

Abstract
Compounds that can protect cells from the effects of radiation are important for clinical use, in the event of an accidental or terrorist-generated radiation event, and for astronauts traveling in space. One of the major concerns regarding the use of radio-protective agents is that they may protect cells initially, but predispose surviving cells to increased genomic instability later. In this study we used WR-1065, the active metabolite of amifostine, to determine how protection from direct effects of high and low-LET radiation exposure influences genomic stability. When added 30 minutes before irradiation and in high concentration, WR-1065 protected cells from immediate radiation-induced effects as well as from delayed genomic instability. Lower, non-toxic concentration of WR-1065 did not protect cells from death, however it was effective in significantly decreasing delayed genomic instability in the progeny of irradiated cells. The observed increase in manganese superoxide dismutase protein levels and activity may provide an explanation for this effect. These results confirm that WR-1065 is protective against both low- and high-LET radiation-induced genomic instability in surviving cells.
Keywords: genomic instability, amifostine, WR-1065, high-LET radiation, delayed effects, radioprotection
INTRODUCTION
For many years there has been an intense search for compounds that protect normal tissues from the deleterious effects of ionizing radiation. Successfully identifying candidate radio-protectors would have obvious applications in the clinic, as well as in cases where humans are accidentally or occupationally exposed to radiation, e.g., the military and the astronaut population. The ideal radioprotector would spare the organism from the immediate effects of radiation exposure, e.g., mutation induction, chromosomal rearrangements and cell death, and also from the delayed effects of radiation, e.g, carcinogenesis. Amifostine is so far the only available agent attaining most of these goals. However, it has at least two inherent properties that do not make it an ideal radioprotector. First, at doses necessary for optimal radioprotection there are toxic side effects in humans including hypotension and nausea. Second, because it primarily works as a free radical scavenger it must be present during radiation exposure to protect against cell killing.
Several different strategies for radioprotection have been proposed including 1) direct scavenging of reactive oxygen species (ROS), 2) inducing/altering the endogenous levels of ROS detoxifying enzymes such as MnSOD and 3) enhancing DNA damage signaling and repair. The majority of current studies concentrate on compounds capable of direct interaction with ROS/free radicals [1]. To be biologically effective, however, free radical scavengers have to be present in the vicinity of DNA at the time of irradiation when ROS are generated and have to be used in relatively high concentrations [1]. In addition, the inherent reactivity of ROS scavengers renders them highly unstable in biological systems, prone to metabolic deactivation, somewhat toxic, and unsuitable for oral administration [1, 2].
Despite many years of research there are surprisingly few radiation protectors in use today and those in use are limited to the clinical treatment of a few specific cancers. Among these molecules are cytokines, growth factors, phytochemicals and antioxidant nutrients including vitamin E, vitamin C and selenium [3, 4]. One of the few radio-protectors in clinical use is the phosphorothioate amifostine (WR-2721). Amifostine is FDA approved for the prevention of cumulative nephrotoxicity associated with high dose cisplatin in patients with advanced ovarian cancer and non-small cell lung cancer, and for reducing the incidence of radiation induced xerostomia in head and neck cancer patients [5]. It is the only cytoprotective agent specifically approved by the FDA as a radio-protector. MedImmune and Schering-Plough currently market it for sale under the trade name Ethyol®.
Grdina and colleagues have shown that a number of thiol containing drugs have well established radio-protective and/or antimutagenic/anticarcinogenic properties [6–8] and that in contrast to protection against cell killing, the anti-mutagenic and anti-carcinogenic effects are not drug concentration dependent. For example, 4 mM WR-1065 is an effective radio-protective dose for most mammalian cells exposed in vitro to X-rays [7, 9], whereas 40 μM WR-1065 is ineffective in protecting cells from the direct killing effects of ionizing radiation, but is as effective as a dose of 4 mM in protecting cells from the mutagenic effects of radiation [7]. Furthermore, both the radioprotectiveness and intracellular concentrations of WR-1065 fall to background levels within one h after the removal of the drug from the culture medium[7]. In this study we have extended these observations to demonstrate WR-1065 can mitigate the delayed effects of exposure to either high or low-LET radiation as measured by hyperrecombination and/or mutations in GFP construct and micronucleus formation in the progeny of surviving cells multiple generations after the initial insult.
MATERIALS AND METHODS
Drugs and reagents
WR-1065 was obtained from the Drug Synthesis and Chemistry Branch, Division of Cancer Treatment, National Institute of Health (Bethesda, MD). The stock solution of 1 M was prepared in sterile PBS immediately before use. Unless otherwise noted, all other materials were purchased from Sigma (St. Louis, MO) or Invitrogen (Carlsbad, CA).
Cell culture
The RKO36 cell line, a derivative of RKO human colorectal carcinoma carrying a green fluorescent protein (GFP) construct pCMV-EGFP2Xho [10], were maintained in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum, at 37 °C in a humidified atmosphere 5% CO2/95% air. G-418 at 1 mg/ml concentration was used as selection agent for maintaining stable transfection with GFP construct. The cells were routinely tested for mycoplasma (Bionique, Saranac Lake, NY) and showed no evidence of infection.
