

Abstract
Aberrant activation of the androgen receptor (AR) by the ErbB2/ErbB3 heterodimer contributes to the development of hormone resistance in prostate cancer. EBP1, an ErbB3-binding protein, acts as an AR corepressor. As EBP1 is decreased in preclinical models of hormone-refractory prostate cancer, we studied the expression of EBP1 in human prostate cancer. We found that the expression of the EBP1 gene was significantly decreased in prostate cancer tissues compared with benign prostate at both mRNA and protein levels. Restoration of EBP1 expression in the hormone-refractory LNCaP C81 cell line led to an amelioration of the androgen-independent phenotype based on established biological criteria and a reduction in the expression of a cohort of AR target genes. The ability of the ErbB3 ligand heregulin (HRG) to stimulate growth and AKT phosphorylation of hormone-refractory prostate cancer cells was abolished. Abrogation of EBP1 expression by short hairpin RNA in hormone-dependent LNCaP cells, which undergo apoptosis in response to HRG, resulted in HRG-stimulated cell growth. Restoration of EBP1 expression decreased the tumorigenicity of C81 xenografts in female mice, whereas elimination of EBP1 expression enhanced the ability of LNCaP cells to grow in female mice. Our data support a role for EBP1 in the development of hormone-refractory prostate cancer via inhibition of both AR- and HRG-stimulated growth and present a novel strategy for treating androgen-refractory prostate cancer.
Introduction
Prostate cancer begins as an androgen-dependent tumor that undergoes clinical regression in response to pharmacologic and surgical strategies that reduce testosterone concentration. Despite this treatment, the cancer eventually regrows as a uniformly fatal hormone-refractory tumor. The androgen receptor (AR) is central to the initiation and growth of prostate cancer and to the therapeutic response to hormones. AR continues to be expressed even in hormone-refractory tumors and aberrant AR signaling is an important mechanism of progression to androgen independence (1). Increases in AR protein levels are associated with hormone-refractory disease clinically (2, 3) and in xenograft models of prostate cancer (4). Thus, new approaches to silence AR signaling are important for the therapy of hormone-refractory prostate cancer.
ErbB family members and their ligands have been implicated in the development of androgen independence due to their stimulatory effects on AR function (5). It was initially shown that androgen-independent sublines of human prostate cancer xenografts expressed higher levels of ErbB2 than androgen-dependent sublines (6). Ectopic expression of ErbB2 sensitized cells to extremely low levels of androgen (7). Inhibition of ErbB2/3 signaling resulted in suppression of AR signaling and cell growth. ErbB2/3 kinase activity stabilized AR protein levels via inhibition of ubiquitin-mediated degradation and increased recruitment of AR to AR-responsive promoters (5). Inhibition of ErbB2 kinase activity also impaired AR recruitment to the prostate-specific antigen (PSA) promoter (8) and triggered apoptosis in androgen-independent C4−2 cells (9). ErbB3 has also recently emerged as an important factor in prostate cancer progression. Overexpression of ErbB3 was linked to a less favorable prognosis in prostate cancer (10). A secreted form of the ErbB3 receptor, MDA-BF-1, was associated with prostate cancer metastases (11). A high frequency of nuclear ErbB3 expression was associated with hormone-refractory prostate cancer (12) and nuclear ErbB3 expression was increased in prostate cancer cells found in lymph node and bone metastases (13). The ErbB3/4 ligand heregulin (HRG), present in normal human adult prostate and in benign prostatic hyperplasia, may function as a paracrine physiologic differentiation factor (14, 15). HRG had opposing effects on growth of hormone-dependent versus hormone-refractory cells lines. HRG inhibited growth and induced differentiation of hormone-sensitive ErbB1-, ErbB2-, and ErbB3-expressing LNCaP prostate cancer cells (14, 15) and induced the expression of the tumor suppressor p53 and the cyclin-dependent kinase inhibitor p21 (16). Tal-Or et al. (17) similarly showed that neuregulin (HRG) activated ErbB2/ErbB3 heterodimers and induced apoptosis of LNCaP cells. In contrast, HRG enhanced growth of CWR-R1 and 22Rv1 hormone-refractory cells and activated AR (18, 19). Such effects were more pronounced in medium with castrate levels of androgens, pointing to the importance of ErbB signaling pathways in androgen-deprived patients.
Our laboratory has cloned EBP1, a protein that binds both the ErbB3 receptor and the AR. EBP1, the product of the PA2G4 gene, is the human homologue of the mouse cell cycle-regulated, DNA-binding protein p38−2G4 (20). EBP1 has a predicted amphipathic helical domain for protein/protein or protein/DNA interactions and an LXXLL motif flanked by basic regions present in many nuclear receptor binding proteins (21). EBP1 physically associated with AR and suppressed AR signaling (22). Overexpression of the EBP1 gene in androgen-dependent LNCaP cells down-regulated expression of AR and its target genes. EBP1 decreased transcription of AR-activated genes in part by binding to an AR response element in AR target genes and recruiting the corepressors Sin3A and HDAC2 to chromatin (23). EBP1 bound AR targets in the presence of bicalutamide, providing evidence of EBP1 function in prostate cancer therapy. The ability of EBP1 to bind AR-regulated promoters and inhibit AR promoter activity was enhanced by HRG (24). The ectopic expression of EBP1 resulted in growth inhibition both in vivo and in vitro (23). As EBP1 expression was reduced in two models of hormone-refractory prostate cancer (25), we postulated that lack of EBP1 function might contribute to growth of cells in an androgen-deprived environment.
In this study, we determined the expression of EBP1 in clinical prostate cancer and the effects of EBP1 on the hormone-refractory phenotype in prostate cancer cells. We found that expression of EBP1 was down-regulated in clinical prostate cancer at both mRNA and protein levels. In addition, increased expression of EBP1 in hormone-refractory cells led to an amelioration of the androgen-independent phenotype. These studies suggest the therapeutic potential of EBP1 in hormone-refractory prostate cancer.
Materials and Methods
Cell Culture and Reagents
The LNCaP-C81 cell line (C81), a hormone-refractory variant of LNCaP cells, was a kind gift of Dr. Ming-Fong Lin (University of Nebraska; ref. 26). All other cell lines were obtained from the American Type Culture Collection and routinely cultured in RPMI 1640 supplemented with 10% fetal bovine serum. HRGh1 was obtained from R&D Systems.
Creation of Stably Transfected Cell Lines
Subconfluent C81 cells in 100 mm tissue culture dishes were transfected with 10 μg CMV-10 or CMV10-EBP1 expression plasmids using Fugene-6 according to the manufacturer's protocol. Cells were selected in G418 (800 μg/mL) for 5 weeks and resistant colonies were expanded. Both individual clones and mass transfectants were obtained.
To generate EBP1-silenced cell lines, LNCaP cells were seeded into 96-well plates and transduced with lentiviral particles (multiplicity of infection = 25) corresponding to different short hairpin RNA (shRNA) constructs targeted to the PA2G4 (EBP1) gene (Mission shRNA; Sigma) according to the manufacturer's protocol. Cell lines were selected in 2 μg/mL puromycin and surviving colonies of cells were expanded as mass cultures. Five individual shRNA lentiviral particle constructs were tested. Only one construct (13C) corresponding to nucleotides 302 to 322 of PA2G4 (Genbank NM_006191.1) inhibited EBP1 expression. Another construct, 16A, did not inhibit EBP1 expression and served as a control.
