

In the United States, the years between 1962 and 1982 were associated with increases in the number of deaths from cancer, in the crude cancer-related mortality rate, in the age-adjusted mortality rate, and in both the crude and the age-adjusted incidence rates, whereas reported survival rates (crude and relative) for cancer patients also increased (1). John Bailar, a coauthor on the report, noted at the time that “despite all the billions of dollars, and the promises and the claims of success, more people are dying of cancer than ever before.” Finally, in 1990, however, the national incidence and mortality of cancer began to decline, and mortality has continued to decline each year since 1990. Furthermore, overall deaths from cancer have declined in 2005 despite the larger and older population, and in 2007, the rate of decline actually doubled (2). Whereas part of the observed decline is likely due to cancer prevention and advances in early diagnosis of malignancies, advances in cancer treatment have contributed substantially, and much of it is due to the inclusion of chemotherapy in treatment programs for many diseases.
For most primary tumors the treatment of choice is surgery and radiotherapy, which measures can be very effective for controlling localized tumors and indeed surgery and radiotherapy dominated the field of cancer therapy into the 1960s. However, at the time of diagnosis the majority of cancers have already microscopically metastasized throughout the body, leading to recurrent disease in the majority of cancer patients. In view of this, systemic chemotherapy is required to control outgrow of metastases. Although for some invasive tumors at advanced disease stages, chemotherapy might be administered up front to allow better surgery, in essence presently the chemotherapy of cancer is the treatment of metastases, either known or assumed, except for hematological malignancies. The current concept that cytotoxic chemotherapeutic agents are administered at a dose to the maximum a patient can tolerate before the onset of severe and even life-threatening toxicity is still in wide clinical use. This approach is based on a series of retrospective analyses which indicated that the greater the dose intensity (ie, the dose delivered over a standard interval of time) of an anticancer drug, the better the outcome. This dosing concept has led to continuous public outrage from prominent scientists -usually working in areas of medicine other than oncology- including Ernst Krebs, commenting in 1988 that “chemotherapy will make the ancient method of drilling holes in a patient's head to permit the escape of demons look relatively advanced” as well as Linus Pauling, remarking in 1986 that “most cancer research is largely a fraud, and the major cancer research organizations are derelict in their duties to the people who support them” (3).
In the last decade, chemotherapy has transitioned from the use of cytotoxic drugs to the era of agents with an apparent selectivity for a cancer-specific target, previously dubbed “targeted therapy”. The story of how this area evolved from the Special Virus Cancer Program in the 1960s and how this program identified oncogenes and signaling pathways essential for developmental biology has been recently discussed (2). This work eventually led to the identification of most of the new drug targets that are currently the focus of cancer drug development. Indeed, an ever increasingly large number of drugs has become available to treat a wide variety of neoplastic diseases (Figure 1). In addition to the current availability of highly innovative and distinct classes of molecules in terms of both mechanism of action and chemical structure, an impressive array of new techniques and ideas is being deployed in approaches to all stages of drug discovery, development and trial design (4). The first and best example of targeted therapy was the development and subsequent approval in 2001 of the Bcr-Abl tyrosine kinase inhibitor imatinib for the treatment of chronic myelocytic leukemia (5). The management and outcome of this disease has been drastically altered as a result of continuous treatment with imatinib, and this consummate success story established proof-of-principle for the therapeutic power of the knowledge of molecular targets in malignant diseases.
Figure 1.
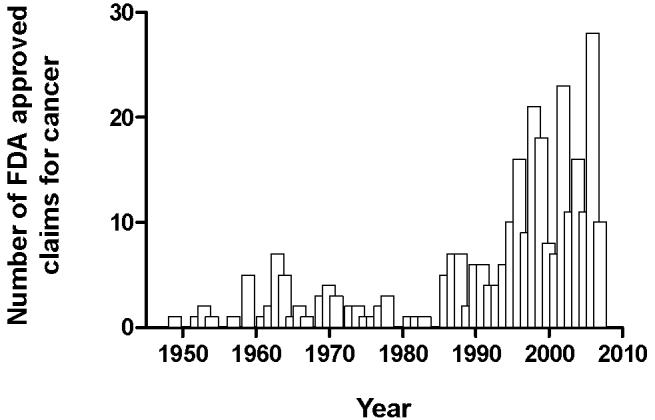
Oncology drug approvals based on approved claims per year between 1949 and 2007. Data was compiled from the U.S. Food and Drug Administration, Center for Drug Evaluation and Research, Oncology Tools <http://www.fda.gov/Cder/cancer/approved.htm> (2008).
