

Abstract
Accumulation of β-amyloid (Aβ) peptide in the brain is a major hallmark of Alzheimer’s disease (AD). Hypercholesterolemia is a risk factor for AD and has been shown by laboratory studies to cause Aβ accumulation. Aβ levels in the brain are governed by its generation from amyloid precursor protein by β-secretase (BACE1), degradation by the insulin degrading enzyme (IDE), clearance from the brain by the low density lipoprotein receptor-related protein (LRP-1), and transport from circulation into the brain by receptor for advanced glycation end products (RAGE). However, the mechanisms by which hypercholesterolemia causes Aβ accumulation in the brain and contributes to the pathogenesis of AD are still to be determined. In the present study, we determined the extent to which hypercholesterolemia-induced Aβ accumulation is associated with alterations in BACE1, IDE, LRP-1, and RAGE expression levels. We show that hypercholesterolemia increases Aβ production, an effect that is associated with increased levels of BACE1 and RAGE and reduced levels of IDE and LRP-1. These results suggest that reducing Aβ accumulation in the brain may require strategies that combine reduction of generation and transport of Aβ in addition to acceleration of degradation and clearance of this peptide.
Keywords: Aβ, BACE1, Cholesterol, IDE, LRP-1
1. Introduction
Alzheimer’s disease (AD) is a major health threat affecting approximately 5% of all individuals over 65 years of age and more than 35% of those over 80 (Brookmeyer et al., 1998; Sjogren et al., 2006). The underlying causes of AD in most cases are unknown. Epidemiological (Jick et al., 2000; Wolozin, 2004), animal (Refolo et al., 2000; Fassbender et al., 2001), and cellular (Racchi et al., 1997; Galbete et al., 2000) studies suggest that disturbances in cholesterol homeostasis may contribute to the etiology of AD by increasing beta-amyloid (Aβ) generation. There is also evidence showing that cholesterol co-localizes with fibrillar Aβ in the amyloid plaques of transgenic mice (Burns et al., 2003). The first indication of a connection between cholesterol and the accumulation of Aβ plaques has been demonstrated in rabbits (Sparks et al., 1994). However, the mechanisms by which cholesterol causes Aβ accumulation and contributes to the pathogenesis of AD are still to be determined.
Aβ is generated from β-amyloid precursor protein (APP) through sequential cleavage by membrane proteases referred to as β- and γ-secretases. The β-secretase, BACE1, first cleaves APP to generate a C-terminus fragment (β-CTF), an immediate substrate for γ-secretase (Busciglio et al., 1993; Haass and Selkoe, 1993), which further cleaves β-CTF to yield Aβ.
The insulin degrading enzyme (IDE), also known as insulin protease, insulysin, or insulinase (Duckworth et al., 1979; Song et al., 2003) is a 110-kDa thiol zinc metalloendopeptidase located in cytosol, peroxisomes, endosomes, and on the cell surface (Goldfine et al., 1984; Seta and Roth, 1997; Duckworth et al., 1998; Vekrellis et al., 2000). This peptide cleaves numerous proteins that share a propensity to form β-pleated sheet–rich amyloid fibrils including insulin, glucagon, atrial natriuretic factor, transforming growth factor-α, β-endorphin, amylin, APP intracellular domain (AICD), and Aβ (Duckworth et al., 1998; Selkoe, 2001) with poor substrate selectivity and specificity. IDE was also the first protease to be implicated in the proteolytic degradation of Aβ (Kurochkin and Goto, 1994). IDE gene was mapped to chromosome 10q23–q25, which made it a candidate gene for the Alzheimer’s disease-6 locus (Affholter et al., 1990; Espinosa et al., 1991).
The low density lipoprotein receptor-related protein (LRP-1) is a member of the LDL receptor family. This receptor functions as a multi-functional scavenger and signaling receptor, a transporter, and a metabolizer of cholesterol and apolipoprotein E (apoE)-containing lipoproteins (Herz and Marschang, 2003). The receptor for advanced glycation end products (RAGE) is a multi-ligand receptor which belongs to immunoglobulin superfamily of cell surface molecules. It binds a wide range of molecules including the products of nonenzymatic glycoxidation (advanced glycation end products, or AGEs), Aβ, proinflammatory cytokine-like mediators, and a DNA-binding protein amphoterin (Yan et al., 2000). RAGE and LRP-1 bind a wide range of ligands including apoE, α2-macroglobulin, amyloid precursor protein, and Aβ (Zlokovic, 2004). While transport of Aβ from the blood into the brain is mediated by RAGE (Deane et al., 2003, 2004), Aβ transport from the brain into the blood is mediated by LRP-1 (Shibata et al., 2000). The net flux of Aβ into or out of the brain depends upon the density and activity of these two receptors (Donahue et al., 2006).
In the present study, we determined the extent to which Aβ accumulation following hypercholesterolemia alters BACE1, IDE, LRP-1, and RAGE, proteins that are involved respectively in the production, degradation, clearance from the brain, and the transport from the systemic circulation into the brain of Aβ.
2. Materials and methods
2.1. Experimental design
New Zealand white male rabbits (1.5–2 years old and 3–5 kg) were assigned randomly to group 1 (n=6) fed with the normal chow and group 2 (n=6) fed with 2% (w:w) cholesterol (Harlan Teklad Global Diets, Madison, WI ) in chow for 12 weeks and then euthanized. At necropsy, animals were perfused with Dulbecco’s phosphate-buffered saline at 37 °C and brains were promptly removed and cut to yield two symmetrical hemispheres; one hemisphere for immunohistochemistry and one for Western blot and ELISA analyses.