Microarray Analysis and Bioinformatics
RNA was prepared using the Trizol reagent as described previously (22). Microarray processing and data analysis was done at Genome Explorations. U133A oligonucleotide arrays (Affymetrix) containing ∼33,000 full-length annotated genes together with additional probe sets designed to represent expressed sequence tags were used. Only genes with a minimum expression level of 500 were included in this analysis. Genes whose expression varied more than 2.9-fold with a P < 0.05 were considered to be significantly different between the two cell lines. Oncomine analysis was done by using Oncomine3.0.6
Real-timeQuantitativeReverse Transcription-PCR
Both cell culture samples and a commercial panel of cDNAs from normal prostate and prostate tumors (TissueScan; Origene) were examined. The method of Nakanishi et al. (27) was used as described previously. Real-time quantitative reverse transcription-PCR was done on the LightCycler (Roche) platform. The following forward and reverse primers were selected using Primer Express software and synthesized by the Core Laboratory of University of Maryland School of Medicine: EBP1 sense 5′-GCACGCCAATAGAAGG-3′ and antisense 5′-GTAAACGGCATGGCATC-3′, kallikrein-2 sense 5′-CATCCAGTCTCGGATTG-3′ and antisense 5′-CTCATATTGTAGAGCGGGT-3′, testis-specific protein, Y-encoded (TSPY) sense 5′-CAGGGCTTCTCATTCCACTC-3′ and antisense 5′-CCATCATATTCAACTCAACAACTGG-3′ (28), AGR2 sense 5′-ATTGGCAGAGCAGTTTCTCC-3′ and antisense 5′-GAGCTGTATCTGCAGGTTCGT-3′ (29), and β-actin sense 5′-GCTATCCAGGCTGTGCTATC-3′ and antisense 5′-TGTCACGCACGATTTCC-3′. A SYBR Green PCR kit was used as per the manufacturer's instructions (Applied Biosystems) and the analyses were done in duplicate or triplicate. Target mRNA values were normalized using β-actin mRNA as an internal control. The relative quantitation of gene expression was done using the comparative ΔΔCt (threshold method) using β-actin as an internal control.
Tissue Microarray and Immunohistochemical Analysis
An intermediate density tissue microarray of prostate cancer and benign prostate tissues was received from the Cooperative Prostate Cancer Tissue Resource (National Cancer Institute). For the benign group, 15 normal prostate tissues were taken from prostate cancer patients and 27 prostate tissues from normals were used. For the malignant group, 153 tumors from patients with both localized and metastatic prostate cancer (Gleason grades 4−10) were evaluated. The Vectastain Elite ABC kit purchased from Vector Laboratories was used for immunohistochemical analysis. Immunohistochemical analysis was done using an EBP1 antibody (Upstate 1:200; pH 7.4) as described previously (30). Immunohistochemical staining was assessed independently by two observers, and a consensus of grading was reached. Immunostaining was evaluated manually and intensity was scored on the following scale: 0 = negative, 1 = weak, 2 = moderate, and 3 = strong. The nonparametric Wilcoxon rank-sum test was conducted to determine significant differences between the benign prostate epithelium and the malignant tissue.
Measurement of PSA Levels
Serum PSA levels were determined using a PSA ELISA kit from DSL as described previously (25).
Western Blot Analysis
Western blot analysis was done as described previously (31). The EBP1 antibody was from Upstate, the polyclonal antibody to actin was from Sigma, and the AKT and phosphorylated AKT antibodies were from Cell Signaling. The AGR2 antibody was described previously (32).
Cell Growth Assays
Cell growth measurement in complete medium was done as described using a hemocytometer (33). For soft-agar growth assays, increasing concentrations of cells (as indicated) were plated in 35 mm Petri dishes in 0.3% agar in complete medium and colonies were counted after 10 days of incubation (33). For studies assessing the effect of DHT, HRG, or drugs on cell growth, cells (5 × 103) were plated in 96-well plates in complete medium. After a 24-h attachment period, the medium was replaced with steroid-free medium [phenol red-free RPMI 1640 and 5% charcoal-stripped serum (Sigma)]. After 48 h of steroid depletion, cells were re-fed with fresh steroid-reduced medium with or without the indicated concentrations of DHT, bicalutamide (Casodex; 10 μmol/L), HRG, or the chemotherapeutic drugs at the concentrations indicated. Relative cell growth was determined using a Promega Proliferation Reagent (Promega) as per manufacturer's instructions with absorbance being read at 490 nm using a Dynexplate reader.
Luciferase Reporter Assays
Cells (5 × 104) were plated in 12-well plates in complete medium. When cells reached 50% to 60% confluence, they were transfected using the Fugene-6 reagent (Roche) according to the manufacturer's instructions. Cells were transfected with 0.5 μg MMTV-luciferase reporter plasmid and 5 ng TK-Renilla plasmid (Promega) as an internal control. Complete medium was replaced 24 h after transfection with phenol red-free RPMI 1640 with charcoal-stripped serum with or without R1881 (10−8 mol/L). Luciferase activity was determined using the Promega dual-luciferase assay kit as described by the manufacturer.
In vivo Studies in Athymic Mice
Female nude athymic NCr-nu/nu mice, ages 4 to 6 weeks, were purchased from the National Cancer Institute. Animals were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum. Cells were suspended in Matrigel (10 mg/mL; Collaborative Research) at 1.5 × 107/mL. Each mouse received s.c. injections at one site on each flank with 300 μL cell suspension. Tumors were measured with calipers. Tumor volumes were calculated by the formula: width2 × length / 2. Mean ± SD tumor volume was calculated and plotted against time. The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Maryland.
Statistical Analysis
Results of growth assays were analyzed using a two-sided Student's t test. Significance was established at P < 0.05. The proportion of developed tumors in control and EBP1 transfectants was compared using Fisher's exact test. All hypothesis tests were two-sided. The different groups were compared at the 0.05 level of significance.
Results
Reduced Expression of the EBP1 Gene in Prostate Cancer Patients
As we have found reduced expression of EBP1 in preclinical models of prostate cancer (25), we first screened a commercially prepared cDNA panel consisting of non-paired benign prostate and prostate tumor samples using quantitative reverse transcription-PCR. EBP1 expression was significantly decreased (P < 0.03) in prostate cancers compared with normal tissue (Fig. 1A). We then determined the expression of the EBP1 (PA2G4) gene in patients using public gene expression data bases. Oncomine analysis of a data set posted by Yu et al. (34) revealed that PA2G4 was significant decreased in localized prostate tumors (n = 64) relative to normal prostate samples (n = 23; P < 0.00001) and further down-regulated in metastases (n = 25; P < 0.00001; Fig. 1B). The studies of Vanaja et al. (35), Singh et al. (36), and LaTulippe et al. (37) also indicate that EBP1 expression is similarly decreased (P < 0.05, P < 0.1, and P < 0.01, respectively; data not shown). To examine the expression of EBP1 protein in human prostate tumor tissues, immunohistochemical analysis was done on a tissue array containing benign (n = 42) and prostate cancer (n = 153) specimens. EBP1 staining intensity was decreased in the cytoplasm of the prostate tumor samples compared with the benign samples (Fig. 1C, 2.73 versus 2.11, P < 0.002). EBP1 staining was predominantly detected in the cytoplasm of both benign epithelial and prostate tumor cells (Fig. 1D). Nuclear and cytoplasmic staining was observed in 30% of prostate cancer specimens (Fig. 1D, middle) but not in benign tissue. This resembles the nuclear and cytoplasmic distribution of EBP1 observed in cell culture (refs. 31, 38, 39; Fig. 1D).
