

Abstract
An analytical method has been developed and validated for the quantitative determination of finasteride, a potent 5 α –reductase inhibitor, in human plasma. Calibration curves are linear in the concentration range of 1 to 100 ng/ml. Sample pretreatment involves a liquid-liquid extraction with ethyl acetate using 0.2 ml aliquots of plasma. Finasteride and the internal standard, beclomethasone, are separated on a Waters Symmetry Shield RP18 column (50 × 2.1 mm, 3.5 μm), and eluted using a gradient mobile phase composed of acetonitrile and 10 mM ammonium acetate with 0.1% formic acid. The column eluant is monitored by mass spectrometry with electrospray ionization. A complete validation of the method has been performed. For quality control samples at three different concentrations that were analyzed in quintuplicate, on six separate occasions, the accuracy and precision range from 95.2 to 101% and 3.4 to 7.3%, respectively. The developed method was subsequently applied to measure the steady state finasteride concentration of patients who participated in the Prostate Cancer Prevention Trial (PCPT).
Keywords: Finasteride, LC-MS, PCPT, steady state
1. Introduction
Benign prostatic hyperplasia (BPH), characterized by an enlarged prostate, lower urinary tract symptoms, and decreased flow of urine, is a common problem among men aged over 50, and its prevalence increases with age (1,2). The development and enlargement of the prostate gland is dependent on the potent androgen, 5α-dihydrotestosterone (DHT) (3,4). Finasteride (Fig 1.), a synthetic 4-azasteroid, inhibits the conversion of testosterone to DHT by blocking the action of the 5α-reductase enzyme (5,6). Treatment with finasteride results in a significant reduction of prostatic and circulating DHT levels compared with baseline (7) and is widely used for the treatment of BPH.
Figure 1.
Due to the role of DHT in the development of prostate cancer, it was hypothesized that finasteride could be used effectively to reduce the risk of this disease. The seven-year long Prostate Cancer Prevention Trial (PCPT), a randomized double-blind comparison of finasteride and placebo, was completed in February 2003 (8). During the study, 18,882 men aged 55 years or older, with a normal digital rectal examination (DRE) and a prostate-specific antigen (PSA) level of 3.0 ng/mL or lower, were randomly assigned to treatment with finasteride (5 mg/day) or placebo. The PCPT demonstrated that long-term finasteride use reduced the incidence of prostate cancer by 24.8%, yet the prevalence of tumors with Gleason score between seven and ten was higher in this group (8). To assist the long term follow-up study, this analytical method was developed and validated for the determination of steady state finasteride concentration in patient plasma.
Several liquid chromatographic methods for determining finasteride concentrations in human plasma have been reported (Table 1), but unfortunately, none were suitable for application to the study of thousands of individual samples arising from the PCPT. Several methods utilize triple-quadrupole mass spectrometry (9-11) which, although very sensitive and specific, is not always affordable for routine measurements in many laboratories. Several methods lacked sensitivity (12-14), involved complicated sample pretreatment procedures (10,13-15), required large sample volumes (9,10,12-15), or lengthy analysis time (12-16) with UV detection. Therefore, we endeavored to develop an analytical method for the detection of finasteride in human plasma, utilizing more readily available single quadrupole mass spectrometric detection, while still achieving 1 ng/ml sensitivity with 200 uL of plasma or less. In addition, we optimized the analysis for speed, using both rapid, simple sample preparation techniques and relatively short run times. Furthermore, the developed assay was validated to very high standards, ensuring the robustness of the method for analysis of large numbers of samples, to be analyzed over a relatively long time-frame.
Table 1.
