

Abstract
In the late 1970s, it was predicted that gene therapy would be applied to humans within a decade. However, despite some success, gene therapy has still not become a routine practise in medicine. In this review, we will examine the problems, both experimental and clinical, associated with the use of viral material for transgenic insertion. We shall also discuss the development of viral vectors involving the most important vector types derived from retroviruses, adenoviruses, herpes simplex viruses and adeno-associated viruses.
This article is part of a themed section on Vector Design and Drug Delivery. For a list of all articles in this section see the end of this paper, or visit: http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: viral vectors, gene therapy, AAV, Ad, HSV, lentivectors, pseudotyping, cell targeting, transcriptional targeting, vectors production
Introduction
The paradigm of in vivo gene transfer is to insert a gene into an organism using a vector that will transfer only the required gene to the desired target cells. Gene transfer is an invaluable tool routinely used in vitro or in animal models to investigate the molecular mechanisms underlying diverse biological functions. However, ‘gene therapy’, which aims to use genes as therapeutic entities in humans, is the ultimate goal of gene transfer. The goal is to deliver genetic information to a target cell, either to replace a defective function (monogenic disease), or to introduce an additional function to treat (as in cancer) or to prevent (as in a vaccine) disease. The introduction of the therapeutic gene into the target cell can be achieved in two ways: ex vivo or in vivo.In ex vivo gene therapy, target cells are first extracted from the patient. The desired gene is then inserted into these cells, and once the transfer is completed, the cells are returned to the patient. This technique has had promising results, but is restricted to a limited number of target cell types and diseases (Aiuti et al., 2002; Hacein-Bey-Abina et al., 2002). This review will therefore focus on in vivo gene therapy protocols, where the vector has to be able to deliver the selected gene directly into the target cells within the whole organism.
The lack of an efficient, non-toxic, gene delivery system, rather than the paucity of therapeutic genes, is the major challenge of in vivo gene therapy. Viruses are naturally very efficient at transducing their own genetic information into host cells for their own replication. By replacing non-essential viral genes with foreign genes of therapeutic interest, recombinant viral vectors can be used to transduce the cell type that they would normally infect (Figure 1). Although viruses may trigger a host immune response, they also have evolved and developed efficient countermeasures, thus enabling them to reach and replicate in their target cells. Using vectors derived from viruses as ‘Trojan horses’ to reach the required cells is to take advantage of millions of years of evolution. The first attempt to use viruses in this way was carried out by Rogers et al. in 1973. Using the Shope papilloma virus, they tried, without success, to induce arginase activity in tissue culture cells of hyper-argininaemic patients (Rogers et al., 1973). However, despite this failure, the use of replication-competent viruses directly as therapeutic agents remains a field of intense research, in particular in the use of oncolytic viruses to treat cancer.
Figure 1.
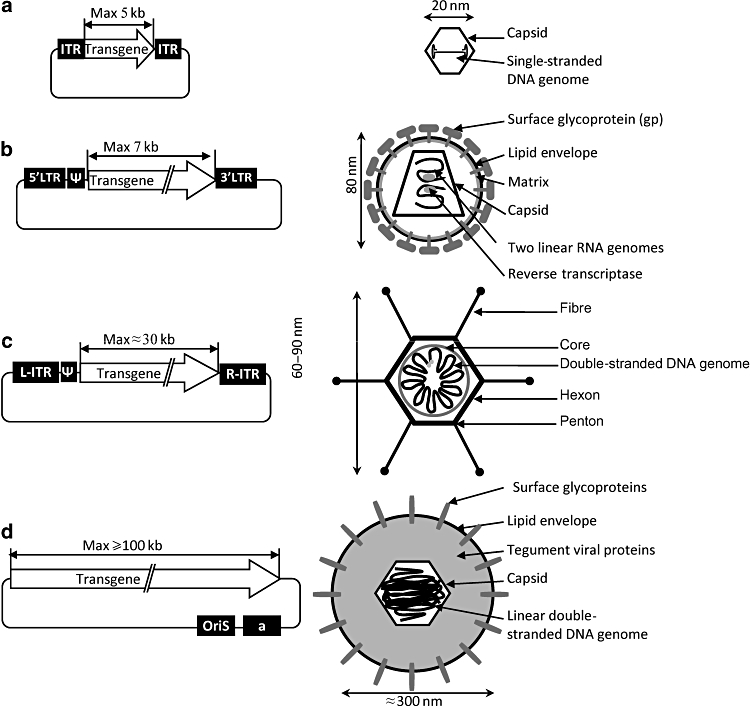
The four main viral vectors: plasmids and viral particles. (a) Recombinant AAV vector. (b) Retroviral vector. (c) Gutless adenoviral vector. (d) Amplicon vector derived from HSV-1. ITR, inverted terminal repeat; L, left; R, right; ψ, a, encapsidation signal; oriS, replication origin.