Drug treatment
RKO36 cells were exposed to WR-1065 at 4 mM final concentration for 30 min directly before irradiation and then washed off immediately before irradiation, or continuously treated with 40 μM final concentration for 24 hours before irradiation. However, while the 40 μM WR-1065 was allowed to “remain” in the culture medium for 24 h, i.e. cells were not washed free of the drug, it is rapidly taken up by cells and no extracellular detectable levels are present following the first h of incubation [7]. Control samples were treated with drug vehicle (PBS) for the same duration.
Irradiation
Immediately before irradiation the WR-1065-containing medium was replaced with pre-warmed, drug-free medium. Cells growing in monolayer cultures were exposed to various doses of radiation at ambient temperature. The control samples were sham-treated according to the same protocol, except for irradiation. For low-LET radiation experiments, X-rays were delivered using Pantak HF320 X-ray machine (250-kV peak, 13 mA; half-value layer, 1.65-mm copper) at a dose-rate of 2.4 Gy/min (Radiation Oncology Research Laboratory, University of Maryland).
For high-LET space radiation studies, cells were exposed to iron ions (56Fe+26, 1 GeV/nucleon) at dose rates of 1 Gy/min (for total doses >1 Gy) or 10 cGy/min (for doses <1 Gy). All high-LET radiation data were obtained at the NASA Space Radiation Laboratory (NSRL, Brookhaven National Laboratory, Upton, NY), in experimental cycles NSRL-06C (2006), NSRL-07A and NSRL-07B (2007). The beam dosimetry was provided by the NSRL and a detailed characterization of this beam has been published previously [11]. Following irradiation cell cultures were returned to 37 °C for 2 h (to allow for media deactivation), then harvested by trypsinization and used in subsequent experiments or frozen in aliquots at −80 °C.
Cell survival assay
The effect of irradiation on cell survival and the cytoprotective activity of WR-1065 were measured by clonogenic cell survival assay. After irradiation cells were harvested by trypsinization, resuspended in fresh DMEM medium and replated into 100-mm-diameter culture dishes at densities calculated to yield 50–100 cell colonies per dish. Following 10–14 days incubation, cells were fixed, stained with crystal violet in 20% ethanol, and colonies >50 cells counted. The number of surviving colonies divided by the number of plated cells was used to calculate the plating efficiency and survival fraction for each treatment.
Growth inhibition assay
RKO36 cells were seeded in 60-mm-diameter dishes at 100 cells per dish and allowed to attach. Subsequently, WR-1065 was added to final concentration ranging between 0 and 10 mM Since the WR-1065 uptake into cells occurs as fast as 5 to 10 min [7], in some experiments the drug containing medium was replaced with fresh, drug-free medium after 30 min of treatment. Following 7–10 days incubation, surviving colonies were fixed and stained with crystal violet in 20% ethanol. Cell growth inhibition was calculated by comparing the number of colonies in treated samples to non-treated controls.
GFP assay for delayed genomic instability
The effect of WR-1065 treatment and irradiation on delayed genomic instability was measured using the previously described GFP assay [10]. Briefly, drug-treated and irradiated RKO36 cells, or sham-treated controls, were plated in 60-mm-diameter culture dishes at densities calculated to yield 50 well-separated colonies per dish at each treatment. Seven to ten days post-irradiation, colonies (>50 cells) were inspected for GFP expression using an inverted fluorescence microscope. At least 20 dishes, containing approximately 1000 colonies total, were examined and the percentage of mixed colonies in each sample was calculated as described [10].
Micronucleus assay
The effects of WR-1065 treatment and irradiation on chromosomal integrity were measured by the frequency of micronucleus formation using the cytokinesis block technique [12]. Briefly, one million drug-treated and irradiated RKO36 cells or sham-treated control cells were plated in T-25 culture flasks in the presence of 1.5 μg/ml cytochalasin B and incubated for 48 h at 37 °C. At the concentration used cytochalasin B is not toxic to RKO36 cells. At the end of the incubation, cells were harvested by trypsinization, fixed with cold methanol : acetic acid (3:1) and stored at −20 °C. The slides were prepared, stained with acridine orange (0.01% in PBS) and examined under a fluorescence microscope (60x, dual-band filter). At least 500 bi-nucleated cells with well-defined cytoplasm were scored for micronuclei formation. The frequency of micronuclei was calculated as a ratio of number of micronucleated cells to total number of bi-nucleated cells scored.
Statistical analysis
Means and standard errors were calculated for all data points from at least three independent experiments unless indicated otherwise. The means were compared between WR-1065-treated and non-treated samples by one-factor ANOVA.
RESULTS
WR-1065 at 4 mM protects RKO36 cells from chromosomal damage and death induced by ionizing radiation
Clonogenic assays were used to determine the radioprotective effect of WR-1065 following exposure to 4 mM drug concentrations 30 min immediately before exposure to X-rays (Fig. 1A) or iron ions (Fig. 1B). As expected, high-LET radiation is more effective in reducing the surviving fraction of irradiated cells, with an RBE ~ 2 as compared to low-LET X-rays. Under both treatments, pre-incubation with WR-1065 provided significant radioprotection to irradiated cells, however the effect was smaller for iron ions. Similar observations were made previously using other high LET radiation including fission-neutrons [9, 13].