Liquid chromatographic methods for the analysis of finasteride in human plasma
| Sample pretreatment | I.S. | Detection (nm) |
Plasma volume (ml) |
LLOQ (ng/mL) |
Run time (min) |
Reference |
|---|---|---|---|---|---|---|
| LLE (hexane:isoamylalcohol, 98:2) | Clobazam | 210 | 1 | 4 | 25 | 12 |
| LLE (methyl-tert.-butyl ether) | Analogue | MS/MS | 1 | 0.025 | 2 | 9 |
| SPE (C18 and CN cartridge) | Analogue | 210 | 1 | 10 | 21 | 13 |
| SPE (CN cartridge) | Analogue | MS/MS | 1 | 0.2 | 3 | 10 |
| SPE (CN cartridge) | Analogue | 210 | 1 | 1 | 23 | 15 |
| SPE (graphitized carbon black) | 4-Androstene -3,17-dione |
215 | 1 | 10 | 20 | 14 |
| Column Switching | No I.S. | 210 | 0.075 | 1 | 38 | 16 |
| LLE (ethyl acetate) | norethindrone | MS/MS | 0.5 | 0.5 | N.R. | 11 |
Abbreviations: I.S., internal standard; LLOQ, lower limit of quantitation; LLE, liquid-liquid extraction; SPE, solid phase extraction; MS, mass spectrometry; CN, cyanonitrile. N.R., not reported.
2. Experimental
2.1. Chemicals
Finasteride, internal standard beclomethasone (99%), and 200 proof ethanol were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ammonium acetate (98%) was obtained from MG Scientific (Pleasant Prairie, WI, USA) and formic acid (98%) from Fluka (through Sigma-Aldrich, St. Louis, MO, USA). Ethyl acetate (Fisher Scientific, Fair lawn, NJ, USA) and acetonitrile (Burdick & Jackson, Mushegon, MI, USA) are of HPLC grade. Deionized water was generated with a Hydro-Reverse Osmosis system (Durham, NC, USA) connected to a Milli-Q UV Plus purifying system (Millipore Corp, Billerica, MA, USA). Drug-free heparinized human plasma was obtained from the National Institutes of Health Clinical Center Blood Bank (Bethesda, MD, USA).
2.2. Preparation of stock solutions and standards
Stock solutions of finasteride were prepared in triplicate by dissolving the drug in absolute ethanol at a concentration of 1 mg/mL and stored in glass tubes at −20 °C. The difference in drug concentration in each of the triplicate stock solutions, estimated from the mean peak area following repeat analysis of a dilution of the stock, was determined to be within 5%. Out of one of the master stock solutions, a working solution containing 100 ug/mL of drug in absolute ethanol was prepared each week and stored at −20 °C between uses. Serial dilutions were prepared from this working solution for the preparation of calibration and quality control (QC) samples. A master stock of the internal standard, beclomethasone, was prepared at a concentration of 1 mg/mL in absolute ethanol. From the master stock, a working solution of the internal standard was prepared by dilution to10 ug/mL. Both the master and working internal standard solutions were stored at −20 °C.
With each analytical run, calibration standards in drug-free human heparinized plasma were prepared freshly in duplicate at finasteride concentrations of 1, 2, 5, 10, 25, 50 and 100 ng/mL, such that the total amount of ethanol added was identical in each sample (5%). QC samples were prepared in batch at concentrations of 3, 40 and 80 ng/mL, by adding plasma to the required amount of working solution in a volumetric flask. After vortexing to ensure complete mixing, these QC samples were subdivided into individual aliquots in polypropylene tubes, and stored at −20 °C. Together with patient samples, a portion of the QC samples were stored at −80 °C for the assessment of long term stability.
2.3. Sample preparation
Samples were prepared by spiking 190 μL of blank human plasma in a 10 ml disposable glass centrifuge tube (Kimble, Vineland, NJ, USA) with 10 μL of the appropriate finasteride working solution so that the total volume for each sample is 200 μL. QC samples were thawed at room temperature, vortex-mixed for 15 s to ensure uniformity, and a volume of 200 μL of each sample was transferred into a glass centrifuge tube. Then, 10 μL of the internal standard working solution and 1.6 mL of extraction solvent, ethyl acetate, were added to each tube. Tubes were then vortex-mixed for 5 min followed by centrifugation for 5 min at 1303 x g. The clear supernatant was transferred to a glass drying tube and evaporated to dryness under desiccated air in a water bath at 40 °C in a Zymark TurboVap LV (Hopkinton, MA, USA). The residue was reconstituted in 80 μL of a mixture of acetontrile/10 mM ammonium acetate with 0.1% formic acid (2:3, v:v), and vortex-mixed for 15 s. Finally, each solution was transferred to a glass vial for injection. A volume of 25 μL of this solution was then injected into the chromatographic system.