2.2. Quantification of Aβ levels by ELISA
Aβ levels were quantified in the cortex and hippocampus of control and cholesterol-fed rabbits by ELISA using the Biosource kit as per the manufacturer’s protocol. Briefly, to measure the amount of Aβ 1–40 and Aβ 1–42, the wet mass of the brain tissue (100 mg) was homogenized thoroughly with 8X mass of cold 5 M guanidine HCl/50mM Tris HCl. The homogenates were mixed for 3–4 hours at room temperature. The samples were diluted with cold reaction buffer (Dulbecco’s phosphate buffered saline with 5% BSA and 0.03% Tween-20 supplemented with 1x protease inhibitor cocktail) and centrifuged at 16,000 × g for 20 minutes at 4°C. The supernatant was decanted, stored in ice until use, diluted at least 1:2 with standard diluent buffer, and quantified by calorimetric sandwich ELISA kits. For aggregated Aβ, the wet mass of the brain tissue (100 mg) was homogenized thoroughly with 10X volume of tissue extraction buffer (25 mM Tris-HCl and 150 mM NaCl, supplemented with protease inhibitors; pH 7.4). The samples were sonicated and centrifuged at 100,000 x g for 1 hour at 4°C. The supernatant was collected and diluted at least 1:2 in standard diluent buffer prior to analysis using the aggregated Aβ ELISA kit from Biosource. The quantity of Aβ in each sample was measured in duplicates. Protein concentrations of all samples were determined by standard BCA assay (Pierce). Aβ levels were normalized to total protein content in the samples.
2.3. Western blot analysis
Cortex and hippocampus from control and cholesterol-fed rabbit brains were homogenized in T-PER tissue protein extraction reagent (Thermo Scientific, Rockford, IL) supplemented with protease and phosphatase inhibitors. Protein concentrations were determined with BCA protein assay. Proteins (10μg) were separated in SDS-PAGE gels followed by transfer to a polyvinylidene difluoride membrane (Millipore, Bedford, MD) and incubation with antibodies to BACE1 (1:100, Chemicon International, Temecula, CA), RAGE (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), IDE (1:100, Chemicon International, Temecula, CA), and LRP-1(1:100, Biodesign International, Saco, Maine). β-actin was used as a gel loading control. The blots were developed with enhanced chemiluminescence (Immmun-star HRP chemiluminescent kit, Bio-Rad, Herculus, CA). Bands were visualized on a polyvinylidene difluoride membrane and analyzed by LabWorks 4.5 software on a UVP Bioimaging System (Upland). Quantification of results was performed by densitometry and the results analyzed as total integrated densitometric values (arbitrary units).
2.4. Confocal microscopy studies
Coronal frozen sections (14 μm) at the level of hippocampus and cortex were air-dried, fixed with cold acetone, blocked with 5% normal goat serum, and reacted overnight at 4°C with antibodies to BACE1 (1:250), RAGE (1:250), IDE (anti-mouse, 1:250, Signet laboratories), LRP-1 (1:500), or 6E10 (1:250, Signet laboratories Inc., Dedham, MA). According to the manufacturer, the region of the Aβ peptide where the 6E10 epitope resides is completely homologous between human and rabbit. Sections were then washed and incubated with secondary antibodies conjugated to Alexa fluor-488 (Molecular Probes, Inc., Eugene, OR) for one hour at room temperature in the dark and washed with PBS. The sections were then incubated in autofluorescence eliminator reagent (Chemicon International, Temecula, CA) for 5 minutes, washed with 70% ethanol, and mounted with vectasheild containing DAPI (Vector laboratories, Inc., Burlingame, CA). The sections were visualized with a Zeiss LSM 510 META confocal system coupled to a Zeiss Axiophot 200 inverted epifluorescence microscope. Imaging was performed with a 63X oil immersion objective. The dimensions of all images displayed on the monitor were 1024 × 1024 pixels. For single scanning detection of the fluorescence of Alexa-fluor(R) 488, a 488 nm Argon laser with wavelength filter, a 488/543 HFT and 545 NFT primary dichroic beam splitter, and a 505–530 nm band pass filter were used. The typical laser power at the sample was around 10% transmission. The DAPI cube was used for both viewing and taking images of the nucleus. The emission was a LP 420, which was combined with the mercury bulb to take the images.
2.5. Statistical Analysis
Quantitative data are presented as mean values ± SEM. The significance of differences between the control and cholesterol-fed group was assessed using the Student’s t test, with p < 0.05 considered statistically significant. Statistical analysis was performed with GraphPad Prism software 4.01.
3. Results
3.1. Cholesterol-enriched diet leads to increase in Aβ levels
ELISA assay was used to determine the extent to which cholesterol-enriched diets influence Aβ levels. We found that the total amount of guanidine-solubilized Aβ1–40 and Aβ1–42 and aggregated Aβ levels were significantly increased in the cortex of cholesterol-fed rabbits in comparison to control rabbits (Fig. 1A). In the cortex, Aβ1–40 levels were 138 ± 5.6 pg/mg of protein in the control animals and 190 ± 37 pg/mg of protein in the cholesterol-fed rabbits, Aβ1–42 levels were 55.7 ± 20 pg/mg of protein in controls and 162 ± 69 pg/mg of protein in the cholesterol-fed rabbits, and aggregated Aβ levels were 3.8 ± 0.7 ng/mg protein in controls and 5.6 ± 1.4 ng/mg protein in the cholesterol-fed rabbits (Fig. 1A). In the hippocampus, Aβ1–40 levels were 480 ± 33 pg/mg of protein in the control animals and 550 ± 41 pg/mg of protein in the cholesterol-fed rabbits, Aβ1–42 levels were 590 ± 80 pg/mg of protein in the controls and 860 ± 204 pg/mg of protein in the cholesterol-fed rabbits, and aggregated Aβ levels were 2.5 ± 0.2 ng/mg protein in the controls and 2.3 ± 0.2 ng/mg protein in the cholesterol-fed rabbits (Fig. 1B).