FIGURE 1.

Direct radio-protective effects of high concentrations of WR-1065 on irradiated RKO36 cells. A) Clonogenic survival of cells pre-treated with 4 mM WR-1065 for 30 min before irradiation (●, WR-1065) or sham-treated (○, sham). B) Clonogenic survival in cells pre-treated with 4 mM WR-1065 for 30 min before irradiation with iron ions (●, WR-1065) or sham-treated (○, sham). C) Percentage of binucleated cells containing micronuclei assayed 48 h after treatment with 4 mM WR-1065 for 30 min before irradiation (■, WR-1065) or sham-treated (□, sham), and then irradiated with X-rays, *** p<0.001 versus sham-treated and irradiated control. D) Frequency of binucleated cells containing micronuclei assayed 48 h after treatment with 4 mM WR-1065 for 30 min before irradiation with iron ion (■, WR-1065) or sham-treated (□, sham), * p<0.05 versus sham-treated and irradiated control. All results are means of at least three independent experiments ±SEM.
Cultures irradiated with X-rays (Fig. 1C) or iron-ions (Fig. 1D) showed up to 30% bi-nucleated cells that produced one or more micronuclei (30+/−2.2 and 28+/−1.0 for X-rays and iron ions, respectively), as compared to 3–5% of micronucleated cells in sham-treated control cultures. Pre-treatment with 4 mM WR-1065 for 30 min before irradiation significantly reduced micronuclei formation, especially in X-ray irradiated cells (p<0.0001; Fig. 1C).
WR-1065 at 4 mM protects irradiated RKO36 cells from delayed genomic instability
To investigate how protection provided by 4 mM WR-1065 might modulate delayed genomic stability in the progeny of irradiated cells, we analyzed the hyperrecombination/deletion sensitive GFP construct previously transfected into these cells. Cells were subjected to 30 min WR-1065 treatment followed by irradiation, re-plated and allowed to establish colonies in fresh, drug-free medium. The GFP expression in every cell in the colony was analyzed using fluorescence microscopy. Figure 2A presents a schematic representation of formation of one-color (stable) and mixed (unstable) colonies, and Fig. 2B shows an example of a representative mixed GFP colony (GFP+/−). Mixed GFP colonies can be only produced by hyperrecombination or deletions/mutation events in the GFP reporter construct in progeny cells, but not the actually irradiated cell, their presence indicates induction of genomic instability. Figure 2C demonstrates that irradiation of cells with 4 Gy X-rays induces significant increase in mixed GFP colonies (open bars), thus suggesting radiation-induced increase in genomic instability. WR-1065 at 4 mM reduces the frequency of mixed colonies to control levels, protecting cells from induced genomic instability induced by ionizing radiation (Fig. 2C, closed bars). Previously, Huang et al., [10] demonstrated that RKO36 cells have a near-diploid chromosome complement and are genetically stable without being subjected to genotoxic stress. Fluorescence in-situ hybridization experiments with probes for chromosomes 4, 13, and 16 in metaphase spreads from 30 unirradiated colonies of RKO36 showed normal patterns in all cases.
FIGURE 2.
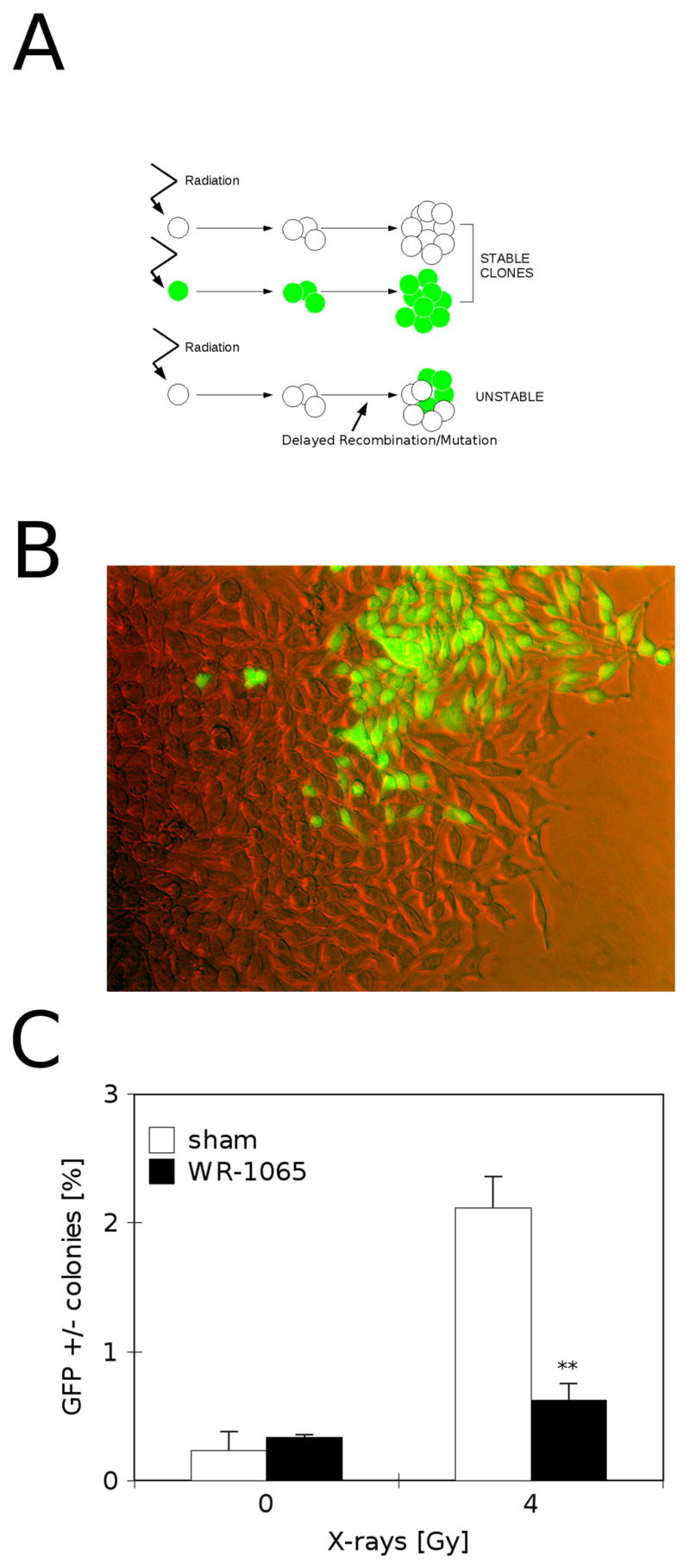