2.4. Equipment
The experiments were conducted on a HP 1100 system (Agilent Technology, Palo Alto, CA, USA) which included a G1312 binary pump, a G1329 refrigerated autosampler, a mobile phase vacuum degassing unit, and a temperature-controlled column compartment, coupled with a single-quadrupole mass spectrometric (MS) detector (Agilent 1100 MSD) equipped with an electrospray ionization source. The autosampler was maintained at 4 °C and the column was at 30 °C. A Waters SymmetryShield column (50 × 2.1 mm I.D.) packed with 3.5-μm RP18 material was employed. Samples were eluted using a gradient at a flow rate of 200 μL/min. Mobile phase A was 10 mM ammonium acetate with 0.1% formic acid. Mobile phase B was HPLC grade acetonitrile. The gradient started with 58% A and 42%B, and the acetonitrile was increased to 50.5% over 3.5 min. Then, the acetonitrile concentration was held at 70% for 2.5 min, followed by original conditions starting at 6.01 min until the end of the 13 min run time to achieve re-equilibration. The MS conditions were as follows: fragmentor, 130 V; gain, 1; drying gas flow, 10.5 L/min; nebulizing gas pressure 45 psi; drying gas temperature, 200 °C; and capillary voltage 2800 V. Selected-ion monitoring was accomplished at m/z 373.3 for finasteride, and m/z 409.2 for the internal standard. The chromatographic data were acquired and analyzed using the Chemstation software package (Agilent).
2.5. Validation procedures
Validation of the method with respect to accuracy and precision was carried out
according to procedures reported in detail previously (17). Calibration standards of 1, 2, 5, 10, 25, 50 and 100 ng/mL were prepared freshly by mixing the working standard solutions of finasteride and drug-free human plasma. Pools of QC samples were prepared at 3, 40 and 80 ng/ml concentrations before the validation process began and stored at −20 °C. Five QC samples at each concentration were thawed at room temperature and analyzed each day. Validation runs included blank (zero concentration) and internal standard only samples, along the calibration curve and QC samples.
Calibration graphs were calculated by least-squares linear regression analysis of the peak area ratio of finasteride and the internal standard versus the drug concentration of the nominal standard. The regression parameters of slope, intercept and correlation coefficient were calculated by least-squares linear regression analysis using a weighting factor of 1/x. The linearity was evaluated by comparing the correlation coefficient (r2), residuals and errors between theoretical and back calculated concentrations of calibration standard samples. The zero concentration (blank), and internal standard only samples were used to visually verify the purity of the reagents and the lack of other potentially interfering substances, but were not considered for the regression anaylsis of standards. This calibration curve was then used to calculate measured QC concentrations by interpolation.
The lower limit of quantitation (LLOQ) of the assay was assessed by determining the concentration of finasteride at which the values for precision and accuracy were less than 20%. At least five different lots of plasma were used and resulted in accurate and reproducible measurement at the concentration of LLOQ, based on the deviation from nominal concentration.
The accuracy and precision of the assay were assessed by the mean relative percentage deviation (DEV) from the nominal concentrations and the within-run and between-run precision, calculated according to previously published equations (18). Estimates of the between-run precision were obtained by one-way analysis of variance (ANOVA) using the run day as the classification variable. The between-groups mean square (MSbet), the within-groups mean square (MSwit), and the grand mean (GM) of the observed concentrations across runs were calculated using Microsoft Excel 2003 (Redmond, WA, USA).
The extraction efficiency for finasteride in human plasma, expressed as a percentage, was determined at two concentration levels, in five replicates by comparing samples spiked to contain 5 and 75 ng/mL before and after extraction. Matrix effects were evaluated at low and high concentration levels, by comparing the detector response of the analyte spiked into the supernatant after blank plasma extraction and to ethyl acetate, the extraction solution, directly. Different lots of plasma were used to prepare QC samples and calibration curve that carried out the same day in order to assess relative matrix. Ion suppression to drug and IS was cross examined by comparing samples spiked with only IS, only finasteride at the ULOD (100ng/mL), or both. Possible interference from two major forms of metabolites, ω-hydroxylated and ω-carboxylated finasteride (16,19), were monitored at m/z 389.3 and 403.3 on the samples obtained from eight patients.
