticals Ireland Limited Daytrana prescribing information. 2006. Rev 04/06.
- Shlomi D, Shitrit D, Bendayan D, Sahar G, Shechtman Y, Kamer MR. Successful lung transplantation for talcosis secondary to intravenous abuse of oral drug. Int J Chron Obstruct Pulmon Dis. 2008;3:327–330. doi: 10.2147/copd.s2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore PA, Brodie BB, Hogben CA. The gastric secretion of drugs: a pH partion hypothesis. J Pharmacol Exp Ther. 1957;119:361–369. [PubMed] [Google Scholar]
- Silva RR. Methylphenidate transdermal system in attention-deficit hyperactivity disorder. Drugs. 2006;66:1127–1128. doi: 10.2165/00003495-200666080-00008. [DOI] [PubMed] [Google Scholar]
- Silva R, Muniz R, McCague K, Childress A, Brams M, Mao A. Treatment of children with attention-deficit/hyperactivity disorder: results of a randomized, multicenter, double-blind, crossover study of extended-release dexmethylphenidate and d,l-methylphenidate and and placebo in a laboratory classroom setting. Psychopharmacol Bull. 2008;41:19–33. [PubMed] [Google Scholar]
- Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998;94:127–152. doi: 10.1016/s0166-4328(97)00175-7. [DOI] [PubMed] [Google Scholar]
- Spencer TJ, Biederman J, Ciccone PE, et al. PET study examining pharmacokinetics, detection and likeability, and dopamine transporter receptor occupancy of short- and long-acting oral methylphenidate. Am J Psychiatry. 2006;163:387–395. doi: 10.1176/appi.ajp.163.3.387. [DOI] [PubMed] [Google Scholar]
- Srinivas NR, Barbhaiya RH, Midha KK. Enantiomeric drug development: issues, considerations, and regulatory requirements. J Pharm Sci. 2001;90:1205–1215. doi: 10.1002/jps.1074. [DOI] [PubMed] [Google Scholar]
- Srinivas NR, Hubbard JW, Quinn D, Midha KK. Enantioselective pharmacokinetics and pharmacodynamics of dl-threo-methylphenidate in children with attention deficit hyperactivity disorder. Clin Pharmacol Ther. 1992;52:561–568. doi: 10.1038/clpt.1992.185. [DOI] [PubMed] [Google Scholar]
- Stein MA, Blondis TA, Schnitzler ER, et al. Methylphenidate dosing twice daily versus three times daily. Pediatrics. 1996;98:748–756. [PubMed] [Google Scholar]
- Stein MA, McGough JJ. The pharamcogenomic ers: promise for personalizing attention deficit hyperactivity disorder therapy. Child Adolesc Psychiatr Clin N Am. 2008;17:475–490. doi: 10.1016/j.chc.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J, Gupta S, Guinta D, et al. Acute tolerance to methylphenidate in the treatment of attention deficit disorder in children. Clin Pharmacol Ther. 1999;66:295–305. doi: 10.1016/S0009-9236(99)70038-X. [DOI] [PubMed] [Google Scholar]
- Swanson J, Gupta S, Lam A, et al. Development of a new once-a-day formulation of methylphenidate for the treatment of attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2003;60:204–211. doi: 10.1001/archpsyc.60.2.204. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Lerner M, Wigal T, et al. The use of laboratory school protocol to evaluate concepts about efficacy and side effects of new formulations of stimulant medications. J Attent Disord. 2002;6:S73–88. doi: 10.1177/070674370200601s10. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Volkow ND. Pharmacokinetic and pharmacodynamic properties of stimulants: implications for design of new treatments for ADHD. Behav Brain Res. 2002;130:73–78. doi: 10.1016/s0166-4328(01)00433-8. [DOI] [PubMed] [Google Scholar]
- Szporny L, Gorog P. Investigations into the correlations between monoamine oxidase inhibition and other effects due to methylphenidate and its stereoisomers. Biochem Pharmacol. 1961;8:263–268. [Google Scholar]
- Teicher MH, Polcari A, Foley M, et al. Methylphenidate blood levels and therapeutic response in children with attention-deficit hyperactivity disorder I. Effects of different dosing regimens. J Child Adolesc Psychopharmacol. 2006;16:416–431. doi: 10.1089/cap.2006.16.416. [DOI] [PubMed] [Google Scholar]
- Teo SK, Stirling DI, Thomas SD, Hoberman AM, Christian MS, Khetani VD. The perinatal and neonatal toxicity of D-methylphenidate and d,l-methylphenidate in rats. Reprod Toxicol. 2002;16:353–366. doi: 10.1016/s0890-6238(02)00044-8. [DOI] [PubMed] [Google Scholar]