Protective effects of 4 mM WR-1065 against genomic instability induced by X-ray irradiation. A) Strategy for detecting delayed genomic instability in the GFP reporter assay. Genetically stable progeny of irradiated cells give uniform GFP+ or GFP− colonies (stable), while cells displaying delayed genomic instability produce mixed GFP+/− colonies (unstable). B) Representative fluorescence microscopy image of mixed GFP+/− colony. C) Frequency of mixed GFP+/− colonies in RKO36 cultures pre-treated with 4 mM WR-1065 for 30 min immediately before irradiation (■, WR-1065) or sham-treated (□, sham), and irradiated with 4 Gy of X-rays. The cultures were scored for GFP fluorescence 7 to 10 days after irradiation. Results are means of four independent experiments ±SEM, ** p<0.01 versus sham-treated and irradiated control.
Milimolar concentrations of WR-1065 are cytotoxic to RKO36 cells
To test the effects of WR-1065 on unirradiated cells, exponentially growing cells were subjected to either short, 30 min drug treatment followed by 7–10 days of recovery in drug-free medium (Fig. 3, closed markers), or incubated continuously with drug for 7–10 days (Fig. 3, open markers). Fig. 3 shows that under conditions used in these experiments WR-1065 is cytotoxic to RKO36 cells, reaching ED50 of 10 μM for continuous treatment (Fig. 3, open markers), and 4 mM after 30 min treatment (Fig. 3, closed markers). The effects observed, especially after continuous treatment, are probably related to topoisomerase IIα inhibition which in turn slows down cellular growth and cell cycle progression [14].
FIGURE 3.

Growth inhibition in cells treated with WR-1065 for 30 min and then returned to drug-free medium for 7–10 days (●, 30 min), or treated continuously for 7–10 days (○, continuous). Results are means of three independent experiments ±SEM.
WR-1065 at 40 μM protects RKO36 cells from delayed genomic instability but not from cell death and immediate chromosomal damage
Figure 4 demonstrates the lack of immediate radio-protective effects of 40 μM WR-1065 in irradiated cells. In our experimental system, at this relatively non-toxic concentration, WR-1065 does not provide statistically significant protection from cell death (Fig. 4A–B, open and closed markers) or chromosomal damage expressed during first metaphase post-irradiation (micronucleus formation, Fig. 4C–D, open and closed bars).
FIGURE 4.

Direct radio-protective effects of non-toxic concentrations of WR-1065 on irradiated cells. A) Clonogenic survival of cells pre-treated with 40 μM WR-1065 for 24 h before X-irradiation (●, WR-1065) or sham-treated (○, sham). B) Clonogenic survival of cells pre-treated with 40 μM WR-1065 for 24 h before iron ion irradiation (●, WR-1065) or sham-treated (○, sham). C) Percentage of binucleated cells containing micronuclei assayed 48 h after irradiation, and 72 h after treatment with 40 μM WR-1065 for 24 h before irradiation (■, WR-1065) or sham-treated (□, sham), and then irradiated with X-rays. D) Percentage of binucleated cells containing micronuclei assayed 48 h after irradiation, and 72 h after treatment with 40 μM WR-1065 for 24 h before irradiation (■, WR-1065) or sham-treated (□, sham), and irradiated with iron ions. Results are means of two independent experiments ±SEM.
Although a low, non-toxic concentration of WR-1065 was not sufficient in protecting cells from radiation-induced death, it was able to provide statistically significant protection from delayed genomic instability as measured by the GFP assay (Fig. 5A–B). Irradiation of cells increased the percentage of mixed colonies from 0.3–0.8% in non-irradiated controls to 1.5–2.0% in cultures irradiated with X-rays (Fig. 5A, open bars) or iron ions (Fig. 5B, open bars). Pre-treatment with 40 μM WR-1065 for 24h before irradiation significantly (p<0.05) reduced percentage of mixed GFP colonies under both conditions (Fig. 5A–B, closed bars).