Finasteride stock solution stability was assessed at different concentration levels, by comparing stock solutions left at room temperature for six hours with freshly prepared solutions. Stability of the drug in the reconstitution solution was measured by reinjection of the validation run after remaining in the autosampler for 24h following initial injection. The stability of finasteride in human plasma was evaluated in triplicate following three freeze-thaw cycles, using QC samples at concentrations of 3 and 80 ng/mL. Long term stability of finasteride in human plasma was evaluated by analysis of five replicates each at 3, 40 and 80 ng/mL after 2.5 and 5 months storage under −80°C.
3. Results and Discussion
3.1. Specificity
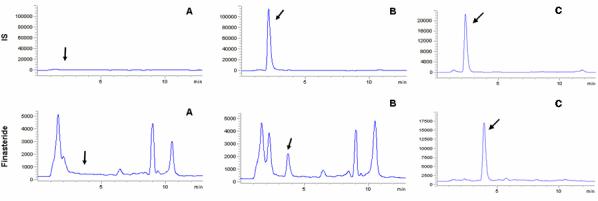
Fig. 2. displays typical chromatograms of an extract of a blank human plasma sample (A), and an extract of a plasma sample spiked with finasteride at a concentration of 1 ng/mL(B). The selectivity of the analysis is shown by symmetrical resolution of the peaks, with no interference around the retention time of the analyte in drug-free plasma obtained from six different individuals. The gradient was designed to elute endogenous materials present in blank plasma to eliminate the possibility of interference with the next run. Overall chromatographic run time was established at 13 min with finasteride eluting at tR=3.60min and internal standard, beclomethasone at tR=2.24 min.
Figure 2.
3.2. Validation characteristics
The calculated detector response of finasteride versus the nominal concentration displayed a linear relationship in the tested range of 1-100ng/mL. However, the measurement of variance over the range increased proportionally with the drug concentration. A weighting factor was applied inversely proportional to the variance at the given concentration level (1/[nominal finasteride concentration]). Using least-squares linear-regression, a mean (± standard deviation) correlation coefficient of 0.9992±0.00055 (range, 0.9983-0.9998) was obtained (Table 2).
Table 2.
Back-calculated concentrations from calibration curves run in duplicate on six occasions
| Nominal (ng/mL) |
GM (ng/mL) |
S.D. (ng/mL) |
DEV(%) |
R.S.D.(%) |
n |
|---|---|---|---|---|---|
| 1.00 | 0.97 | 0.041 | -3.34 | 4.27 | 12 |
| 2.00 | 1.98 | 0.149 | -0.78 | 7.49 | 12 |
| 5.00 | 4.93 | 0.228 | -1.37 | 4.62 | 12 |
| 10.0 | 10.34 | 0.470 | 3.36 | 4.55 | 12 |
| 25.0 | 25.41 | 0.950 | 1.63 | 3.74 | 12 |
| 50.0 | 51.12 | 1.842 | 2.25 | 3.60 | 12 |
| 100 | 98.25 | 2.266 | -1.75 | 2.31 | 12 |
Abbreviations: GM, grand mean; S.D., standard deviation; DEV, percent deviation from nominal value; R.S.D., relative standard deviation; n, total number of replicate observations within the validation runs.
In blank human plasma spiked with finasteride at a concentration of 1 ng/mL, all of the 12 samples run on six separate days were within ±12% deviation of the nominal value, significantly below the acceptable ±20% deviation limits for accuracy. The mean percentage deviation from nominal value for these 12 samples was −3.34% (Table 2), together with within and between-run variability of 4.04% and 1.43% (not shown in the table), respectively. Samples at 1ng/mL prepared on the same day from different lots of plasma were back calculated from the same calibration curve which gave the following results: 1.07, 0.95, 0.98, 0.85, 0.94 ng/mL, all within 15% deviation of the nominal value. Based on these results, the lower limit of quantitation for finasteride was established at 1 ng/mL.