Figure 1.
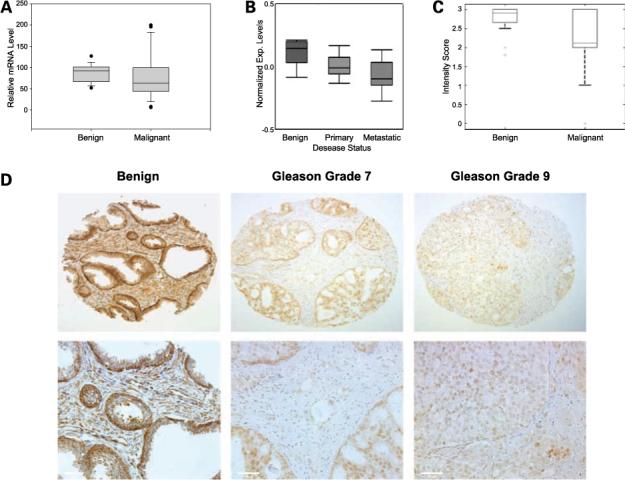
EBP1 expression is decreased with prostate cancer progression. A, quantitative reverse transcription-PCR was done for EBP1 mRNA expression on a panel of benign (7 normal and 11 benign prostatic hyperplasia), prostate cancer stage I and II (21), and prostate cancer stage III and IV (9) tissues obtained from Origene. The relative levels of all test mRNAs were normalized to β-actin. B, a public data set was obtained by Yu et al. and processed by Oncomine 3.0 for EBP1 expression. The average gene expression level at three different stages (benign n = 23, primary n = 64, and metastatic n = 25) of the disease is presented. C, box plots of immunohistochemical staining intensity of normal prostate tissue (n = 42) or tissue from patients with prostate cancer (n = 153). D, representative staining of (left) normal prostate epithelium, (middle) grade 7 Gleason showing nuclear staining of prostate cancer cells, and (right) Gleason grade 9 at both low (top) and high (bottom) magnification.
Restoration of EBP1 Expression in Hormone-Refractory C81 Cells Ameliorates the Hormone-Refractory Phenotype
The C81 LNCaP subline has been made androgen independent by continuous long-term passage in complete medium (26). To analyze whether the increased expression of EBP1 could ameliorate the hormone-refractory pheno-type, we established FLAG-EBP1 or empty vector stably transfected C81 cell lines. EBP1 expression was increased approximately 3-fold (Fig. 2A). We then examined EBP1 transfectants based on established biological criteria for androgen independence: growth rate, response to hormones, and chemotherapy. The growth rate of the C81-EBP1 transfectants in complete medium was significantly decreased (P < 0.05) compared with that of the vector control (Fig. 2B, left) and resembled that of low-passage LNCaP cells. Ectopic expression of EBP1 in the C81 cell line also decreased colony growth in soft agar approximately 50% at the highest cell concentration tested (Fig. 2B, right) and was similar to that of low-passage LNCaP cells.
Figure 2.
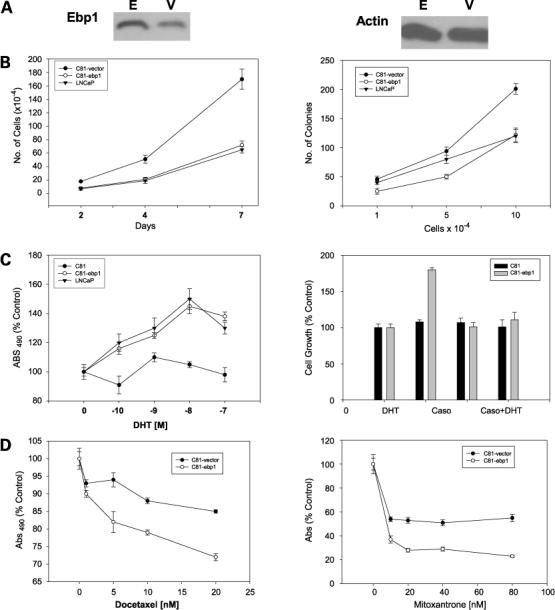
EBP1 restoration ameliorates the hormone-refractory phenotype. A, Western blot analysis of C81 cells transfected with a vector control (V) or EBP1 (E) expression plasmid. B, left, growth in monolayer culture. Equal numbers (5 × 104) of vector or EBP1 stably transfected cells were plated at day 0 in complete medium and viable cell numbers were determined at the indicated times. Points, mean of three wells; bars, SE. Representative of three experiments (right). Colony growth in soft agar. C81cells stably transfected with either pcDNA or EBP1 were plated in soft agar at the cell densities indicated and colony numbers were assessed at day 10. Point, mean of three dishes; bars, SE. Representative of three experiments. C, left, response of EBP1- and vector-transfected C81 cells to DHT. EBP1 and vector control C81-transfected cells and early-passage LNCaP cells were plated in complete medium at 5 × 103 per well in 96-well plates for 1 d. The next day, cells were switched to steroid-reduced medium for 2 d as described in Materials and Methods. DHT was then added at the indicated concentrations. Cells were re-fed medium every 3 d. Total cell numbers were determined 7 d later by MTT assays. Points, mean of six wells; bars, SE. Representative of two experiments. Right, effect of bicalutamide on DHT-stimulated growth. Cells were plated as described in Materials and Methods. After 2 d in steroid-reduced medium, cells were fed with fresh steroid-reduced medium with or without 10 μmol/L bicalutamide (Casodex) in the presence or absence of 10 nmol/L DHT. Total cell numbers were assessed 7 d later by MTT assay. Cells were re-fed medium every 3 d. Points, mean of six wells; bars, SE. Similar results were found in two independent experiments. D, EBP1 expression sensitizes C81 cells to chemotherapeutic drugs. C81 vector control or EBP1 transfectants were plated in 96-well plates in complete medium and allowed to attach overnight. The cells were then placed in steroid-reduced medium for 2 d and then re-fed with steroid-reduced medium in the presence of the indicated concentrations of docetaxel (left) or mitoxantrone (right). Total cell numbers were determined by MTT assays 5 d later. Points, mean of six wells; bars, SE. Representative of two experiments.
Androgen-independent cells in culture can be either refractory to hormone stimulation or sensitized to low doses of hormone. In comparison with the parental LNCaP cells, C81 cells are no longer growth stimulated by DHT (26). We found that the growth of EBP1 transfectants was reduced to one-third of that of the vector controls in the absence of androgens (data not shown). The vector control C81 cells were unresponsive to androgen (Fig. 2C, left)as reported previously (26). In contrast, EBP1-transfected C81 cells responded to androgen in a similar manner as the original low-passage LNCaP androgen-sensitive cells (Fig. 2C, left; ref. 26). To determine if the androgen-stimulated growth of the EBP1-transfected cells was mediated via the AR, we examined the effect of the antiandrogen bicalutamide (Casodex) on DHT-stimulated growth. The DHT-stimulated growth of the EBP1-transfected cells was suppressed by 10 μmol/L bicalutamide (Fig. 2C, right), suggesting that the increased growth in the presence of androgens was mediated via the AR.