Subsequent data from the human genome sequence suggested that many of the abnormalities associated with cancer beyond chronic myelocytic leukemia are also due to the abnormal function of protein kinases, and a major thrust of the current oncology drug development era has been to develop a series of small molecule tyrosine kinase inhibitors. Indeed, already eight of such agents have now been approved by the U.S. Food and Drug Administration for the treatment of a variety of diseases that were previously essentially resistant to standard chemotherapy. Clearly, these agents hold promise to treat a broad range of solid tumors and hematologic malignancies. Nonetheless, many of the recent advances in cancer care have been relatively modest and many of the novel therapeutics, like their cytotoxic counterparts, have rather limited efficacy combined with a significant degree of unexpected and unexplained toxicity (6), and most are anything but “magic bullets” (7). The major reasons for this are presumably still the lack of sufficiently detailed knowledge in tumor cell biology, the absence of appropriate preclinical models for the identification and testing of compounds, and the notion that, unlike in the case of chronic myelocytic leukemia where a unique single molecular abnormality drives the disease, most cancers are affected by multiple abnormalities that must be targeted simultaneously (8, 9). Moreover, as with conventional cytotoxic agents, the novel therapeutics are susceptible to intrinsic resistance or the emergence of resistance through various mechanisms recently reviewed in this journal (10). A number of additional critical issues surrounding the development and recent implementation of these advanced therapeutics in cancer care are highlighted in the current issue of Clinical Pharmacology & Therapeutics.
As discussed by Collins (11), molecularly-targeted therapy has accelerated a trend toward the incorporation of major shifts in goals and approaches to first-in-human studies in oncology. Indeed, many early human studies are now specifically designed to test mechanistic hypotheses which can drive important development decisions. One type of exploratory study, the so-called Phase 0 trial, may help provide more confidence in the selection of drug candidates early in clinical development by allowing dosing to pharmacologic response and limited multiple dosing of investigational agents in patients with cancer. This strategy has the potential to reduce attrition in clinical drug development, although its value as a major retooling of the drug development process has been questioned by some (12, 13). Phase 0 trials are currently being extensively explored as a platform to establish the feasibility of assays for target modulation as well as to evaluate the potential utility of biomarkers. The utilization of appropriate biomarkers in for example Phase I clinical trials of novel anticancer agents, discussed by Carden et al. (14), has the potential to increase the possibility of patient benefit, accelerate the drug development process, and maximize the ability to generate important biological information about human cancer and further decrease the risk of late and costly drug attrition. However, as outlined by Glassman et al. (15), the enthusiasm for widespread adoption of biomarker studies in early drug development is not entirely justified due to statistical and cost considerations, as well as a current lack of historic evidence for their utility.
Beyond first-in-human studies, shifts in the classical Phase I, II and III trial approaches-as they have historically been applied to conventional cytotoxic chemotherapeutic agents-are being pursued to meet the requirements for the clinical development of novel therapeutics. As discussed by Schellens et al. (16), multiple strategies are being explored to develop alternative strategies for identification of an optimal dose, as opposed to a maximum-tolerable dose. Furthermore, enrichment of patient populations based on molecular pathology of the tumor, implementation of novel imaging techniques (17, 18), adapted designs for Phase II and III trials as well as health technology assessments in parallel with Phase III studies may all help to further optimize clinical development of targeted agents.
In spite of the revolutionary changes in the various stages of drug development employed for advanced therapeutics, the standard strategy still in use for dose selection is to establish a therapeutic dose in Phase II trials and subsequently, at best, only modify it for individual differences in body-surface area. However, there is already a wealth of experimental data indicating that both the efficacy and safety of novel therapeutics might be optimized if dosing strategies would take into consideration individual patient characteristics as they relate to the agent's pharmacokinetic, pharmacodynamic or pharmacogenetic profiles. As discussed by Baker et al. (19), we have slowly come to the important realization that although these drugs offer possibly a number of important advantages over conventional cytotoxic agents, they are still afflicted by some of the same problems, including an extensive interindividual pharmacokinetic variability (Figure 2) and the existence of a really rather narrow therapeutic window. In view of the present opportunity to identify patients likely to benefit from a certain drug or drug combination, it will be imperative to make an effort to ensure that sufficiently high local drug concentrations are reached in order to maximize efficacy without exacerbating toxicity. Hence, optimizing individualized measures of systemic exposure and understanding of the underlying factors involved in intersubject variability are critical issues that need to be addressed prospectively in adequately powered trials.