In 1978, 3 years after being awarded the Nobel Prize for the discovery of reverse transcriptase and the mechanisms that retroviruses use to infect cells, David Baltimore claimed that gene therapy would be applied to humans within the next 5 years. It, nevertheless, took more than 10 years before Stephen Rosenberg became the first person to successfully insert foreign genes into humans and conduct clinical studies of gene therapy for cancer (Rosenberg et al., 1990). Unfortunately, few successes have been reported since then. Researchers have, however, learnt a great deal from early mistakes and important milestones have been reached in recent years. The first-generation retroviral vectors, modified to carry a gene to transfer (or ‘transgene’), instead of their own viral genes, into a cell, were engineered in the early 1980s (Shimotohno and Temin, 1981; Wei et al., 1981; Tabin et al., 1982). Furthermore, despite the setbacks, the number of gene therapy clinical trials has increased (Figure 2).
Figure 2.
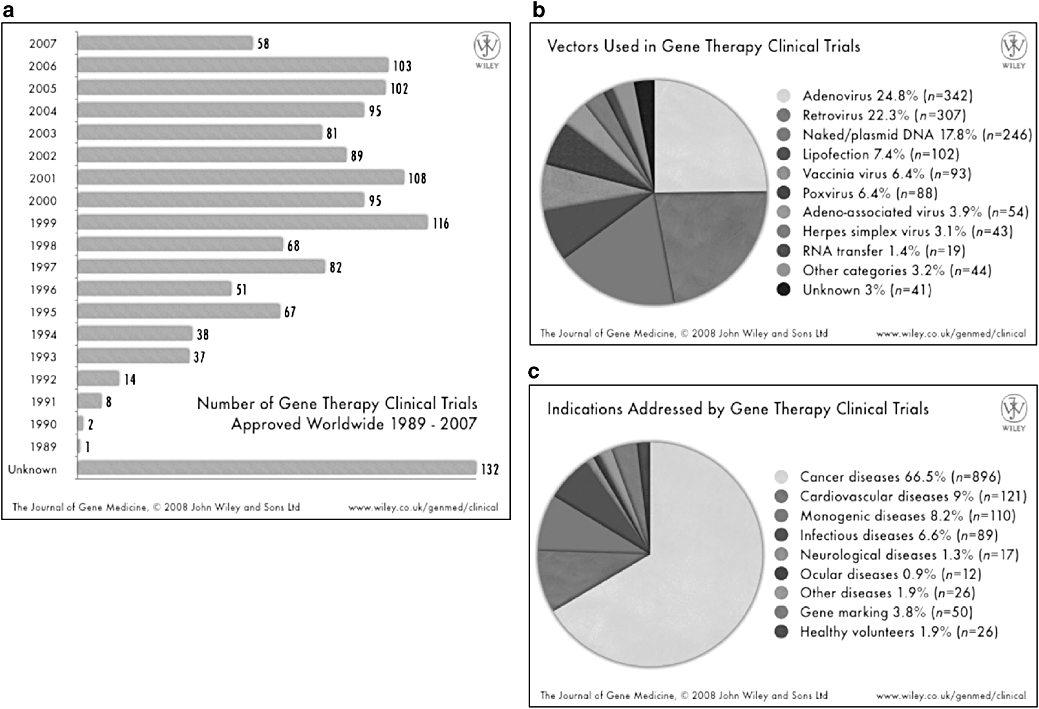
Overview of gene therapy clinical trials worldwide. (a) Number of trials initiated per year, (b) vectors used and (c) indications addressed. Updated information available from the Journal of Gene Medicine: http://www.wiley.co.uk/genmed/clinical. Reproduced with the permission from Wiley and Son Ltd.
What, at present, are the critical issues in in vivo gene therapy?
First, being derived from a number of different viruses, vectors inherit specific properties from their wild-type parent viruses (Figure 3, Table 1), which require thorough investigation of their replication mechanisms and interaction with their natural hosts.
Figure 3.
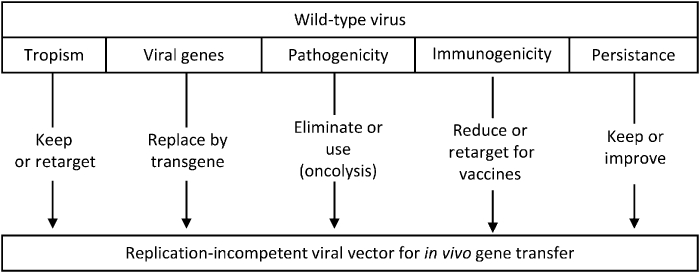
From wild-type virus to replication-defective viral vectors. Outline of the possible modifications of viral properties required to transform a virus into a gene transfer vector.