Mitoxantrone or docetaxel is currently the standard palliative treatment in hormone-refractory prostate cancer patients (40). C81 cells were relatively refractory to docetaxel in androgen-deprived conditions as reported previously (41), but EBP1-transfected cells were significantly more sensitive to 5 to 20 nmol/L docetaxel under these same conditions (Fig. 2D, left). The sensitivity of both vector control and EBP1 transfectants to mitoxantrone plateaued at 20 nmol/L. However, growth of EBP1 transfectants was decreased approximately 80% as opposed to 40% for the vector controls at this concentration (Fig. 2D, right). In contrast, the sensitivity of the EBP1 transfectants to both drugs was the same as vector controls when cells were grown in complete medium (data not shown).
Restoration of EBP1 Protein Levels Significantly Reduces Expression of Molecules Associated with the Androgen-Refractory Phenotype
To elucidate how restoration of EBP1 gene expression in C81 cells mitigates the hormone-refractory phenotype, we examined the expression levels of proteins that play a key role in androgen independence. Western blot analysis of two individual C81 clones stably transfected with a FLAG-tagged EBP1 or vector controls (Fig. 3A) showed that ectopic expression of EBP1 markedly reduced the protein levels of AR, consistent with our previous report for LNCaP cells (25), and BCL-2, another molecule associated with the androgen-independent phenotype. In contrast, the level of the proapoptotic protein BAD remained the same as the vector controls.
Figure 3.
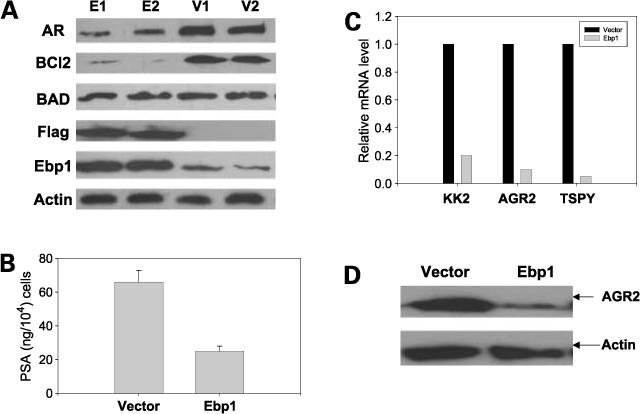
Expression of genes associated with androgen independence is decreased in C81 EBP1 transfectants. A, lysates of logarithmically growing clones of C81 EBP1 transfectants (E1 and E2) or vector controls (V1 and V2) were resolved by SDS-PAGE and immunoblotted for AR, BCL-2, BAD, FLAG, EBP1, and actin as indicated. Representative of three experiments. B, secreted PSA as measured by ELISA. EBP1 and vector control cells were plated at 1 × 10 6 per 100 mm dish in complete medium. Forty-eight hours later, conditioned medium was collected and frozen. PSA activity was assessed by ELISA. Levels of PSA were adjusted to total cell number. Representative of two experiments. C, validation of mRNA levels in EBP1 transfectants. Quantitative reverse transcription-PCR was done for the kallikrein-2, AGR2, and TSPY genes in EBP1 and vector transfectants. The relative levels of all test mRNAs were normalized to β-actin. Representative of three experiments using different sets of cells. D, expression of AGR2 protein in vector and EBP1 C81 transfectants. Lysates of vector control or EBP1-transfected cells grown in complete medium were resolved by SDS-PAGE and analyzed by Western blotting with antibody to AGR2.
We next conducted a microarray analysis of mass-transfected EBP1 and vector-transfected C81 cell lines to determine the spectrum of differentially expressed genes contributing to the hormone-refractory phenotype (Supplementary Tables 1 and 2).7 Six AR target genes potentially involved in androgen-independent growth were found to be down-regulated at least 3-fold in EBP1-overexpressing cells compared with controls (Table 1). These include PSA (kallikrein 3), kallikrein 2, TMPRSS2, and prostate differentiation factor as reported previously for LNCaP cells (25). In addition, the expression of the AR-regulated gene AGR2 (32) and the candidate oncogene TSPY (28) was significantly down-regulated in the EBP1 transfectants.
Table 1.
Androgen-regulated genes decreased in EBP1-transfected C81 cells
| Accession no. | Fold change | Gene name | |
|---|---|---|---|
| NM_003308 | TSPY | 90 | 0.002 |
| AF088867 | AGR2 | 50 | 0.002 |
| NM_003527 | POV-1 | 4.8 | 0.001 |
| U17040 | PSA | 30 | 0.001 |
| BC005196 | Prostatic kallikrein 2 | 2.9 | 0.004 |
| AF003934 | Prostate differentiation factor | 7.0 | 0.0002 |
| AF2700487 | TMPRSS2 | 4.0 | 0.01 |
NOTE: A microarray analysis of EBP1-transfected and vector control cells was done as described in Materials and Methods. The expression of 60 genes (minimum of 2.9-fold change) was found to differ significantly between the two cell lines. Nine genes were induced and 51 were repressed. AR signaling-associated genes are presented.
We next validated the expression of four representative genes associated with prostate cancer progression. Secreted PSA protein levels were decreased 60% in EBP1 transfectants compared with C81 vector controls (Fig. 3B). Expression of the kallikrein 2 gene, a potential marker for prostate cancer progression (42), was down-regulated over 80% in EBP1 transfectants compared with vector controls (Fig. 3C). TSPY mRNA was also down-regulated over 90%. AGR2, the human homologue of the Xenopus anterior gradient 2 gene, is regulated by AR and its overexpression is associated with enhanced metastasis (43). Real-time reverse transcription-PCR indicated that AGR2 mRNA was down-regulated over 90% in EBP1 transfectants compared with vector controls (Fig. 3C). Western blot analysis detected decreased AGR2 protein in C81-EBP1 transfectants (Fig. 3D).
Restoration of EBP1 Expression Blocked HRG-Activated Phosphatidylinositol 3-Kinase/AKT Signaling
HRG-activated pathways contribute to ligand-independent activation of AR (44). We therefore determined if EBP1 could affect the responsiveness of C81 androgen-independent cells to HRG. Growth of EPB1 transfectants in androgen-depleted medium was reduced by two-thirds compared with vector-transfected controls (Fig. 4A). HRG induced the growth of vector control C81 cells in androgen-free medium as expected (19). However, the ectopic expression of EBP1 completely abrogated the ability of C81 cells to grow in response to HRG (Fig. 4A). In contrast, the response of C81 EBP1 transfectants to epidermal growth factor was unchanged (Supplementary Fig. S1).7
Figure 4.