FIGURE 5.

Protective effects of milimolar concentrations of WR-1065 against delayed genomic instability induced by irradiation in RKO36 cells. A) Frequency of mixed GFP+/− colonies in cultures pre-treated with 40 μM WR-1065 for 24 h before X-irradiation (■, WR-1065) or sham-treated (□, sham). The cultures were scored for GFP fluorescence 7 to 10 days after irradiation. Results are means of two independent experiments ±SEM, * p<0.05 versus sham-treated and irradiated control. B) Percentage of mixed GFP+/− colonies in cultures pre-treated with 40 μM WR-1065 for 24 h before iron ion irradiation (■, WR-1065) or sham-treated (□, sham). The cultures were scored for GFP fluorescence 7 to 10 days after irradiation. Results are means of two independent experiments ±SEM, * p<0.05 versus sham-treated and irradiated control. C) Percentage of binucleated cells containing micronuclei in cultures assayed 7 days after treatment with 40 μM WR-1065 for 24 h before X-irradiation (■, WR-1065) or sham-treated (□, sham). Results are means of four independent experiments ±SEM. D) Percentage of binucleated cells containing micronuclei in cultures assayed 7 days after treatment with 40 μM WR-1065 for 24 h before iron ion irradiation (■, WR-1065) or sham-treated (□, sham). Results are means of two independent experiments ±SEM, * p<0.05 versus sham-treated and irradiated control.
In addition to protecting from delayed genomic instability, WR-1065 at 40 μM concentration decreased the level of prolonged chromosomal damage in cells irradiated with iron ions (Fig. 5D). Following drug-treatment (40 μM for 24h), the cells were irradiated with 2 Gy of X-rays (Fig. 5C) or 1 Gy of iron ions (Fig. 5D), and allowed to recover for 7 days in drug-free medium. Fig. 5C shows that cells exposed to X-rays recovered completely after 7 days, reducing the micronuclei level to control values. However, chromosomal damage induced by iron ions was more persistent reaching the level of 7.9+/−0.45% micronucleated cells after 7 days recovery (Fig. 5D, open bars), as compared to 3.0+/−0.41% control level and 28+/−1.0% initial damage level (Fig. 1D, open bars). In the case of iron ions induced prolonged chromosomal damage, pre-treatment with 40 μM WR-1065 for 24h before irradiation significantly (p<0.01) reduced micronucleus induction (Fig. 5D, closed bars).
DISCUSSION
A significant question when developing novel radio-protectors is whether protecting a cell or tissue from the lethal effects of radiation predisposes that irradiated volume to significantly elevated risks for genomic instability, which could lead to neoplastic change. Genomic instability is defined as the increased rate of alterations to the genome, including gene mutations, chromosomal abnormalities, micronuclei formation, reduced plating efficiency and cellular transformation. Genomic instability has been detected following both low and high-LET radiation exposure [15]. Currently, the mechanism(s) responsible for initiating and perpetuating delayed genomic instability are unknown, however several pathways have been suggested [16, 17]. In particular, the intracellular increase in reactive oxygen species (ROS) was indicated and in our previous work we have demonstrated that preventing ROS from damaging DNA (through e.g. free radical scavenging), and promoting chemical or enzymatic repair of DNA lesions can reduce the subsequent onset of X-ray irradiation-induced genomic instability [18].
In the present study we evaluated the efficacy of WR-1065 in reducing direct and delayed effects of both high and low-LET radiation exposures. As expected the high-LET iron ion radiation was more effective than X-rays, achieving similar levels of chromosomal damage, delayed genomic instability and cell death (Fig. 1). The observed relative biological efficiency (RBE) confirms previously published results showing that when genomic damage (chromosomal aberrations and micronuclei formations) induced by high-LET iron ions was compared to that from low-LET γ-rays, iron ion radiation was up to 3.3 times more effective than γ-rays [19, 20].
In agreement with previously published observations, WR-1065 at high milimolar concentrations protected cells from immediate X-ray-induced effects (e.g. DNA damage and cell death) (Fig. 1). Similar, although less pronounced radioprotective effects were observed in cells irradiated with iron ions consistent with previous published work describing the effects of WR-1065 on cells irradiated with high LET fission spectrum neutrons [6, 13]. This difference can be explained by differences in the mechanisms of initial DNA damage induction between iron ions and X-rays. The ROS/free radical-mediated effect is responsible for ~ 66% of cellular damage following low-LET irradiation, while in case of high mass – high energy ions the direct deposition of energy from ions to DNA is predominant, creating complex and/or clustered DNA lesions [21]. The persistence of iron ions induced chromosomal damage, which is still visible as increase in micronucleus formation in cells cultivated for 7 days post-irradiation (Fig. 5D) suggests that the complex lesions are also more difficult to repair by cellular systems. Since WR-1065’s direct free radical scavenging activity is limited to between 30 min to an h after intracellular uptake [7] and depends mostly on detoxifying highly reactive hydroxyl radicals (OH -) [22] the drug was more effective in the case of low LET X-ray-irradiated cells. However, other properties of the drug including its ability to participate in polyamine mediated reactions related to genomic stabilization [23–25] and the induction of endogenous anti-oxidant enzymes such as MnSOD (SOD2) [26–30] may have contributed to its overall genomic protective effects. MnSOD levels in particular are known to rise significantly in cells within 2 h following WR-1065 exposure at doses ranging from 40 μM to 4 mM and to persist up to 30 h later. This extends the effective radical scavenging period to times much longer than can be accounted for by the kinetics of WR-1065 uptake and metabolism alone [7, 26–30]. The period of time during which genomic instability processes are occurring following irradiation are unknown. However, radiation-induced free radicals along with the delayed post-radiation-metabolically mediated elevation in intracellular free radical formation [31, 32], suggest WR-1065 exposure induced elevation of MnSOD activity plays a significant role in modulating these deleterious processes.