- Teo SK, Stirling DI, Hoberman AM, Christian MS, Thomas SD, Khetani VD. D-methylphenidate and D,L-methylphenidate are not developmental toxicants in rats and rabbits. Birth Defects Res. 2003b;68:162–171. doi: 10.1002/bdrb.10018. [DOI] [PubMed] [Google Scholar]
- Teo SK, Stirling DI, Thomas SD, Khetani VD. Neurobehavioral effects of racemic threo-methylphenidate and its d and l enantiomers in rats. Pharmacol Biochem Behav. 2003a;74:747–754. doi: 10.1016/s0091-3057(02)01073-0. [DOI] [PubMed] [Google Scholar]
- Tuerck D, Wang Y, Maboudian M, et al. Similar bioavailability of dexmethylphenidate extended (bimodal) release, dexmethylphenidate immediate release and racemic extended (bimodal) release formulations in man. Int J Clin Pharmacol Ther. 2007;45:662–668. doi: 10.5414/cpp45662. [DOI] [PubMed] [Google Scholar]
- Vasiloff N, Cohen D, Dillon JB. Effective treatment of hiccup with intravenous methylphenidate. Can Anaesth Soc J. 1965;12:306–310. doi: 10.1007/BF03004146. [DOI] [PubMed] [Google Scholar]
- Vastag B. Pay attention: Ritalin acts much like cocaine. JAMA. 2001;286:296–296. doi: 10.1001/jama.286.8.905. [DOI] [PubMed] [Google Scholar]
- Vetter VL, Elia J, Erickson C, et al. Cardiovascular monitoring of children and adolescents with heart disease receiving stimulant drugs. Circulation. 2008;117:2407–2423. doi: 10.1161/CIRCULATIONAHA.107.189473. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Swanson JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry. 2003;160:1909–1918. doi: 10.1176/appi.ajp.160.11.1909. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G-F, Fowler JS. ‘Nonhedonic’ food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this. Synapse. 2002b;44:175–180. doi: 10.1002/syn.10075. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Fowler JS, et al. Relationship between blockade of dopamine transporters by oral methylphenidate and the increases in extracellular dopamine: therapeutic implication. Synapse. 2002a;43:181–187. doi: 10.1002/syn.10038. [DOI] [PubMed] [Google Scholar]
- Wagner MW, Markowitz JS, Patrick KS. Methylphenidate ER lodging in esophagus. J Am Acad Child Adolesc Psychiatry. 2001;40:1244–1245. doi: 10.1097/00004583-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Weiss M, Hechtman L, Turay A, et al. Once-daily multilayer-release methylphenidate in a double-blind, crossover comparison to immediate-release methylphenidate in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2007;17:675–688. doi: 10.1089/cap.2006.0101. [DOI] [PubMed] [Google Scholar]
- Wigal S, Swanson JM, Feifel D, et al. A double-blind, placebo-controlled trial of dexmethylphenidate hydrochloride and d,l-threomethylphenidate hydrochloride in chidren with attention deficit-hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2004;43:1406–1414. doi: 10.1097/01.chi.0000138351.98604.92. [DOI] [PubMed] [Google Scholar]
- Wilens TE, Adler LA, Adams J, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. J Am Acad Child Adolesc Psychiatry. 2008a;47:21–31. doi: 10.1097/chi.0b013e31815a56f1. [DOI] [PubMed] [Google Scholar]
- Wilens T, Boellner SW, Lopez FA, et al. Varying the wear time of the methylphenidate transdermal system in children with attention-deficit-hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2008b;47:700–708. doi: 10.1097/CHI.0b013e31816bffdf. [DOI] [PubMed] [Google Scholar]
- Williard RL, Middaugh LD, Zhu H, Patrick KS. Methylphenidate and its ethanol transesterification metabolite ethylphenidate: brain disposition, monoamine transporters and motor activity. Behav Pharmacol. 2007;18:39–52. doi: 10.1097/FBP.0b013e3280143226. [DOI] [PubMed] [Google Scholar]
- Yang PB, Swann AC, Dafny N. Chronic administration of methylphenidate produces neurophysiological and behavioral sensitization. Brian Res. 2007;1145:66–80. doi: 10.1016/j.brainres.2007.01.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabik JE, Levine RM, Maikel RP. Drug interactions with brain biogenic amines and the effects of amphetamine isomers on locomotor activity. Pharmacol Biochem Behav. 1978;8:429–435. doi: 10.1016/0091-3057(78)90081-3. [DOI] [PubMed] [Google Scholar]
- Zhu H-J, Patrick KS, Yuan H-J, et al. Two CES1 gene mutations lead to dysfunctional carboxylesterase-1 activity in man: clinical significance and molecular basis. Am J Hum Genet. 2008;82:1–8. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]