Fig. 1.
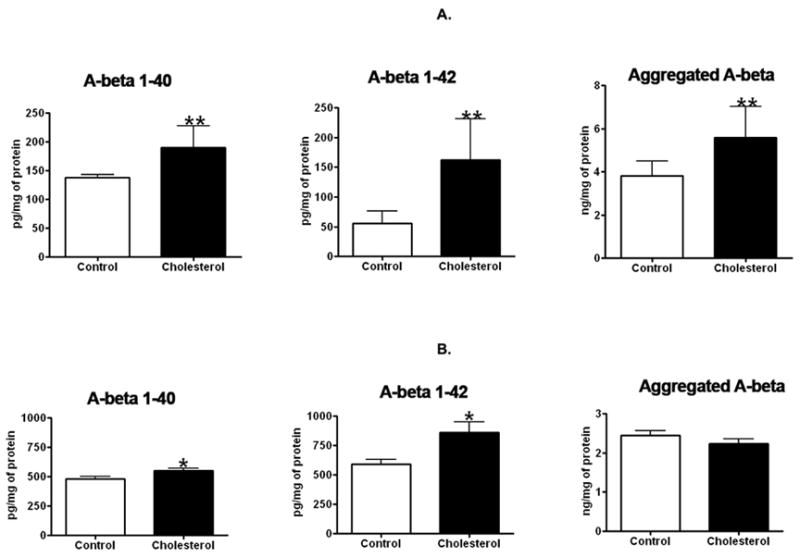
ELISA assay demonstrated that the 2% cholesterol-enriched diet significantly increased Aβ40and Aβ42 in the cortex (A) and in the hippocampus (B); Aggregated Aβ levels were significantly increased in cortex and unchanged in hippocampus. Values from control (n=6) and cholesterol-fed (n=6) rabbits were expressed as mean ± standard error: Aβ1–40 (p = 0.005), Aβ1–42 (p = 0.002), and aggregated Aβ (p = 0.004) in cortex. Aβ1–40 (p =0.0461) and Aβ1–42 (p =0.0431) in hippocampus tissue. *p<0.05; **p<0.01 (Student t test)
Immunohistochemistry and confocal microscopy using 6E10 antibody showed an increase in the immunoreactivity to Aβ antibody in both hippocampus and cortex of cholesterol-fed rabbits in comparison to control brains (Fig. 2). The immunostaining imaging in the hippocampus was carried out in CA1, CA2, and CA3. Results in CA1 and adjacent cortex are shown throughout the results section. Aβ staining in the CA1 appears as perinuclear dots and also in a filamentous pattern suggestive of axonal staining. In the cortex, most of Aβ staining is confined to small aggregates.
Fig. 2.
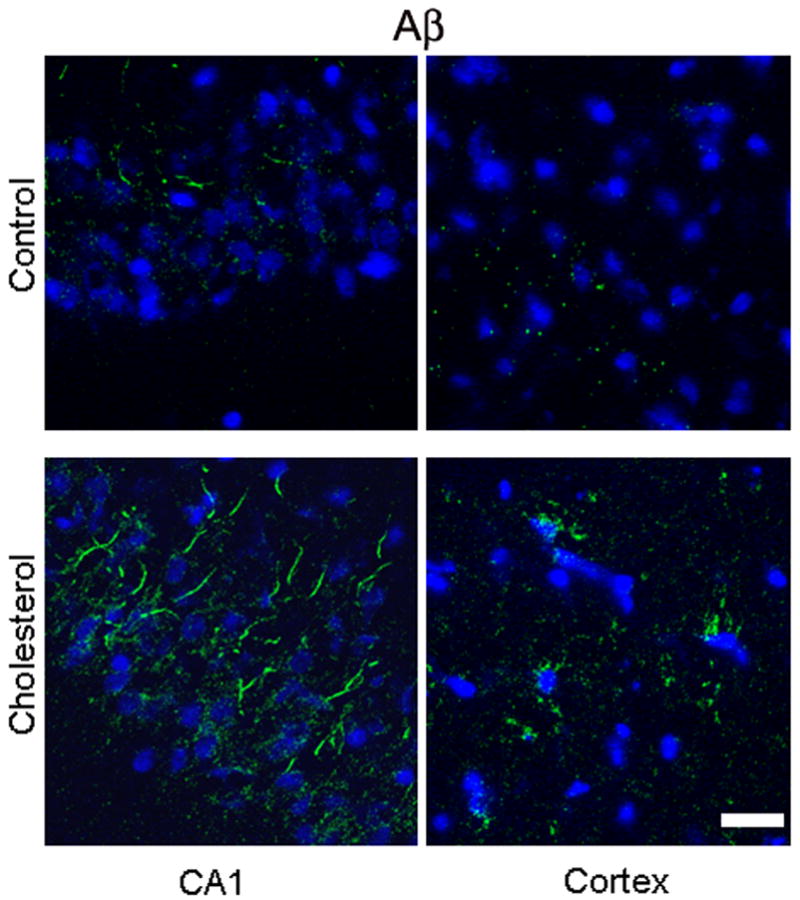
Immunofluorescence staining showing increased expression of Aβ levels (green) as detected by 6E10 antibody in the CA1 region of the hippocampus and in the adjacent cortex of a cholesterol-fed rabbit brain compared to a control rabbit. DAPI (blue) was used to stain nuclei. Bar= 20μm.