As toxicity (Fig. 3) and other side effects of high doses of amifostine are important in case of human use, the effects of low, non-toxic drug dose was also assessed. In contrast to high doses, a lower non-toxic concentration of WR-1065 (40 μM) did not provide significant immediate radioprotection as measured by cell survival or micronucleus formation (Fig. 4). However, when added 24 h before irradiation, it was effective in mitigating genomic instability in progeny of irradiated cells (Fig. 5A–B). Interestingly, pre-treatment with the low (40 μM) WR-1065 concentrations also protected cells from long-term chromosomal damage as detected by micronucleus formation assay (Fig. 5D). WR-1065 directly concentrates within the nucleus and mitochondria of cells. Therefore, irradiation of cells 30 min following exposure of cells to a high dose of WR-1065 would insure that both the nucleus and mitochondria are protected from radiation-induced free radical damage. In contrast, because of the relatively short time for direct radical scavenging by WR-1065 [7], cells irradiated 24 h following WR-1065 exposure would be essentially protected only by elevated levels of MnSOD, an anti-oxidant enzyme that is localized only within the mitochondria [33]. These data would therefore suggest that the mitochondria are important targets as a potential source for metabolically induced free radicals that could initiate or propagate genomic instability as evidenced by hyper-recombination as detected in our novel GFP RKO36 model system. Support for this comes from observations that mitochondria can exhibit elevated ROS production following a ROS insult that leads to ROS propagation. This phenomenon has been termed ROS-induced ROS release (RIRR) [34, 35]. In this manner radiation-induced ROS stress to the mitochondria could not only initiate but also promote genomic instability for a prolonged period of time. Successful interruption of the propagation of mitochondrial produced ROS by an anti-oxidant enzyme localized within the mitochondria such as MnSOD would therefore be an effective means to inhibit the RIRR effect and its role in propagating an intracellular environment conducive to the development of genomic instability.
The superoxide dismutases are crucial to protect oxygen-dependent cells from the toxicity of metabolically produced ROS, as well as external stress-generated ROS. We have reported previously that WR-1065 at 40 μM and 4 mM (30 min treatment followed by 24 h post-incubation) enhanced significantly expression of active MnSOD in human endothelial [28], mouse sarcoma cells [29], as well as in RKO36 human colorectal carcinoma cells [26]. The ability to enhance MnSOD activity in cells and tissues is not limited to thiol containing drugs such as WR-1065 alone. Recent reports suggest that resveratrol, a polyphenol with anti-oxidant properties, is a potent inducer of MnSOD expression and activity in mouse brain and normal human lung fibroblasts (MRC5) [36, 37]. These investigators suggest that modulation of MnSOD activity may be an important factor in the protective properties of resveratrol. Modulation of intracellular MnSOD activity may be a here-to-fore underappreciated element in the overall cytoprotective consequences of exogenously administrated anti-oxidant drugs.
Cancer cells usually have relatively low MnSOD protein levels and enzymatic activity, and it has been suggested that their elevation can suppress tumor development [38, 39]. The recent report demonstrated that the increased levels of MnSOD gene significantly decrease chromosomal instability and the onset of tumor formation in mice [40]. Based on these observations we propose that WR-1065-induced increase in MnSOD activity could be the main pathway of cell protection from genomic instability. However, the increase in MnSOD activity in our experimental system did not persist for more than 24 – 72 h after drug treatment (data not shown and [30]), while the protection from genomic instability was still clearly visible in the progeny of surviving cells, 7 days post drug treatment and irradiation. This discrepancy could be explained in two ways. First, the drug-induced protection of parental cell from the initial cellular damage and/or ROS increases during or immediately after irradiation is sufficient to prevent the development of genomic instability in its progeny. Second, there is another drug-induced cellular mechanism(s) protecting from perpetuating genomic instability in progeny of irradiated cell. For example, Mitchell et al. reported that WR-1065 affected normal polyamine metabolism, increasing putrescine and spermidine synthesis in treated cells [24]. It was suggests that this elevated polyamine synthesis and therefore increased DNA/chromatin stabilization probably accounts for WR-1065’s anti-mutational effect but not its effects on cell survival [23, 24]. However, regardless of the underlying mechanism of action, the inability of WR-1065 to directly scavenge free radicals for periods of time greater than one h following exposure [7] suggests that inducible mechanisms must be at work to account for the long term protective effectiveness against genomic instability. The MnSOD effect is one such inducible mechanism that can account for an extended period of protection against post-radiation-metabolically-induced free radical formation and damage.