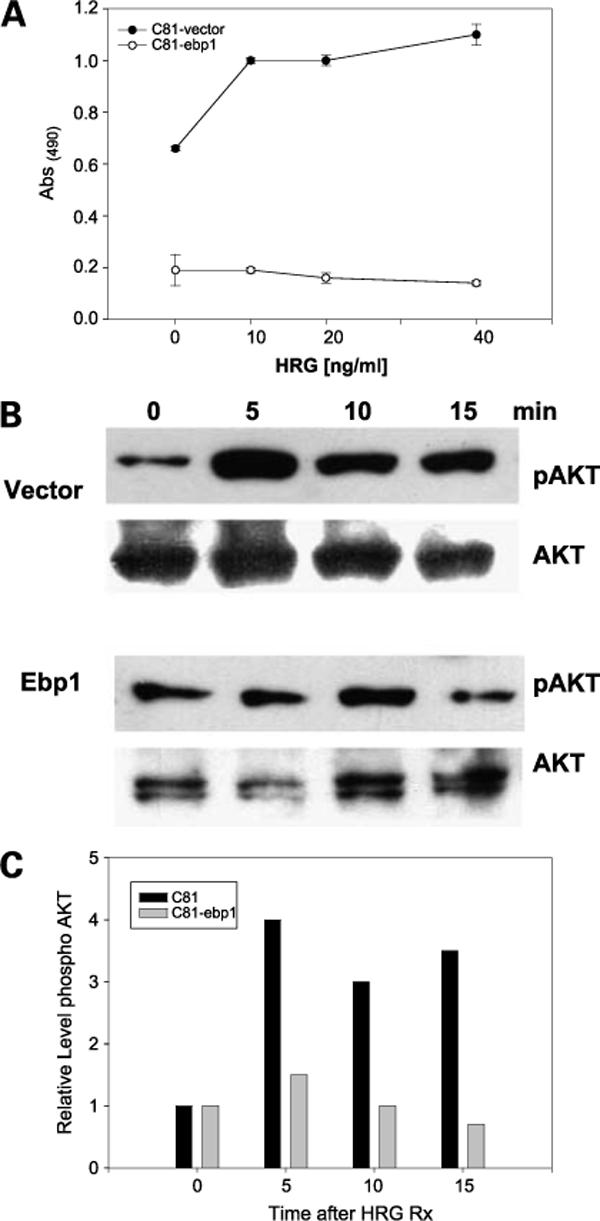
EBP1 overexpression inhibits the response to HRG. A, EBP1 and vector control transfected cells were plated in complete medium at 5 × 103 per well for 1 d. The next day, cells were switched to steroid-reduced medium and HRG was added at the indicated concentrations. Total cell numbers were determined 7 d later by MTT assay. Points, mean of six wells; bars, SE. Representative of three experiments. B, AKT activation in response to HRG C81-EBP1 or vector control cells were serum starved overnight and then treated with HRG (20 ng/mL) for the indicated times (min). Cell lysates were immunoblotted with antibody to phosphorylated AKT or total AKT as indicated. C, densitometric evaluation of AKT phosphorylation. Representative of two independent experiments.
As restoration of EBP1 expression inhibited HRG-induced growth, we examined if EBP1 could attenuate HRG signaling. We examined the AKT pathway as HRG-induced AKT phosphorylation and activation is important in growth of androgen-independent cells (45) and inhibition of AKT phosphorylation delays the transition to androgen independence (46). AKT was basally phosphorylated in C81 vector- and EBP1-transfected cells as expected (Fig. 4B) presumably due to loss of PTEN expression (47). Densitometry indicated that HRG induced a 4-fold increase in AKT phosphorylation at 5 min that gradually declined in vector control cells (Fig. 4C). In contrast, HRG failed to induce an increase in AKT phosphorylation in EBP1 transfectants.
Abrogation of EBP1 Protein Expression by shRNA in LNCaP Cells Results in a Hormone-Refractory Phenotype
To further test the hypothesis that EBP1 is important in the reversal of the hormone-refractory phenotype, we created LNCaP cells in which EBP1 expression has been ablated by transduction with a lentivirus targeting the EBP1 gene. EBP1 expression was knocked down in the C13 but not in the A16 cell line (Fig. 5A). HRG inhibited the growth of A16 cells as it does for LNCaP cells (17). In contrast, HRG stimulated the growth of C13 LNCaP cells in which EBP1 expression has been ablated, similar to its effects on androgen-independent cell lines (Fig. 5B, left). To examine the potential role of phosphatidylinositol 3-kinase/AKT signaling pathway in this phenomenon, we treated EBP1 knockdown cells with HRG and measured phosphorylated AKT. AKT was basally phosphorylated in both LNCaP and EBP1 knockout cells. However, HRG induced a 2-fold increase in pAKT protein levels in LNCaP cells and a 3.5-fold increase in pAKT levels in EBP1-depleted LNCaP cells as determined by densitometry (Fig. 5B, right).
Figure 5.
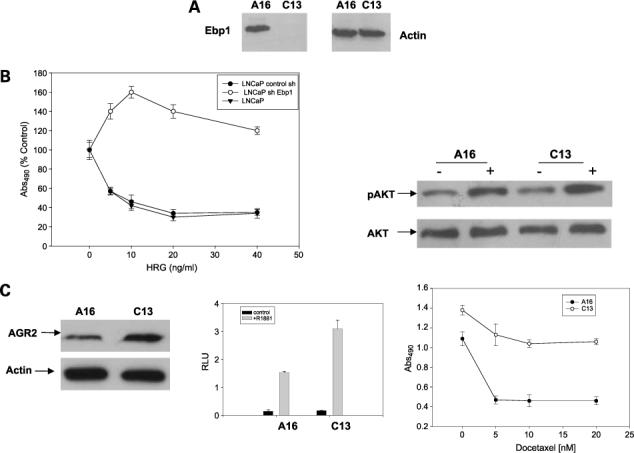
Suppression of EBP1 by shRNA promotes a hormone-refractory phenotype. A, Western blot analysis of EBP1. Lysates of LNCaP cells stably transduced with a control lentivirus (A16) or an EBP1-targeted shRNA (13C) were collected and resolved by SDS-PAGE. Western blots were probed for endogenous EBP1 or actin as indicated. B, inhibition of EBP1 expression abrogates the ability of HRG to arrest growth. Left, control and shRNA EBP1-targeted cells were plated in complete medium at 5 × 103 per well for 1 d. The next day, cells were switched to steroid-reduced medium and HRG was added at the indicated concentrations. Total cell numbers were determined 7 d later by MTT assay. Points, mean of six wells; bars, SE. Representative of three experiments. Right, AKT phosphorylation in EBP1 knockout cells. Control (A16) or EBP1 knockout cells (C13) were treated with HRG (20 ng/mL) for 15 min and cell lysates were assessed for the presence of phosphorylated and total AKT as indicated. C, left, AGR2 is overexpressed in C13 LNCaP cells that do not express EBP1. Lysates of logarithmically growing A16 and C13 cells were resolved by SDS-PAGE and analyzed by Western blotting using antibodies to AGR2 and actin as indicated. Middle, elimination of EBP1 expression enhances the activity of an AR-regulated promoter. A16 or C13 cells were transfected with a MMTV-luciferase reporter plasmid. Twenty-four hours after transfection, cells were switched to phenol red-free RPMI 1640 with 1% charcoal-stripped serum containing R1881 (10−8 mol/L) or vehicle control. Sixteen hours later, luciferase activity was measured. Points, mean of triplicate wells; bars, SE. Representative of three experiments. Right, elimination of EBP1 expression affects the response to docetaxel. C13 or A16 cells were plated in 96-well plates in complete medium and allowed to attach overnight. The cells were then placed in steroid-reduced medium for 2 d and then re-fed with steroid-reduced medium in the presence of the indicated concentrations of docetaxel. The viability of the cells was determined by MTT assays 5 d later.
We further showed that the expression of two genes associated with hormone-refractory growth was altered in EBP1 knockout cells. Levels of secreted PSA in conditioned medium of log-phase cells were 12.5 ng/104 cells for A16 cells and 75 ng/104 cells for the shEBP1-transduced C13 cells. Abrogation of the expression of the EBP1 gene also led to an up-regulation of AGR2 (Fig. 5C, left). We next tested the transcriptional response of an AR-regulated MMTV promoter to R1881. The response was significantly (P < 0.05) enhanced in EBP1 knockout cells (Fig. 5C, middle). Finally, we examined the response of A16 and C13 cells to docetaxel. The A16 control cells were sensitive to docetaxel with a 65% reduction in growth observed at 5 nmol/L of the drug, In contrast, C13 cells were relatively resistant to docetaxel with a maximum of 15% growth inhibition at 20 nmol/L (Fig. 5C, right).