Figure 2.
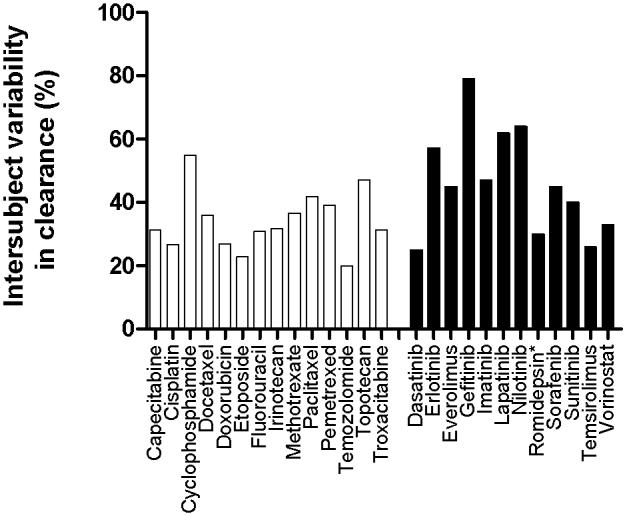
Interindividual pharmacokinetic variability of select cytotoxic agents (white bars) and advanced therapeutics (black bars), expressed as percent coefficient of variation in apparent (oral) clearance. Data was compiled from Mathijssen et al. (24) and publicly available prescribing information. *Denotes investigational new drug with FDA-granted Orphan Drug Designation and Fast Track.
The arsenal of anticancer drugs used in pediatric malignancies is in general similar to that available for the treatment of adult diseases (20). Although conventional cytotoxic anticancer drugs historically have had their greatest impact in the treatment of childhood cancers, as discussed by Balis et al. (21), there is a continued need to evaluate new agents in pediatric oncology, in particular for intrinsically resistant diseases and in an attempt to reduce acute and long-term side effects associated with the use of conventional agents. The clinical development of new anticancer drugs in children is surrounded by a number of important issues, including an impact of ontogeny on drug disposition and differential disease pathogenesis and manifestation that warrant alternate approaches compared with studies performed in adults. One additional important consideration that has received little more than cursory interest from pharmaceutical companies and that has substantially hampered the development of novel therapeutics in pediatric oncology, is the lack of suitable pediatric formulations of approved oncology products. This is particularly problematic for those agents that are administered orally, since capsule or tablet sizes used in adults may be too large for accurate dosing in children.
The last few decades have provided evidence that cancer chemotherapy can be curative in subsets of patients with advanced disease, including Hodgkin's lymphoma, certain leukemias, and testicular cancer, and the expectation is that the list of cancers effectively treated and possibly even cured usingmodalities that include novel therapeutics will continue to expand in the near future (2). It should be pointed out, however, that in countries that include an analysis of cost-effectiveness as part of the approval process, many of the novel therapeutics are frequently not approved (22), owing to a marginal benefit at an extremely high cost, as discussed in this issue by Sleijfer et al. (23). Nonetheless, it is anticipated that incorporation of pharmacologic principles in drug development and the continued exploration and optimization of innovative trial designs with an especially careful and imaginative choice of dosage regimens for novel therapeutics will ultimately result in the discovery of new, cutting edge treatment modalities for multiple malignant diseases.
Footnotes
CONFLICT OF INTEREST The authors declared no conflict of interest.