3.2. Increased Aβ levels are associated with increased BACE1 and RAGE
Levels of BACE1 and RAGE are significantly increased in the hippocampus and cortex of the cholesterol-fed rabbits in comparison to control levels. Western blot (Fig. 3a) and densitometric analysis (Fig. 3b) show an increase in BACE1 and RAGE in the hippocampus and 2cortex of cholesterol-fed rabbits compared to controls.
Fig. 3.
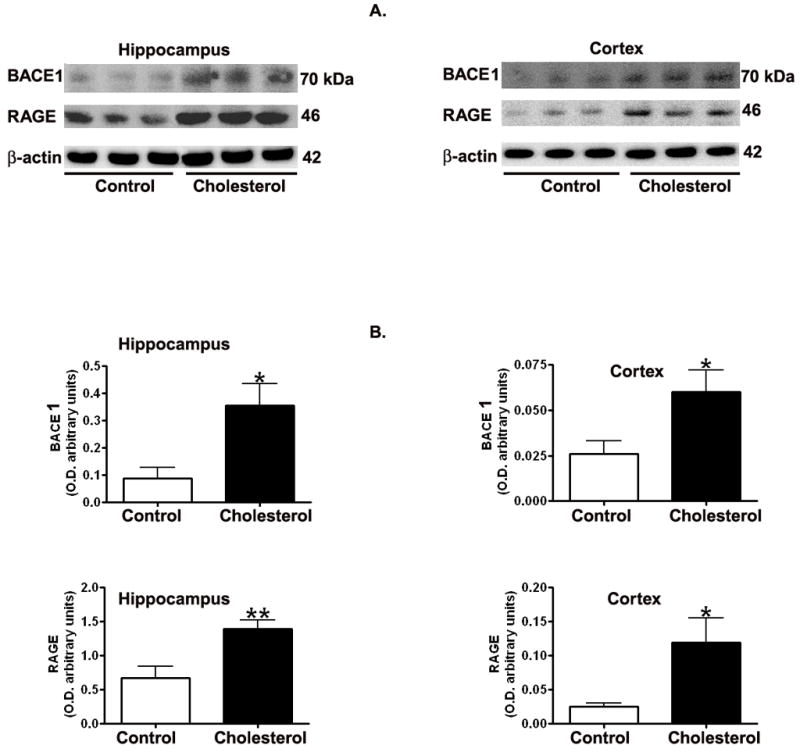
Representative Western blots (A) and densitometric analysis (B) showing increased levels of BACE1 and RAGE in hippocampus and cortex from cholesterol-fed rabbits in comparison to control animals. *p<0.05; **p<0.01 (Student t test).
The increased levels of BACE1 and RAGE proteins is confirmed by confocal microscopy, where expression levels of BACE1 (Fig. 4a) and RAGE (Fig. 4b) increased dramatically in the hippocampus and moderately in the cortex of cholesterol-fed rabbits compared to control brains.
Fig. 4.
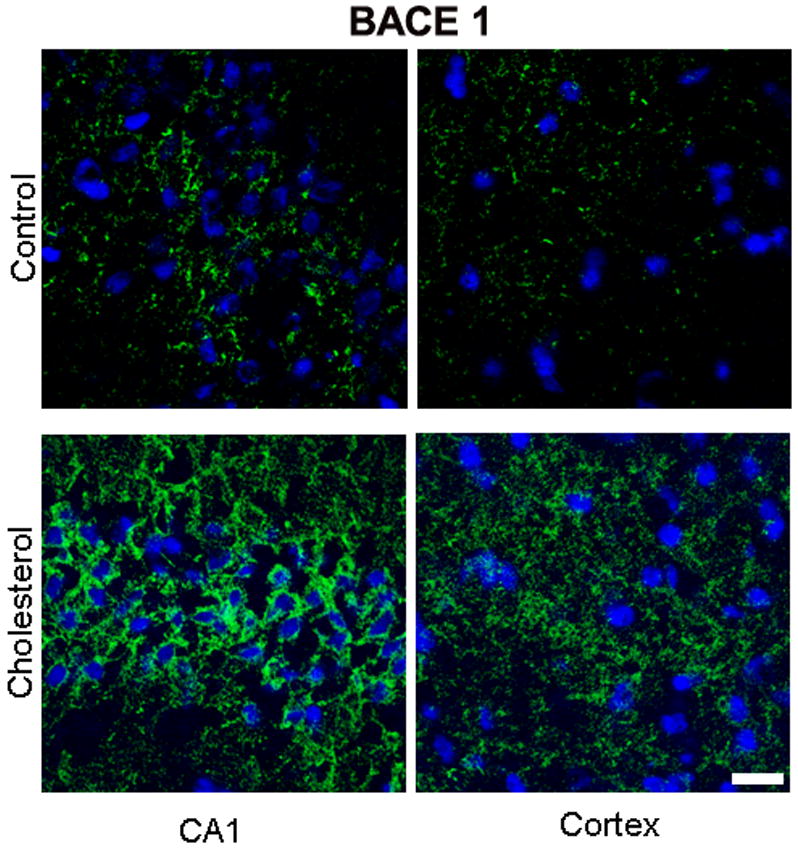

(A) Immunofluorescence staining demonstrating a dramatic increase in expression of BACE1 (green) in the CA1 region of the hippocampus and adjacent cortex in sections from a cholesterol-fed rabbit brain compared to section from a control rabbit. DAPI (blue) was used as nuclear counterstain. Bar= 20μm.