In summary, using our unique reporter based assay for investigation of delayed genomic instability we demonstrated that WR-1065, the active metabolite of amifostine, can reduce both the direct and delayed detrimental effects of both high and low-LET radiation exposures. These radioprotective effects of WR-1065 are complex and may be related in part to its free radical scavenging ability, polyamine like properties and effects on polyamine biosynthesis, and to the induction of increases in endogenous MnSOD protein levels and activity. Our findings support the usefulness of amifostine, its free thiol metabolite WR-1065 in particular, as an effective radio-protective drug capable of protecting against radiation induced genomic instability and the risk of delayed carcinogenesis.
Acknowledgments
We thank Drs. Marcelo Vazquez, Betsy M. Sutherland, Peter Guida and Adam Rusek, and the numerous other individuals at the Brookhaven National Laboratory who helped us with these studies. We wish also to thank Ms. Dinah Pyles, Mr. Umut Aypar and Mr. Yi-Jun Li for help with experiments. This work was supported in part by NASA grant NNJ05HE73G (for WFM), DOE Low Dose grant DE-FG-01-04ER04-21 (for DJG), and NIH grant P30-CA086862 (for DRS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grdina DJ, et al. Radioprotectors: current status and new directions. Radiat Res. 2005;163(6):704–5. [PubMed] [Google Scholar]
- 2.Weiss JF. Pharmacologic approaches to protection against radiation-induced lethality and other damage. Environ Health Perspect. 1997;105 Suppl 6:1473–8. doi: 10.1289/ehp.97105s61473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh VK, V, Yadav S. Role of cytokines and growth factors in radioprotection. Exp Mol Pathol. 2005;78(2):156–69. doi: 10.1016/j.yexmp.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Weiss JF, Landauer MR. Protection against ionizing radiation by antioxidant nutrients and phytochemicals. Toxicology. 2003;189(1–2):1–20. doi: 10.1016/s0300-483x(03)00149-5. [DOI] [PubMed] [Google Scholar]
- 5.Brizel DM, et al. Phase III randomized trial of amifostine as a radioprotector in head and neck cancer. J Clin Oncol. 2000;18(19):3339–45. doi: 10.1200/JCO.2000.18.19.3339. [DOI] [PubMed] [Google Scholar]
- 6.Carnes BA, Grdina DJ. In vivo protection by the aminothiol WR-2721 against neutron-induced carcinogenesis. Int J Radiat Biol. 1992;61(5):567–76. doi: 10.1080/09553009214551381. [DOI] [PubMed] [Google Scholar]
- 7.Grdina DJ, et al. Thiol and disulfide metabolites of the radiation protector and potential chemopreventive agent WR-2721 are linked to both its anti-cytotoxic and anti-mutagenic mechanisms of action. Carcinogenesis. 1995;16(4):767–74. doi: 10.1093/carcin/16.4.767. [DOI] [PubMed] [Google Scholar]
- 8.Kataoka Y, et al. Antimutagenic effects of amifostine: clinical implications. Semin Oncol. 1996;23(4 Suppl 8):53–7. [PubMed] [Google Scholar]
- 9.Hill CK, et al. 2-[(Aminopropyl)amino]ethanethiol (WR-1065) is anti-neoplastic and anti-mutagenic when given during 60Co gamma-ray irradiation. Carcinogenesis. 1986;7(4):665–8. doi: 10.1093/carcin/7.4.665. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, et al. Ionizing radiation induces delayed hyperrecombination in mammalian cells. Mol Cell Biol. 2004;24(11):5060–8. doi: 10.1128/MCB.24.11.5060-5068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeitlin C, Heilbronn L, Miller J. Detailed characterization of the 1087 MeV/nucleon iron-56 beam used for radiobiology at the alternating gradient synchrotron. Radiat Res. 1998;149(6):560–9. [PubMed] [Google Scholar]
- 12.Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455(1–2):81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 13.Grdina DJ, Sigdestad CP, Carnes BA. Protection by WR-1065 and WR151326 against fission-neutron-induced mutations at the HGPRT locus in V79 cells. Radiat Res. 1989;117(3):500–10. [PubMed] [Google Scholar]
- 14.Grdina DJ, Murley JS, Roberts JC. Effects of thiols on topoisomerase-II alpha activity and cell cycle progression. Cell Prolif. 1998;31(5–6):217–29. doi: 10.1111/j.1365-2184.1998.tb01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Limoli CL, et al. Genomic instability induced by high and low LET ionizing radiation. Adv Space Res. 2000;25(10):2107–17. doi: 10.1016/s0273-1177(99)01062-5. [DOI] [PubMed] [Google Scholar]
- 16.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiationinduced genomic instability and bystander effects in vitro. Radiat Res. 2003;159(5):567–80. doi: 10.1667/0033-7587(2003)159[0567:nadeoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 17.Hall EJ, Hei TK. Genomic instability and bystander effects induced by high-LET radiation. Oncogene. 2003;22(45):7034–42. doi: 10.1038/sj.onc.1206900. [DOI] [PubMed] [Google Scholar]