Validation data for the analytical method in terms of accuracy and precision are summarized in Table 2 and 3. Table 2 displays the data calculated from duplicate calibration curves on six separate days. Shown in Table 3 is data from the QC samples that were run in quintuplicate at each concentration on these six days. Values were back-calculated from the calibration curve from the same run. The assay was found to be accurate, within 5% at all three concentrations, and precise with within-run precision error of less than 8%. Only minor between-run variance was observed at the low QC concentration of 3 ng/mL, with between-run precision error at 3.43%. No further between-run variance was observed at 40 and 80 ng/mL QC samples.
Table 3.
Assessment of accuracy and precision from quality control samplesa
| Nominal (ng/mL) |
GM (ng/mL) |
S.D. (ng/mL) |
DEV (%) |
BRP (%) |
WRP (%) |
n |
|---|---|---|---|---|---|---|
| 3.00 | 2.86 | 0.161 | -4.76 | 3.43 | 4.63 | 30 |
| 40 | 40.40 | 2.714 | 0.99 | NA | 7.31 | 30 |
| 80 | 79.54 | 4.079 | -0.58 | NA | 5.19 | 30 |
Abbreviations: GM, grand mean; S.D., standard deviation; DEV, percent deviation from nominal value; WRP, within-run precision; BRP, between-run precision; n, total number of replicate observations within the validation runs. NA, not applicable, in this case it means no further between-run variance was observed.
Five samples at each concentration were run on six different occasions.
The mean overall extraction efficiency for finasteride was approximately 92% (Table 4), independent of the spiked concentration. Absolute recovery for finasteride was estimated to be 88%. Finasteride, at concentrations of 5 and 75ng/mL, was spiked into the supernatant after blank plasma extraction or to the extraction solution in five replicates each. The matrix effect was estimated to be 5.7% and −0.8% (Table 4), respectively, leading to the conclusion that no significant matrix effect is present. QC samples run in quintiplicate along the calibration curve prepared from different lots of plasma were back calculated to be 3.1, 39.7 and 80.2 ng/mL, indicating no plasma to plasma variation. This result is also consistent with the slopes of calibration curves obtained from different lots of plasma with a mean (± of standard deviation) of 0.02000 ± 0.00099 (range, 0.01889-0.02156).
Table 4.
Assessment of recovery and matrix effect.
| Sample concentration |
Peak Area | Extraction Efficiencye |
Matrix Effectf |
Drying Lossg |
Absolute Recoveryh |
|||
|---|---|---|---|---|---|---|---|---|
| Aa | Bb | Cc | Dd | |||||
| 5 ng/ml | 47119.5 | 50466.8 | 53537.1 | 54282.8 | 93.4% | 5.7% | 1.4% | 86.8% |
| 75 ng/ml | 735281.4 | 812394.8 | 805937.2 | 819856.8 | 90.5% | -0.8% | 1.7% | 89.7% |
Experiments were conducted in five replicates for each different sample condition:
Finasteride spiked into blank plasma, extracted, dried and reconstituted
Finasteride spiked into supernatant from blank plasma extraction, dried and reconstituted
Finasteride spiked into ethyl acetate, dried and reconstituted
Finasteride spiked into mobile phase directly
Extraction Efficiency =A/B*100%
Matrix Effect =(1-B/C)*100%
Drying Loss =(1- C/D)*100%
Absolute Recovery = A/D*100%
Possible crossover interference was also evaluated by comparing the direct instrument responses to the following three sets of experiments in triplicate: samples spiked with only IS, only 100 ng/ml finasteride, and both. No interference between finasteride and the IS was found, as the response of each analyte did not vary with the addition of the other.
The samples obtained from eight patients were monitored at m/z 389.3 and 403.3 for possible interference from two major form of metabolites, ω-hydroxylated and ω-carboxylated finasteride. No peaks were present in these two channels at the finasteride retention time 3.60min.