(B) Immunostaining for RAGE is low in sections from CA1 region of hippocampus or adjacent cortex in a control rabbit. In section from CA1 and adjacent cortex from a cholesterol-fed rabbit, immunoreactivity to RAGE (green) is increased in hippocampus and in cortex. Nuclei were stained with DAPI (blue). Bar= 20μm
3.3. Increased Aβ levels are associated with decreased IDE and LRP-1 levels
Hypercholesterolemia caused a substantial decrease in levels of IDE and LRP-1 in the hippocampus and cortex as shown by Western blot (Fig. 5a) and densitometric analysis (Fig. 5b). Confocal studies further confirmed the changes in the expression of LRP-1 and IDE showing a reduced immunoreactivity to LRP-1(Fig. 6) and IDE (Fig. 7) antibodies in both hippocampus and cortex of cholesterol-fed rabbits.
Fig. 5.
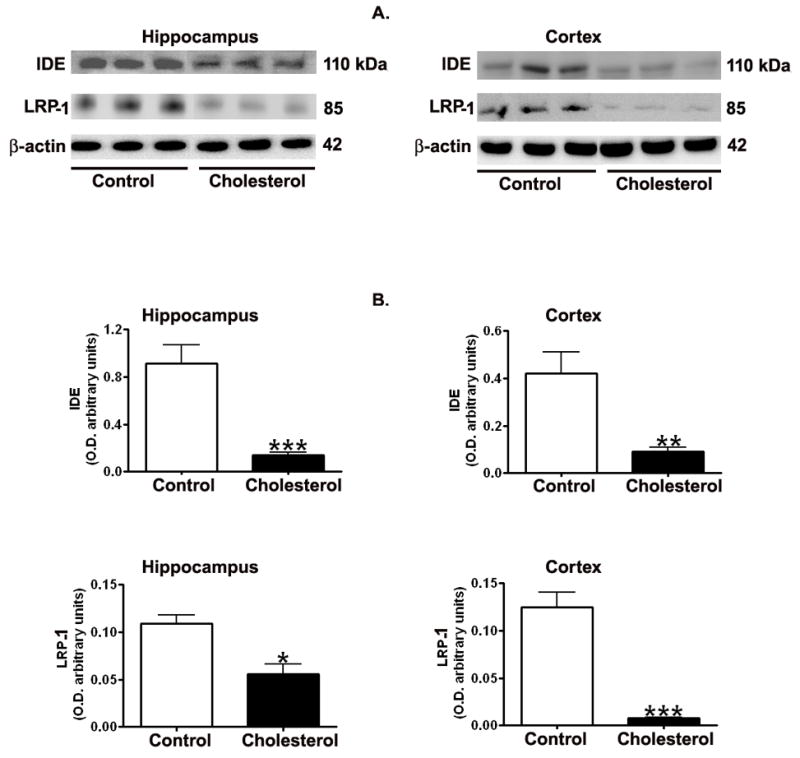
Representative Western blots (A) and densitometric analysis (B) showing decreased levels of IDE and LRP-1 in hippocampus and cortex from cholesterol-fed rabbits in comparison to control animals. *p<0.05; **p<0.01; ***p<0.001 (Student t test).
Fig. 6.
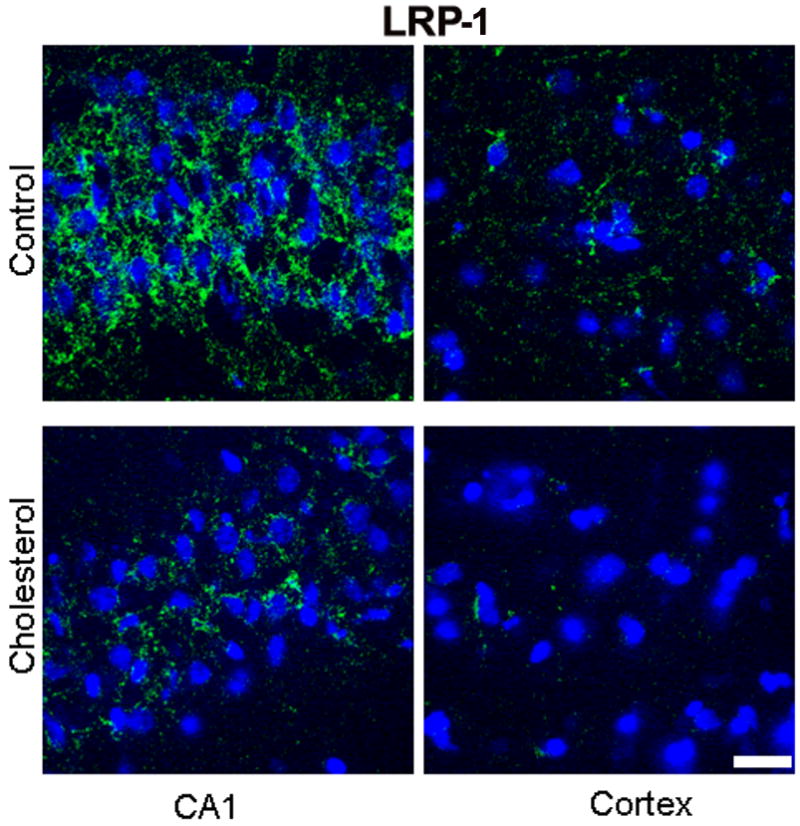
Immunofluorescence staining for LRP-1 (green) showing a marked immunoreactivity in the CA1 region of the hippocampus and a lesser reactivity in cortex from a control rabbit. In a cholesterol-fed rabbit, the immunoreactivity to LRP-1 antibody is greatly reduced in both hippocampus and cortex. DAPI (blue) was used as nuclear marker. Bar=20μm.