Manipulation of EBP1 Expression Affects Growth in Animal Models
To corroborate if the effects observed in vitro are observed in vivo, we examined the consequence of expression of EBP1 on the tumorigenicity of C81 cells in female nude mice. EBP1- and vector-transfected cells were injected s.c. into female nude mice and tumor growth was monitored in two independent experiments (Fig. 6). The first measurable tumor derived from C81 vector transfectants was detected on day 8. In contrast, tumor development of EBP1 transfectants was much slower, with the first tumor being observed on day 20. On day 28 post-transplantation, tumors were observed at only 1 of 26 of the EBP1 inoculation sites as opposed to 24 of 26 of the sites for vector controls. At the end of both studies, tumors had developed at 25 of 26 of the sites injected with the vector control cells, whereas 10 of 26 of the sites inoculated with EBP1 transfectants developed tumors (Fig. 6A; P = 0.00001, Fisher's exact test, two-sided). Average tumors volumes for the EBP1 transfectants observed at the end of the first study (3 animals per group, 6 sites per group) were 104 ± 70 mm3 compared with 998 ± 206 mm3 (mean ± SE) for the vector controls (P = 0.01). In the second experiment (10 animals per group, 20 sites per group), the average tumor volume was 206 ± 54 for the EBP1 transfectants and 561 ± 47 for the vector controls (P = 0.0001; Fig. 6B).
Figure 6.
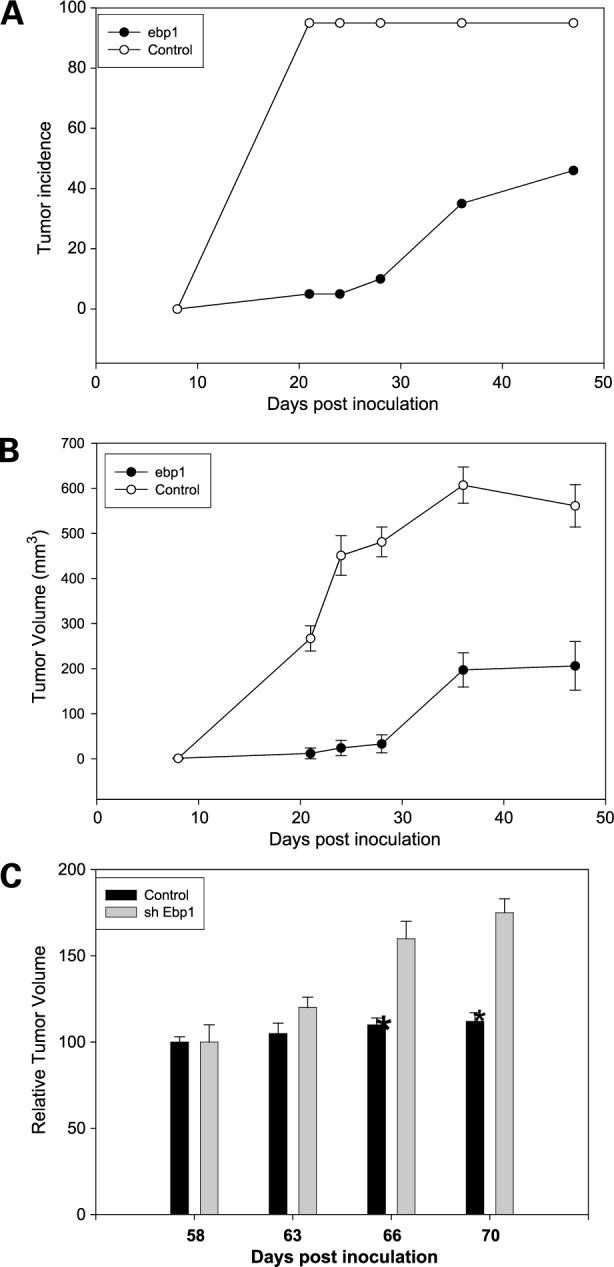
Manipulation of EBP1 expression affects growth of prostate cancer cells in vivo. A and B, ectopic expression of EBP1 reduces the growth of androgen-independent cells in athymic female mice. C81 vector-or EBP1-transfected cells were injected s.c. into female athymic mice. A, tumor development with time (n = 10 animals, 20 sites per group). B, tumor volumes and growth rate. Tumor volumes were calculated as described in Materials and Methods. Points, mean (10 mice per group injected at 20 sites); bars, SE. C, elimination of EBP1 expression enhances growth of androgen-dependent LNCaP cells in vivo. A16 lentivirus control or C13 EBP1 shRNA-transduced LNCaP cells were injected s.c. into female athymic mice. Tumor volumes were calculated as described in Materials and Methods. Points, mean (10 mice per group injected at 20 sites); bars, SE. *, P = 0.04.
We also examined the ability of A16 and C13 cells (in which EBP1 expression had been decreased) to form tumors in female nude mice. At the concentrations used, we found that parental low-passage LNCaP cells formed tumors in the same manner as the A16 cells, with measurable growth being observed at all sites at day 50 (data not shown). However, growth of LNCaP controls and A16 cells leveled off at day 58. In contrast, C13 cells continued to grow. At the termination of the experiment at day 68 post-inoculation, the average tumor volume was 307 ± 64 mm3 for the A16 controls and 542 ± 67 mm3 for the C13 EBP1 knockout cells (P = 0.04; Fig. 6C). Wet weights of the tumors were also significantly different: A16 = 0.150 ± 0.03 versus C13 = 0.288 ± 0.05 g (P = 0.03).
Discussion
Emerging studies indicate the importance of ErbB receptor signaling in hormone-refractory prostate cancer (44). EBP1, an ErbB3-binding protein, also functions as an AR corepressor, suggesting its role in suppression of ErbB-mediated activation of AR. In these studies, we found that restoration of EBP1 expression ameliorated the hormone-refractory phenotype of C81 cells and that elimination of EBP1 expression resulted in a hormone-refractory pheno-type in hormone-dependent LNCaP cells. These studies suggest the therapeutic potential of EBP1, an endogenous AR corepressor, for treatment of advanced prostate cancer.
Restoration of EBP1 gene expression in hormone-refractory C81 cells led to a reversal of the androgen-independent phenotype based on a series of established criteria. These biological changes were likely due in part to the decreased expression of proteins contributing to the androgen-independent phenotype. AR protein levels were decreased in C81-EBP1 transfectants, consistent with our previous finding obtained from low-passage hormone-dependent LNCaP cells (25). The decreased expression of these AR target genes may have restored the physiologic response of C81 cells to DHT as we observed and resulted in the growth inhibition induced by Casodex. The increased dependence of C81 EBP1-transfected cells on androgens was also evidenced by the fact that EBP1-transfected cells grew poorly in female mice.
Expression of the androgen-regulated genes AGR2 and TSPY was decreased in C81-EBP1 transfectants. Further, inhibition of EBP1 protein expression in LNCaP cells led to up-regulation of AGR2. AGR2, the human homologue of the Xenopus anterior gradient 2 gene, is normally expressed in LNCaP cells only in response to androgen treatment and overexpressed in primary prostate adenocarcinoma (32). Elevated AGR2 expression is significantly associated with poor survival of prostate cancer patients (48). As there is no effective therapy for metastatic prostate cancer, the possible role of EBP1 as a modulator of AGR2 gene expression warrants further investigation.