3.3. Stability
The freshly made finasteride stock solution and the same stock that was kept at room temperature for six hours were measured in quintuplicate. The difference in the stock concentration determined from the mean peak area, was within 2.85%, lower than the experimental error range. Therefore, no degradation was observed. The reinjection measurements was consistent with the initial run, allowing samples to be reanalyzed on the following day when necessary (for example, in the case of machine failure). For the freeze-thaw stability test, no degradation was observed at the 80 ng/mL samples. However, lower concentration (3 ng/mL) samples showed approximately 5% degradation after two freeze-thaw cycles, and 10% after the third cycle, compared to the regular QC samples used in the analysis. But the degradation observed was still within the 15% experimental error range as reported previously (15). QC samples of 3, 40 and 80 ng/mL stored at −80°C were analyzed in quintiplicate at 2.5 and 5 months time point. Back calculated values were within 15% of the corresponding nominal values, so that no degradation was observed.
4. Application
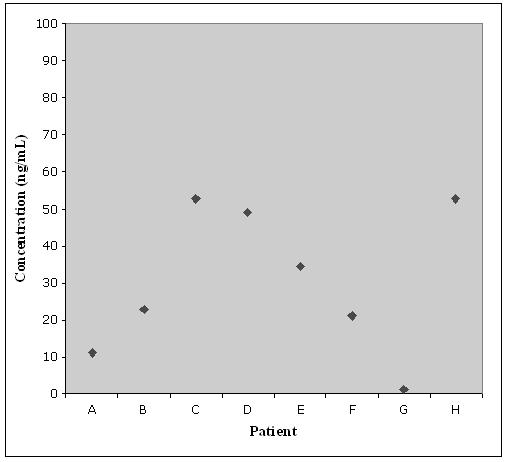
After completion of the validation process, the assay was utilized to determine the steady state plasma concentration of finasteride in patients who participated in the PCPT. The chromatogram of a patient sample is shown in Fig 2.(C) and has a calculated finasteride concentration of 52.8 ng/mL. These patients had been continuously treated with 5 mg/day finasteride during their participation in the PCPT. All eight patients who are reported here self-claimed to be 90% compliant for the six months before sample collection. Observed concentrations are shown in Fig 3. The low concentration detected in plasma from patient G suggests that adherence to the dosing schedule may be questionable.
Figure 3.
5. Conclusion
In conclusion, a robust chromatographic method with mass-spectrometric detection has been developed for the quantitative determination of finasteride in human plasma. This method is specific, accurate and precise, and can be easily implemented into routine practice. The sample pretreatment procedure, which involves liquid-liquid extraction, is simple and efficient, thereby eliminating the need for solid phase extraction, column switching procedures, and/or the use of large volumes of plasma for sample clean-up. No proprietary internal standard was used, and all the compounds used are commercially available. This comprehensively validated method is ideal for routine analysis of large numbers of samples arising from the PCPT, detecting as low as 1 ng/mL using 200 uL of plasma with a rapid extraction process.
6. Acknowledgements
The authors thank Southwest Oncology Group (SWOG) for the support during this study. This work is supported by National Cancer Institute, NIH, Department of Health and Human Services grant P01CA106451.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400.* The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. *ER Gardner
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Girman CJ. Population-based studies of the epidemiology of benign prostatic hyperplasia. Br. J. Urol. 1998;82(Suppl 1):34–43. doi: 10.1046/j.1464-410x.1998.0820s1034.x. [DOI] [PubMed] [Google Scholar]
- 2.Guess HA, Arrighi HM, Metter EJ, Fozard JL. Cumulative prevalence of prostatism matches the autopsy prevalence of benign prostatic hyperplasia. Prostate. 1990;17:241–6. doi: 10.1002/pros.2990170308. [DOI] [PubMed] [Google Scholar]
- 3.Bruchovsky N, Wilson JD. The conversion of testosterone to 5-alpha-androstan-17-beta-ol-3-one by rat prostate in vivo and in vitro. J. Biol. Chem. 1968;243:2012–21. [PubMed] [Google Scholar]