Fig. 7.
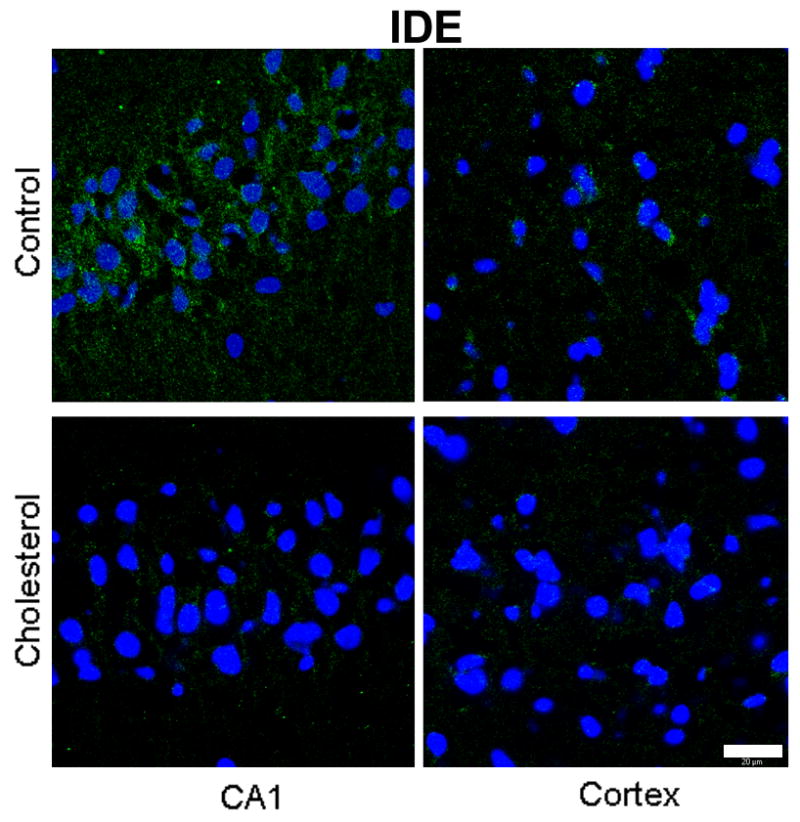
Immunofluorescence staining for IDE (green) is higher in the CA1 region of the hippocampus than the cortex of a control rabbit. IDE immunostaining is greatly reduced in both hippocampus and cortex from a cholesterol-fed rabbit. DAPI (blue) was used as nuclear marker. Bar=20μm.
4. Discussion
The present study was designed to determine the extent to which hypercholesterolemia leads to alterations in levels of Aβ and proteins that are involved in the accumulation of this peptide. We demonstrated that hypercholesterolemia increases Aβ levels, an effect that is associated with changes in levels of proteins that are involved in production, degradation, clearance, and transport processes of Aβ. Our experiments were carried out in the cholesterol-fed rabbit as this model system displays pathologies consistent with certain features of the AD brain (Sparks, 2004).
Our data showed increased levels of all forms of Aβ (i.e. soluble Aβ1–40, soluble Aβ1–42 and aggregated Aβ) in the cortex of cholesterol-fed rabbit brains. In hippocampus, soluble Aβ1–40 and soluble Aβ1–42 were increased but not aggregated Aβ as shown by ELISA assay. Currently, the molecular mechanisms by which dietary cholesterol promotes Aβ peptide accumulation are not fully understood. The BACE1 and γ-secretase complexes, that sequentially cleave amyloid precursor protein to yield Aβ, are dependent on cholesterol metabolism. BACE1 appears to be particularly sensitive to membrane cholesterol content and is located in lipid rafts. BACE1 activity has also been demonstrated to be increased in lipid microdomains (Marlow et al., 2003; Cordy et al., 2003), and is upregulated in sporadic cases of Alzheimer’s disease (Holsinger et al., 2002; Yang et al., 2003). Our previous studies showed co-localization of cholesterol with BACE1 in neurons and a dramatic increase in BACE1 levels and activity in brains from cholesterol-fed rabbits (Ghribi et al., 2006). The increased Aβ levels caused by hypercholesterolemia may be due, at least in part, to increased expression levels of BACE1 that we demonstrated in the present study. In Tg2576 transgenic mice however, although a cholesterol diet influenced APP metabolism, BACE1 levels were not different in these mice compared to mice on a control basal diet (George et al., 2004).
We also show that high cholesterol levels led to increased expression of RAGE, a receptor that transports Aβ from the blood to the brain. This suggests that increased transportation of Aβ from the circulation may be also involved in accumulation of this peptide in the brain. Plasma RAGE levels were decreased in hypercholesterolemic patients compared to normocholesterolemic subjects (Santilli et al., 2007). However, whether reduced RAGE levels in plasma were associated with increased levels of RAGE in the brain in the latter study remain to be determined.
We also demonstrated that levels of LRP-1, a receptor that clears Aβ out of the brain, are decreased in cholesterol-fed rabbits, suggesting that reduced clearance may play a role in the increased levels of Aβ in the brain. Several studies have provided strong support for the hypothesis that RAGE and LRP-1 perform opposite functions in transporting Aβ (Shibata et al., 2000; Deane et al., 2003). The fact that RAGE transports Aβ into the brain and LRP-1 transports Aβ out of the brain (Donahue et al., 2006) demonstrates that AD is associated with significant changes in the relative distribution of RAGE and LRP-1 receptors at the BBB in the human hippocampus.