References
- 1.Bailar JC, Smith EM. Progress against cancer? N. Engl. J. Med. 1986;314:1226–1232. doi: 10.1056/NEJM198605083141905. [DOI] [PubMed] [Google Scholar]
- 2.DeVita VT, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 3.IVU Quotes and Poetry 2008 http://www.ivu.org/people/quotes/health.html
- 4.Vallance P, Levick M. Drug discovery and development in the age of molecular medicine. Clin. Pharmacol. Ther. 2007;82:363–366. doi: 10.1038/sj.clpt.6100333. [DOI] [PubMed] [Google Scholar]
- 5.Sherbenou DW, Druker BJ. Applying the discovery of the Philadelphia chromosome. J. Clin. Invest. 2007;117:2067–2074. doi: 10.1172/JCI31988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saltz LB. Progress in cancer care: the hope, the hype, and the gap between reality and perception. J. Clin. Oncol. 2008;26:5020–5021. doi: 10.1200/JCO.2008.17.6198. [DOI] [PubMed] [Google Scholar]
- 7.Maitland ML, Ratain MJ. Terminal ballistics of kinase inhibitors: there are no magic bullets. Ann. Intern. Med. 2006;145:702–703. doi: 10.7326/0003-4819-145-9-200611070-00015. [DOI] [PubMed] [Google Scholar]
- 8.Stommel JM, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 9.Fojo T. Novel therapies for cancer: why dirty might be better. Oncologist. 2008;13:277–283. doi: 10.1634/theoncologist.2007-0090. [DOI] [PubMed] [Google Scholar]
- 10.Mimeault M, Hauke R, Batra SK. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin. Pharmacol. Ther. 2008;83:673–691. doi: 10.1038/sj.clpt.6100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins JM. Phase zero clinical studies in oncology. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.246. this issue. [DOI] [PubMed] [Google Scholar]
- 12.Twombly R. Slow start to phase 0 as researchers debate value. J. Natl. Cancer Inst. 2006;98:804–806. doi: 10.1093/jnci/djj249. [DOI] [PubMed] [Google Scholar]
- 13.Boyd RA, Lalonde RL. Nontraditional approaches to first-in-human studies to increase efficiency of drug development: will microdose studies make a significant impact? Clin. Pharmacol. Ther. 2007;81:24–26. doi: 10.1038/sj.clpt.6100058. [DOI] [PubMed] [Google Scholar]
- 14.Carden CP, Banerji U, Kaye SB, Workman P, De Bono JS. From darkness to light with biomarkers in early clinical trials. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.223. this issue. [DOI] [PubMed] [Google Scholar]
- 15.Glassman RH, Ratain MJ. Biomarkers in early cancer drug development: limited utility. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.231. this issue. [DOI] [PubMed] [Google Scholar]
- 16.Schellens JHM, Beijnen JH. Novel clinical trial designs for innovative therapies. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.247. this issue. [DOI] [PubMed] [Google Scholar]
- 17.Han Z, et al. Noninvasive assessment of cancer response to therapy. Nature Med. 2008;14:343–349. doi: 10.1038/nm1691. [DOI] [PubMed] [Google Scholar]
- 18.McLennan G, Clarke L, Hohl R. Imaging as a biomarker for therapy response: cancer as a prototype for the creation of research resources. Clin. Pharmacol. Ther. 2008;84:433–436. doi: 10.1038/clpt.2008.171. [DOI] [PubMed] [Google Scholar]
- 19.Baker SD, Hu S. Pharmacokinetic considerations of new targeted therapies. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.242. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paolucci P, Jones KP, Del Carmen Cano Garcinuno M, Catapano M, Iolascon A, Ceci A. Challenges in prescribing drugs for children with cancer. Lancet Oncol. 2007;9:176–183. doi: 10.1016/S1470-2045(08)70030-5. [DOI] [PubMed] [Google Scholar]
- 21.Balis FM, Fox E, Widemann BC, Adamson PC. Clinical drug development for childhood cancers. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.237. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messersmith WA, Ahnen DJ. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008;359:1834–1836. doi: 10.1056/NEJMe0806778. [DOI] [PubMed] [Google Scholar]
- 23.Sleijfer S, Verweij J. The price of success: cost-effectiveness of molecularly targeted agents. Clin. Pharmacol. Ther. 2009 doi: 10.1038/clpt.2008.245. this issue. [DOI] [PubMed] [Google Scholar]
- 24.Mathijssen RHJ, et al. Flat-fixed dosing versus body-surface area-based dosing of anticancer drugs in adults: does it make a difference? Oncologist. 2007;12:913–923. doi: 10.1634/theoncologist.12-8-913. [DOI] [PubMed] [Google Scholar]