Table 1.
Viral vectors and their main properties
| Vectors | Vector yield (transducing units mL1) | Inflammatory potential | Tropism re-targeting | Entry pathway | Vector genome forms | Transgene expression | Genotoxicity | Shedding |
|---|---|---|---|---|---|---|---|---|
| Retrovirus | Moderate (1010) | Low | Dividing cells | Receptor-binding, membrane fusion | Integrated | Long term (years) | Integration might induce oncogenesis | Low |
| Lentivirus HSV-1 | High (1012) | High | Broad Neurons | Receptor-binding, membrane fusion | Episomal | Transient | Low | ND |
| Adenovirus | High (1012) | High | Broad | CAR-mediated endocytosis endosomal escape | Episomal | Short term (weeks) | Low | Moderate |
| AAV | High (1012) | Low | Broad | Receptor-mediated endocytosis endosomal escape | Episomal (90%) Integrated (10%) | Medium to long term (year) | Should not be underestimated. Needs more investigation | Moderate |
Second, although small-scale production can be sufficient for laboratory use, there is still an urgent need for the development of production and purification procedures that generate batches of vector in increasing yield and of sufficient quality necessary for routine clinical use. Industrial-scale production of vectors is outside the scope of this review, but for further information on the considerable research in this area see Merten (2004).
Third, being parasites, viruses have evolved to evade the immune system while at the same time the immune system has evolved to destroy viruses; hence, if viral vectors are destroyed or if the immune response recognizes and eliminates transduced cells, all potential benefit is lost (Nunes et al., 1999). We will discuss here the engineering of gene transfer vectors to circumvent this problem.
Fourth, cell targeting: Any vector must be able to circumvent most biological barriers and reach only the desired cells. Massive infection of non-target cells would severely decrease bioavailability of the vector and, moreover, would increase the potential risk of unwanted clinical consequences. If infection of non-target cells cannot be prevented, harmful consequences may be limited by ‘transcriptional targeting’, that is the restriction of transgene expression to the target cells.
Fifth, maintenance of transgene expression and integration, and the consequences of this integration for the cell are important issues to be addressed.
Finally, viral vectors must be accepted not only by both ethical and regulatory authorities but also by the general public. Vectors derived from viruses that are not pathogenic to humans may be preferred in this respect, for example AAV, feline immunodeficiency virus or spumaviruses (Loewen et al., 2003a b; Rethwilm, 2007). But as a counter-example, chimeric human immunodeficiency virus-based vectors pseudotyped with Ebola virus glycoproteins have been shown to transduce primary cultures of human airway epithelia, but such vectors would, in all probability, not be acceptable to the public (Sinn et al., 2003; Sutherland, 2003; Sanders, 2004).
To date, up to 1000 clinical gene therapy trials have been initiated using a limited number of viruses as vectors, namely adenoviruses (Ads), retroviruses (γ-retroviruses and lentiviruses), poxviruses, adeno-associated viruses (AAV) and herpes simplex virus (HSV). The main indication addressed by these clinical trials is cancer, followed by cardiovascular and monogenic diseases (for updated information see http://www.wiley.co.uk/genmed/clinical/). The number of viruses that are under development as gene-therapy vectors is increasing, but we will focus on the four most promising viruses, namely retroviridaes (including, γ-retroviruses and lentiviruses), Ads, AAV and HSV type-1 (HSV-1), as shown in Figure 3.
We will first examine the production of vectors and their physicochemical stability, before discussing cell and transcriptional targeting.
Vector particle production and stability
Viral vector production systems and biosafety
Promising results obtained in pre-clinical studies using animal models have led to the rapid development of clinical trials using viral vectors.
One of the major concerns in viral vector design and production is biosafety and vector safety. Vectors and cell-based vector production systems have to be approved by regulatory agencies. Vectors discussed in this review tend to be devoid of as many viral sequences as possible, to be nonpathogenic or replication-defective, and therefore they are likely to be of low toxicity.