Proteolysis of Aβ peptide by degrading enzymes is one way to lower peptide levels in the brain. There is ample evidence of the role of IDE in Aβ degradation. Kurochkin and Goto (1994) showed the first evidence that IDE is involved in Aβ degradation. IDE was found to be the main soluble β-amyloid degrading enzyme at neutral pH in rat brain and in normal human brain (Kurochkin and Goto, 1994). The highest β-amyloid degrading activity occurs at pH 4–5, indicating that IDE may act as an “amyloidase” that prevents the accumulation of amyloidogenic derivatives (McDermott and Gibson, 1997). In rat cortical cell system, IDE was shown to eliminate the neurotoxic effects of both Aβ 1–40 and Aβ 1–42 (Mukherjee et al., 2000; Hoyer, 2002). We demonstrate in the present work that the expression level of IDE is decreased by hypercholesterolemia in both the hippocampus and cortex. Our data suggests that the reduction in IDE levels may lead to the accumulation of Aβ in the brain.
In summary, our results demonstrated that hypercholesterolemia-induced Aβ accumulation is associated with (1) an increase in levels of BACE1, an enzyme that initiates Aβproduction; (2) a decrease in levels of IDE, an enzyme that degrades Aβ; (3) a decrease in levels of LRP-1, a protein that transports Aβ from brain to blood; and (4) an increase in levels of RAGE, a receptor that transports Aβ from blood to brain. The changes in expression levels of these proteins may have contributed to the cholesterol-induced Aβ accumulation. The potential mechanisms by which high cholesterol diet may increase Aβ levels in cholesterol-fed rabbit brains are summarized in Fig 8. We propose that maintaining normal blood cholesterol levels would prevent changes in proteins that regulate Aβ levels, thus precluding Aβ accumulation and potentially protecting against Alzheimer’s disease.
Fig. 8.
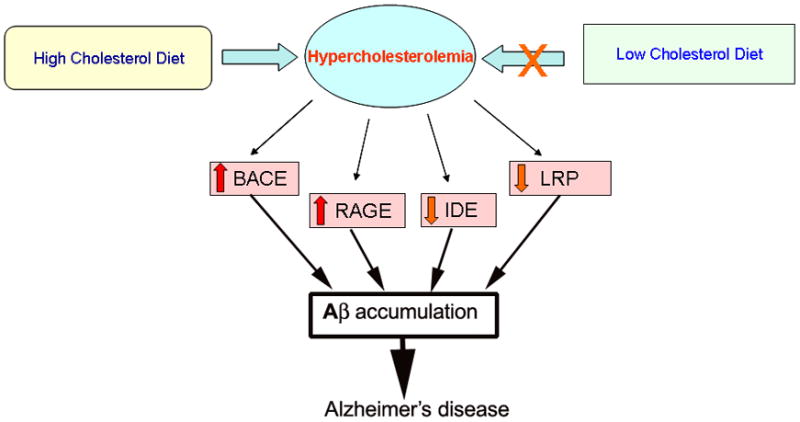
A schema showing the possible mechanisms involved in hypercholesterolemia-induced Aβ accumulation. A high cholesterol diet increases blood cholesterol levels (hypercholesterolemia) and leads to increased levels of BACE1 and RAGE, and decreased levels of IDE and LRP-1. Increased levels of BACE1 accelerate processing of APP to Aβ, and increased levels of RAGE result in increased transport of Aβ from the circulation into the brain. Decreased levels of IDE in the brain lead to a decreased cleavage of Aβ and decreased levels of LRP-1 result in reduced clearance of Aβ from the brain out to the circulation. All these changes triggered by hypercholesterolemia may contribute to increased levels of Aβ in the brain. Accumulation of Aβ potentially causes neurodegeneration and may ultimately contribute to the pathogenesis of Alzheimer’s disease. Reducing hypercholesterolemia or maintaining normal levels of cholesterol in the blood would prevent changes in BACE1, RAGE, IDE and LRP-1, thereby precluding Aβ accumulation and subsequent deleterious effects.