- 18.Limoli CL, et al. Attenuation of radiation-induced genomic instability by free radical scavengers and cellular proliferation. Free Radic Biol Med. 2001;31(1):10–9. doi: 10.1016/s0891-5849(01)00542-1. [DOI] [PubMed] [Google Scholar]
- 19.Brooks A, et al. Relative effectiveness of HZE iron-56 particles for the induction of cytogenetic damage in vivo. Radiat Res. 2001;155(2):353–9. doi: 10.1667/0033-7587(2001)155[0353:reohip]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Hada M, et al. mBAND analysis of chromosomal aberrations in human epithelial cells exposed to low- and high-LET radiation. Radiat Res. 2007;168(1):98–105. doi: 10.1667/RR0759.1. [DOI] [PubMed] [Google Scholar]
- 21.Belli M, et al. DNA fragmentation induced in human fibroblasts by accelerated (56)fe ions of differing energies. Radiat Res. 2006;165(6):713–20. doi: 10.1667/RR3574.1. [DOI] [PubMed] [Google Scholar]
- 22.Marzatico F, et al. In vitro antioxidant properties of amifostine (WR-2721, Ethyol) Cancer Chemother Pharmacol. 2000;45(2):172–6. doi: 10.1007/s002800050026. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell JL, et al. Involvement of the polyamine transport system in cellular uptake of the radioprotectants WR-1065 and WR-33278. Carcinogenesis. 1995;16(12):3063–8. doi: 10.1093/carcin/16.12.3063. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell JL, et al. Mammalian cell polyamine homeostasis is altered by the radioprotector WR-1065. Biochem J. 1998;335(Pt 2):329–34. doi: 10.1042/bj3350329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savoye C, et al. Thiol WR-1065 and disulphide WR-33278, two metabolites of the drug ethyol (WR-2721), protect DNA against fast neutron-induced strand breakage. Int J Radiat Biol. 1997;71(2):193–202. doi: 10.1080/095530097144319. [DOI] [PubMed] [Google Scholar]
- 26.Murley JS, et al. Manganese superoxide dismutase (SOD2)-mediated delayed radioprotection induced by the free thiol form of amifostine and tumor necrosis factor alpha. Radiat Res. 2007;167(4):465–74. doi: 10.1667/RR0758.1. [DOI] [PubMed] [Google Scholar]
- 27.Murley JS, et al. Delayed radioprotection by NFkappaB-mediated induction of Sod2 (MnSOD) in SA-NH tumor cells after exposure to clinically used thiol-containing drugs. Radiat Res. 2004;162(5):536–46. doi: 10.1667/rr3256. [DOI] [PubMed] [Google Scholar]
- 28.Murley JS, et al. Activation of NFkappaB and MnSOD gene expression by free radical scavengers in human microvascular endothelial cells. Free Radic Biol Med. 2001;30(12):1426–39. doi: 10.1016/s0891-5849(01)00554-8. [DOI] [PubMed] [Google Scholar]
- 29.Murley JS, et al. Delayed cytoprotection after enhancement of Sod2 (MnSOD) gene expression in SA-NH mouse sarcoma cells exposed to WR-1065, the active metabolite of amifostine. Radiat Res. 2002;158(1):101–9. doi: 10.1667/0033-7587(2002)158[0101:dcaeos]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 30.Murley JS, et al. Maintenance of manganese superoxide dismutase (SOD2)-mediated delayed radioprotection induced by repeated administration of the free thiol form of amifostine. Radiat Res. 2008;169(5):495–505. doi: 10.1667/RR1194.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 2006;66(21):10377–83. doi: 10.1158/0008-5472.CAN-05-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Limoli CL, et al. Apoptosis, reproductive failure, and oxidative stress in Chinese hamster ovary cells with compromised genomic integrity. Cancer Res. 1998;58(16):3712–8. [PubMed] [Google Scholar]
- 33.Epperly MW, et al. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res. 2003;160(5):568–78. doi: 10.1667/rr3081. [DOI] [PubMed] [Google Scholar]
- 34.Brady NR, et al. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid Redox Signal. 2006;8(9–10):1651–65. doi: 10.1089/ars.2006.8.1651. [DOI] [PubMed] [Google Scholar]
- 35.Leach JK, et al. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61(10):3894–901. [PubMed] [Google Scholar]
- 36.Robb EL, et al. Molecular mechanisms of oxidative stress resistance induced by resveratrol: Specific and progressive induction of MnSOD. Biochem Biophys Res Commun. 2008;367(2):406–12. doi: 10.1016/j.bbrc.2007.12.138. [DOI] [PubMed] [Google Scholar]
- 37.Robb EL, et al. Dietary resveratrol administration increases MnSOD expression and activity in mouse brain. Biochem Biophys Res Commun. 2008;372(1):254–9. doi: 10.1016/j.bbrc.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 38.Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother. 2005;59(4):143–8. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Weydert CJ, et al. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic Biol Med. 2006;41(2):226–37. doi: 10.1016/j.freeradbiomed.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 40.van de Wetering CI, et al. Manganese superoxide dismutase gene dosage affects chromosomal instability and tumor onset in a mouse model of T cell lymphoma. Free Radic Biol Med. 2008;44(8):1677–86. doi: 10.1016/j.freeradbiomed.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]