Mitoxantrone or docetaxel is currently the standard palliative treatment in hormone-refractory prostate cancer patients. BCL-2, which is overexpressed in human refractory prostate cancers (49), is a potent inhibitor of apoptosis induced by these drugs. Unexpectedly, the protein level of BCL-2 was decreased in EBP1-transfected C81 cells. The mechanism of the inhibition of BCL-2 expression in C81-EBP1 transfectants is not known. Our microarray data showed no changes in BCL-2 mRNA levels in EBP1 transfectants compared with vector controls. EBP1 can directly bind BCL-2 mRNA, suggesting that EBP1 may have effects on translation efficiency (50). Alternatively, EBP1 may alter the stability of the BCL-2 protein. We suggest that the increase in sensitivity to docetaxel and mitoxantrone observed in EBP1-transfected cells in the absence of androgens was due to the down-regulation of BCL-2. It is of interest that changes in sensitivity were only observed in androgen-deprived conditions. Studies of drug sensitivity of LNCaP C33 and C81 variants were similarly conducted in steroid-free medium (41). It is possible that activation of alternative pathways, such as ErbB signaling, enhances cell survival after drug treatment in the androgen-deprived environment and EBP1, by interfering with such pathways, inhibits growth.
A growing literature indicates that ErbB signaling in androgen-deprived conditions is vital to androgen-refractory growth and that hormone-naive and hormone-refractory cells respond differently to such signaling. The ErbB2/ErbB3 ligand HRG augments the growth of CWR-R1 hormone-refractory cells in androgen-deprived conditions (19) but inhibits growth of hormone-dependent cells (17). It has been postulated that changes in the profiles of ErbB receptors and downstream signaling pathways account for such differences (19). We found that ectopic expression of EBP1, a member of the ErbB3 signal transduction pathway, completely abrogated the ability of HRG to induce growth of C81 androgen-independent cells in androgen-deprived medium. Knockdown of EBP1 expression resulted in HRG-induced growth stimulation, rather than growth inhibition, in hormone-dependent LNCaP cells. We suggest that EBP1 may be one member of an ErbB signal transduction pathway that is important in regulating the response of prostate epithelial cells to HRG. Thus, HRG-induced signals that would ordinarily inhibit growth in hormone-dependent cells because of EBP1 or similar proteins may increase growth when these proteins are nonfunctional. Of interest, the inhibition of HRG-induced growth appeared to be specific, as the response to epidermal growth factor was not affected.
Although androgen-independent survival of prostate cancer cells through the AKT pathway has been shown (45), the role that the inhibition of AKT activation observed may have played in the EBP1-induced growth suppression in response to HRG is not yet clear. We initially examined the AKT pathway due to its role as a mediator of HRG-induced cell survival. In addition, both phosphatidylinositol 3-kinase, an activator of AKT, and EBP1 bind the cytoplasmic domain of ErbB3. This raised the possibility that EBP1 might affect recruitment of phosphatidylinositol 3-kinase to ErbB3 after HRG stimulation. Although AKT signaling was suppressed in EBP1 transfectants, changes in other downstream signaling pathways may also have contributed to EBP1-induced growth inhibition. For example, ongoing studies in our laboratory indicate that EBP1 expression affects mitogen-activated protein kinase signaling in a complex pattern that is dependent on cell type, ligand concentration, and duration of exposure. Thus, at this time, there is uncertainty regarding the importance of AKT modulation versus other downstream pathways. In addition, we do not know if the biological effects of EBP1 are due to its direct effects on AR, modification of the effects of ErbB on AR, or modulation of ErbB effects that are unrelated to AR signaling.
In summary, we show that overexpression of EBP1, an AR corepressor that interacts with ErbB3, modulated the hormone-refractory phenotype both in vitro and in vivo. These studies support the role of EBP1 as an endogenous negative regulator of AR signaling and provide a rationale for the design of EBP1-based approaches for the treatment of hormone-refractory prostate cancer.
Supplementary Material
Acknowledgments
We thank Dr. Lin for permission to use the C81 cells, Dr. Yun Qiu for providing these cells, and Drs. Joseph Fondell (University of Medicine and Dentistry of New Jersey) and Xin-Wei Wang (National Cancer Institute) for a careful reading of the article.
Grant support: NIH grants R01 CA76047 and R21 088882-01, Department of Pathology (A.W. Hamburger), and Maryland Technology Corporation (June 6, 2006) and Department of Defense grant W81XWH-07-0267 (Y. Zhang).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material for this article is available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–90. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 2.Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89:552–6. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–8. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–27. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 6.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280–5. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 7.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci U S A. 1999;96:5458–63. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Majumder S, McCall W, et al. Inhibition of HER-2/neu kinase impairs androgen receptor recruitment to the androgen responsive enhancer. Cancer Res. 2005;65:3404–9. doi: 10.1158/0008-5472.CAN-04-4292. [DOI] [PubMed] [Google Scholar]
- 9.Murillo H, Schmidt LJ, Tindall DJ. Tyrphostin AG825 triggers p38 mitogen-activated protein kinase-dependent apoptosis in androgen-independent prostate cancer cells C4 and C4-2. Cancer Res. 2001;61:7408–12. [PubMed] [Google Scholar]
- 10.Leung HY, Weston J, Gullick WJ, Williams G. A potential autocrine loop between heregulin-α and erbB-3 receptor in human prostatic adenocarcinoma. Br J Urol. 1997;79:212–6. doi: 10.1046/j.1464-410x.1997.30412.x. [DOI] [PubMed] [Google Scholar]
- 11.Vakar-Lopez F, Cheng CJ, Kim J, et al. Up-regulation of MDA-BF-1, a secreted isoform of ErbB3, in metastatic prostate cancer cells and activated osteoblasts in bone marrow. J Pathol. 2004;203:688–95. doi: 10.1002/path.1568. [DOI] [PubMed] [Google Scholar]
- 12.Koumakpayi IH, Diallo JS, Le PC, et al. Expression and nuclear localization of ErbB3 in prostate cancer. Clin Cancer Res. 2006;12:2730–7. doi: 10.1158/1078-0432.CCR-05-2242. [DOI] [PubMed] [Google Scholar]
- 13.Cheng CJ, Ye XC, Vakar-Lopez F, et al. Bone microenvironment and androgen status modulate subcellular localization of ErbB3 in prostate cancer cells. Mol Cancer Res. 2007;5:675–84. doi: 10.1158/1541-7786.MCR-06-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyne JC, Melhem MF, Finley GG, et al. Tissue expression of neu differentiation factor/heregulin and its receptor complex in prostate cancer and its biologic effects on prostate cancer cells in vitro. Cancer J Sci Am. 1997;3:21–30. [PubMed] [Google Scholar]