Acknowledgments
This work was supported by a Grant from the National Center for Research Resources (5P20RR017699, Centers of Biomedical Research Excellence).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Affholter JA, Hsieh CL, Francke U, Roth RA. Insulin degrading enzyme: stable expression of the human complementary DNA, characterization of its protein product, and chromosomal mapping of the human and mouse genes. Molecular Endocrinology. 1990;4(8):1125–1135. doi: 10.1210/mend-4-8-1125. [DOI] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, Mathews PM, Noble W, Matsuoka Y, Duff K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci. 2003;23:5645–5649. doi: 10.1523/JNEUROSCI.23-13-05645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of beta-amyloid in the secretory pathway in neuronal and non-neuronal cells. Proc Natl Acad Sci U S A. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition upregulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deane R, Wu Z, Zlokovic BV. RAGE (Yin) Versus LRP (Yang) balance regulates Alzheimer amyloid β-peptide clearance through transport across the blood–brain barrier. Stroke. 2004;35:2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 7.Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain. Nature Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 8.Donahue JE, Flaherty SL, Johanson CE, Duncan JA, III, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 9.Duckworth WC, Bennett RG, Hamel FG. Insulin acts intracellularly on proteasomes through insulin degrading enzyme. Biochemical and Biophysical Research communications. 1998;224(2):390–394. doi: 10.1006/bbrc.1998.8276. [DOI] [PubMed] [Google Scholar]
- 10.Duckworth WC, Stentz FB, Heinemann M, Kitabchi AE. Initial site of insulin cleavage by insulin protease. Proc Natl Acad Sci USA. 1979;76(2):635–639. doi: 10.1073/pnas.76.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Espinosa R, 3rd, Lemons RS, Perlman RK, Kuo WL, Rosner MR, Le Beau MM. Localization of the gene encoding insulin degrading enzyme to human chromosome 10, bands q23— q25. Cytogenetics and Cell Genetics. 1991;57(4):184–186. doi: 10.1159/000133142. [DOI] [PubMed] [Google Scholar]
- 12.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galbete JL, Martin TR, Peressini E, Modena P, Bianchi R, Forloni G. Cholesterol decreases secretion of the secreted form of amyloid precursor protein by interfering with glycosylation in the protein secretory pathway. Biochem J. 2000;348:307–313. [PMC free article] [PubMed] [Google Scholar]
- 14.George AJ, Holsinger RM, McLean CA, Laughton KM, Beyreuther K, Evin G, Masters CL, Li QX. APP intracellular domain is increased and soluble Abeta is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiol Dis. 2004;16:124–132. doi: 10.1016/j.nbd.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Ghribi O, Larsen B, Schrag M, Herman MM. High cholesterol content in neurons increases BACE, β-amyloid, and phosphorylated tau levels in rabbit hippocampus. Experimental Neurology. 2006;200:460–467. doi: 10.1016/j.expneurol.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 16.Goldfine ID, Williams JA, Bailey AC, Wong KY, Iwamoto Y, Yokono K, Baba S, Roth RA. Degradation of insulin by isolated mouse pancreatic acini. Evidence for cell surface protease activity. Diabetes. 1984;33:64–72. doi: 10.2337/diab.33.1.64. [DOI] [PubMed] [Google Scholar]
- 17.Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- 18.Herz J, Marschang P. Coaxing the LDL receptor family into the fold. Cell. 2003;112:289–292. doi: 10.1016/s0092-8674(03)00073-4. [DOI] [PubMed] [Google Scholar]
- 19.Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 20.Hoyer S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J Neural Transm. 2002;109:341–360. doi: 10.1007/s007020200028. [DOI] [PubMed] [Google Scholar]
- 21.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 22.Kurochkin IV, Goto S. Alzheimer’s β amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Letters. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 23.Marlow L, Cain M, Pappolla MA, Sambamurti K. Beta-secretase processing of the Alzheimer’s amyloid protein precursor (APP) J Mol Neurosci. 2003;20:233–239. doi: 10.1385/JMN:20:3:233. [DOI] [PubMed] [Google Scholar]
- 24.McDermott JR, Gibson AM. Degradation of Alzheimer’s beta-amyloid protein by human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochemical Research. 1997;22:49–56. doi: 10.1023/a:1027325304203. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee A, Song E, Kihiko-Ehmann M, Goodman JP, Jr, Pyrek JS, Estus S, Hersh LB. Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. The Journal of Neuroscience. 2000;20(23):8745–8749. doi: 10.1523/JNEUROSCI.20-23-08745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Racchi M, Baetta R, Salvietti N, Ianna P, Franceschini G, Paoletti R, Fumagalli R, Govoni S, Trabucchi M, Soma M. Secretory processing of amyloid precursor protein is inhibited by increase in cellular cholesterol content. Biochem J. 1997;322:893–898. doi: 10.1042/bj3220893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia upon differentiation-associated increases in tau and cyclin-dependent kinase accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 28.Santilli F, Bucciarelli L, Noto D, Cefalu AB, Davi V, Ferrante E, Pettinella C, Averna MR, Ciabattoni G, Davi G. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43:1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32(2):177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 30.Seta KA, Roth RA. Overexpression of insulin degrading enzyme: cellular localization and effects on insulin signaling. Biochemical and Biophysical Research Communications. 1997;231(1):167–171. doi: 10.1006/bbrc.1997.6066. [DOI] [PubMed] [Google Scholar]
- 31.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer’s amyloid-β1–40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sjogren M, Mielke M, Gustafson D, Zandi P, Skoog I. Cholesterol and Alzheimer’s disease – is there a relation? Mech Ageing Dev. 2006;127:138–147. doi: 10.1016/j.mad.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 33.Song ES, Juliano MA, Juliano L, Hersh LB. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. The Journal of Biological Chemistry. 2003;278(50):49789–49794. doi: 10.1074/jbc.M308983200. [DOI] [PubMed] [Google Scholar]
- 34.Sparks DL. Cholesterol, copper, and accumulation of thioflavine S-reactive Alzheimer’s-like amyloid beta in rabbit brain. J Mol Neurosci. 2004;24:97–104. doi: 10.1385/jmn:24:1:097. [DOI] [PubMed] [Google Scholar]
- 35.Sparks DL, Scheff SW, Hunsaker JC, III, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol. 1994;126:88–94. doi: 10.1006/exnr.1994.1044. [DOI] [PubMed] [Google Scholar]
- 36.Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin degrading enzyme. The Journal of Neuroscience. 2000;20(5):657–1665. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolozin B. Cholesterol and the biology of Alzheimer’s disease. Neuron. 2004;41:7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- 38.Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, Yu J, Soto C, Schmidt AM, Stern D, Kindy M. Receptor dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. 2000;6:643–651. doi: 10.1038/76216. [DOI] [PubMed] [Google Scholar]
- 39.Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 40.Zlokovic BV. Clearing amyloid through the blood–brain barrier. J Neurochem. 2004;89:807–811. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]