- 4.Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5alpha-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186:1213–5. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- 5.Stoner E. The clinical effects of a 5 alpha-reductase inhibitor, finasteride, on benign prostatic hyperplasia. The Finasteride Study Group. J Urol. 1992;147:1298–302. doi: 10.1016/s0022-5347(17)37547-x. [DOI] [PubMed] [Google Scholar]
- 6.Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, McConnell JD, Andriole GL, Geller J, Bracken BR, Tenover JS, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N. Engl. J. Med. 1992;327:1185–91. doi: 10.1056/NEJM199210223271701. [DOI] [PubMed] [Google Scholar]
- 7.Wilde MI, Goa KL. Finasteride: an update of its use in the management of symptomatic benign prostatic hyperplasia. Drugs. 1999;57:557–81. doi: 10.2165/00003495-199957040-00008. [DOI] [PubMed] [Google Scholar]
- 8.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA., Jr. The influence of finasteride on the development of prostate cancer. N. Engl. J. Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 9.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: a method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998;70:882–9. doi: 10.1021/ac971078+. [DOI] [PubMed] [Google Scholar]
- 10.Constanzer ML, Chavez CM, Matuszewski BK. Picogram determination of finasteride in human plasma and semen by high-performance liquid chromatography with atmospheric-pressure chemical-ionization tandem mass spectrometry. J. Chromatogr. B. 1994;658:281–7. doi: 10.1016/0378-4347(94)00250-9. [DOI] [PubMed] [Google Scholar]
- 11.Almeida A, Almeida S, Filipe A, Gagnon S, Mirapeix A, Girard B, Tanguay M. Bioequivalence study of two different coated tablet formulations of finasteride in healthy volunteers. Arzneimittelforschung. 2005;55:218–22. doi: 10.1055/s-0031-1296848. [DOI] [PubMed] [Google Scholar]
- 12.Ptacek P, Macek J, Klima J. Determination of finasteride in human plasma by liquid-liquid extraction and high-performance liquid chromatography. J. Chromatogr. B. 2000;738:305–10. doi: 10.1016/s0378-4347(99)00543-5. [DOI] [PubMed] [Google Scholar]
- 13.Carlin JR, Christofalo P, Vandenheuvel WJ. High-performance liquid chromatographic determination of N-(2-methyl-2-propyl)-3-oxo-4-aza-5 alpha-androst-1-ene-17 beta-carboxamide, a 4-azasteroid, in human plasma from a phase I study. J. Chromatogr. 1988;427:79–91. doi: 10.1016/0378-4347(88)80106-3. [DOI] [PubMed] [Google Scholar]
- 14.Carlucci G, Mazzeo P. Finasteride in biological fluids: extraction and separation by a graphitized carbon black cartridge and quantification by high-performance liquid chromatography. J. Chromatogr. B. 1997;693:245–8. doi: 10.1016/s0378-4347(97)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.Constanzer ML, Matuszewski BK, Bayne WF. High-performance liquid chromatographic method for the determination of finasteride in human plasma at therapeutic doses. J. Chromatogr. 1991;566:127–34. doi: 10.1016/0378-4347(91)80117-u. [DOI] [PubMed] [Google Scholar]
- 16.Takano T, Hata S. High-performance liquid chromatographic determination of finasteride in human plasma using direct injection with column switching. J. Chromatogr. B. 1996;676:141–6. doi: 10.1016/0378-4347(95)00399-1. [DOI] [PubMed] [Google Scholar]
- 17.U.S. Department of Health and Human Services Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER) Center for Veterinary Medicine (CVM) Guidance for Industry Bioanalytical Method Validation. 2001 May; http://www.fda.gov/cder/guidance/4252fnl.htm
- 18.Lepper ER, Hicks JK, Verweij J, Zhai S, Figg WD, Sparreboom A. Determination of midazolam in human plasma by liquid chromatography with mass-spectrometric detection. J. Chromatogr. B. 2004;806:305–10. doi: 10.1016/j.jchromb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Carlin JR, Hoglund P, Eriksson LO, Christofalo P, Gregoire SL, Taylor AM, Andersson KE. Disposition and pharmacokinetics of [14C]finasteride after oral administration in humans. Drug Metab. Dispos. 1992;20:148–55. [PubMed] [Google Scholar]