- 15.Grasso AW, Wen D, Miller CM, Rhim JS, Pretlow TG, Kung HJ. ErbB kinases and NDF signaling in human prostate cancer cells. Oncogene. 1997;15:2705–16. doi: 10.1038/sj.onc.1201447. [DOI] [PubMed] [Google Scholar]
- 16.Bacus SS, Yarden Y, Oren M, et al. Neu differentiation factor (heregulin) activates a p53-dependent pathway in cancer cells. Oncogene. 1996;12:2535–47. [PubMed] [Google Scholar]
- 17.Tal-Or P, Di Segni A, Lupowitz Z, Pinkas-Kramarski R. Neuregulin promotes autophagic cell death of prostate cancer cells. Prostate. 2003;55:147–57. doi: 10.1002/pros.10200. [DOI] [PubMed] [Google Scholar]
- 18.Mendoza N, Phillips GL, Silva J, Schwall R, Wickramasinghe D. Inhibition of ligand-mediated HER2 activation in androgen-independent prostate cancer. Cancer Res. 2002;62:5485–8. [PubMed] [Google Scholar]
- 19.Gregory CW, Whang YE, McCall W, et al. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005;11:1704–12. doi: 10.1158/1078-0432.CCR-04-1158. [DOI] [PubMed] [Google Scholar]
- 20.Radomski N, Jost E. Molecular cloning of a murine cDNA encoding a novel protein, p38-2G4, which varies with the cell cycle. Exp Cell Res. 1995;220:434–45. doi: 10.1006/excr.1995.1335. [DOI] [PubMed] [Google Scholar]
- 21.Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors [see comments]. Nature. 1997;387:733–6. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 22.Zhang YX, Fondell JD, Wang QB, et al. Repression of androgen receptor mediated transcription by the ErbB-3 binding protein, Ebp1. Oncogene. 2002;2:5609–18. doi: 10.1038/sj.onc.1205638. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Akinmade D, Hamburger AW. The ErbB3 binding protein Ebp1 interacts with Sin3A to repress E2F1 and AR-mediated transcription. Nucleic Acids Res. 2005;33:6024–33. doi: 10.1093/nar/gki903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Hamburger AW. Specificity and heregulin regulation of Ebp1 (ErbB3 binding protein 1) mediated repression of androgen receptor signalling. Br J Cancer. 2005;92:140–6. doi: 10.1038/sj.bjc.6602257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Wang XW, Jelovac D, et al. The ErbB3-binding protein Ebp1 suppresses androgen receptor-mediated gene transcription and tumori-genesis of prostate cancer cells. Proc Natl Acad Sci U S A. 2005;102:9890–5. doi: 10.1073/pnas.0503829102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Igawa T, Lin FF, Lee MS, Karan D, Batra SK, Lin MF. Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate. 2002;50:222–35. doi: 10.1002/pros.10054. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi T, Karp JE, Tan M, et al. Quantitative analysis of breast cancer resistance protein and cellular resistance to flavopiridol in acute leukemia patients. Clin Cancer Res. 2003;9:3320–8. [PubMed] [Google Scholar]
- 28.Krick R, Jakubiczka S, Arnemann J. Expression, alternative splicing and haplotype analysis of transcribed testis specific protein (TSPY) genes. Gene. 2003;302:11–9. doi: 10.1016/s0378-1119(02)01104-6. [DOI] [PubMed] [Google Scholar]
- 29.Fritzsche FR, Dahl E, Pahl S, et al. Prognostic relevance of AGR2 expression in breast cancer. Clin Cancer Res. 2006;12:1728–34. doi: 10.1158/1078-0432.CCR-05-2057. [DOI] [PubMed] [Google Scholar]
- 30.Santegoets SJ, Schreurs MW, Reurs AW, et al. Identification and characterization of ErbB-3-binding protein-1 as a target for immunotherapy. J Immunol. 2007;179:2005–12. doi: 10.4049/jimmunol.179.3.2005. [DOI] [PubMed] [Google Scholar]
- 31.Xia X, Lessor TJ, Zhang Y, Woodford N, Hamburger AW. Analysis of the expression pattern of Ebp1, an ErbB-3-binding protein. Biochem Biophys Res Commun. 2001;289:240–4. doi: 10.1006/bbrc.2001.5942. [DOI] [PubMed] [Google Scholar]
- 32.Zhang JS, Gong A, Cheville JC, Smith DI, Young CY. AGR2, an androgen-inducible secretory protein overexpressed in prostate cancer. Genes Chromosomes Cancer. 2005;43:249–59. doi: 10.1002/gcc.20188. [DOI] [PubMed] [Google Scholar]
- 33.Lessor TJ, Yoo JY, Xia X, Woodford N, Hamburger AW. Ectopic expression of the ErbB-3 binding protein ebp1 inhibits growth and induces differentiation of human breast cancer cell lines. J Cell Physiol. 2000;183:321–9. doi: 10.1002/(SICI)1097-4652(200006)183:3<321::AID-JCP4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 34.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 35.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–82. [PubMed] [Google Scholar]
- 36.Singh D, Febbo PG, Ross K, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 37.LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–506. [PubMed] [Google Scholar]
- 38.Squatrito M, Mancino M, Donzelli M, Areces LB, Draetta GF. EBP1 is a nucleolar growth-regulating protein that is part of pre-ribosomal ribonucleoprotein complexes. Oncogene. 2004;23:4454–65. doi: 10.1038/sj.onc.1207579. [DOI] [PubMed] [Google Scholar]
- 39.Ahn JY, Liu X, Liu Z, et al. Nuclear Akt associates with PKC-phosphorylated Ebp1, preventing DNA fragmentation by inhibition of caspase-activated DNase. EMBO J. 2006;25:2083–95. doi: 10.1038/sj.emboj.7601111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tannock IF, Berry WR, Horti J, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 41.Zelivianski S, Spellman M, Kellerman M, et al. ERK inhibitor PD98059 enhances docetaxel-induced apoptosis of androgen-independent human prostate cancer cells. Int J Cancer. 2003;107:478–85. doi: 10.1002/ijc.11413. [DOI] [PubMed] [Google Scholar]
- 42.Partin AW, Catalona WJ, Finlay JA, et al. Use of human glandular kallikrein 2 for the detection of prostate cancer: preliminary analysis. Urology. 1999;54:839–45. doi: 10.1016/s0090-4295(99)00270-8. [DOI] [PubMed] [Google Scholar]
- 43.Innes HE, Liu D, Barraclough R, et al. Significance of the metastasis-inducing protein AGR2 for outcome in hormonally treated breast cancer patients. Br J Cancer. 2006;94:1057–65. doi: 10.1038/sj.bjc.6603065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 45.Wen Y, Hu MC, Makino K, et al. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000;60:6841–5. [PubMed] [Google Scholar]
- 46.Miyamoto H, Altuwaijri S, Cai Y, Messing EM, Chang C. Inhibition of the Akt, cyclooxygenase-2, and matrix metalloproteinase-9 pathways in combination with androgen deprivation therapy: potential therapeutic approaches for prostate cancer. Mol Carcinog. 2005;44:1–10. doi: 10.1002/mc.20121. [DOI] [PubMed] [Google Scholar]
- 47.Hermans KG, van Alewijk DC, Veltman JA, van Weerden W, van Kessel AG, Trapman J. Loss of a small region around the PTEN locus is a major chromosome 10 alteration in prostate cancer xenografts and cell lines. Genes Chromosomes Cancer. 2004;39:171–84. doi: 10.1002/gcc.10311. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y, Forootan SS, Liu DS, et al. Increased expression of anterior gradient-2 is significantly associated with poor survival of prostate cancer patients. Prostate Cancer Prostatic Dis. 2007;10:293–300. doi: 10.1038/sj.pcan.4500960. [DOI] [PubMed] [Google Scholar]
- 49.Colombel M, Symmans F, Gil S, et al. Detection of the apoptosis-suppressing oncoprotein bc1−2 in hormone-refractory human prostate cancers. Am J Pathol. 1993;143:390–400. [PMC free article] [PubMed] [Google Scholar]
- 50.Bose SK, Sengupta TK, Bandyopadhyay S, Spicer EK. Identification of Ebp1 as a component of cytoplasmic bcl-2 mRNP (messenger ribonucleo-protein particle) complexes. Biochem J. 2006;396:99–107. doi: 10.1042/BJ20051548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.