Both Ad-and herpes virus-derived vectors can be produced easily at high titres. In addition to the transgene(s) of interest, these modified vectors (gutted Ad) could only contain/express the viral sequences required for the initiation of viral DNA replication and packaging. They are therefore also known ‘helper-dependent’ vectors, as their production requires that all the necessary structural proteins be provided. This was originally achieved by co-infection with a helper virus, but contamination of the desired vector stock with unwanted helper virus was a major problem. In the newer-generation helper viruses, the packaging signal was flanked by loxP recognition sites, which can be cleaved by the Cre recombinase encoded in the supporting cell line. This neutralizes the packaging signal of the helper virus and ultimately leads to the selective encapsidation only of the vector sequences (Parks et al., 1996; Zaupa et al., 2003). Despite these modifications, small amounts of helper particles were still produced, such that the helper viruses were further modified by gene deletion to render them replication-incompetent and, hence, to prevent them from spreading to non-target cells and tissues (Cuchet et al., 2007). However, the presence of contaminating particles, defective or not, continues to represent a limitation of this approach and thus the system still needs further improvement.
Providing helper activities in the form of a single helper plasmid encoding for a full-length defective helper herpes virus genome was proved to successfully replace helper virus requirement (Saeki et al., 2001). However, although this system allows for the production of entirely helper-free amplicon stocks, the yield of amplicon vectors is limited. Therefore, further research is still required for both Ad and HSV-1 vector syntheses that will increase, yield yet eliminate helper contamination.
Production of retrovirus and recombinant AAV (rAAV) vectors requires two main components, namely the vector genome containing a transgene expression cassette flanked by the cis-acting long terminal repeat of retroviruses or inverted terminal repeat of AAV sequences and the proteins necessary for vector replication and encapsidation provided by the helper constructs. Unwanted recombination between the helper constructs and the vector has been minimized by using non-viral regulatory sequences to control their expression (Cosset et al., 1995). Packaging and producer cell lines providing gag pol and envelope glycoprotein have been developed for retroviruses, but have proved to be more challenging for rAAV (reviewed in Blouin et al., 2004). Indeed, in addition to the cytotoxicity of AAV Rep proteins, rAAV vector production also requires helper activity from a heterologous virus, that is adenovirus or herpes virus. Helper activities were originally provided by infecting cells with Ad, but were advantageously replaced by the use of plasmids encoding only the adenoviral helper activities (Grimm and Kleinschmidt, 1999). Stocks of rAAV free of any detectable Ad particles can therefore be produced at high titres, but the production method based on the transfection of a minimum of two plasmids remains difficult to scale up. This limitation could, however, soon be overcome, thanks to the use of baculovirus-mediated gene delivery in mammalian cells and bioreactors, which represent a promising direction towards mass production (Huang et al., 2007).
Purification, concentration and storage
Often, vector production systems are quantitatively insufficient to raise the appropriate numbers of vector particles required for therapeutic applications (reviewed in McTaggart and Al-Rubeai, 2002). Although concentration procedures can be used, most of them have the disadvantage of also concentrating impurities from cell lysates (such as proteoglycans) or from pretreatment reagents. Depending on the vectors and their physical properties, centrifugation, tangential flow filtration and ultrafiltration can be used, as well as polyethyleneglycol precipitation, two-phase extraction, membrane filtration, liquid chromatography or adsorption chromatography. Viruses such as Ad, HSV, rAAV are purified from cell debris. Conventional methods rely on the use of density gradients (cesium chloride or iodixanol), and are therefore difficult to scale up. In contrast, a two-step chromatography process based on the use of ion-exchange resins can be easily scaled-up and requires no pretreatment of cells and results in pure rAAV stocks (reviewed in Blouin et al., 2004). This technique has the advantage over affinity chromatography resins of not being serotype-dependent, but requires the careful modification of a few parameters between serotypes, such as pH and salt concentration.
Viral vector stocks are usually stored at −150°C or at −80°C. Several studies have shown that retroviral vectors can be frozen with variable decay (Lee et al., 1996; Kaptein et al., 1997). Some authors have investigated the effects of lyophilization (Kotani et al., 1994) and, more recently, ambient temperature vacuum dehydration for vector preservation to try to generate stable yet easy to use viral stocks (Andreadis et al., 1999).
Immunostability of vectors
One of the first barriers that gene therapy vectors have to circumvent in vivo is the immune response, in particular the complement system and other components of innate immunity as well as pre-existing antibody-mediated immunity. An extreme example of immune response to viral vectors occurred in a patient with ornithine transcarbamylase deficiency who died of systemic inflammatory response syndrome after hepatic arterial injection of an Ad vector (Raper et al., 2003). Immune system sensitization by pre-exposure to the wild-type virus or direct immunostimulation by the vector may cause clinical problems.
But the long-term correction of rare monogenetic diseases will require sustained transgene expression from non-integrative viral vectors upon a single inoculation, which is difficult and may require frequent re-administration of the vector. The Ad vectors were initially susceptible to immune system attack (complement system and pre-existing antibodies) but progress was made by engineering ‘gutted’ or ‘helper-dependent’ vectors stripped of all viral genes, thereby limiting the production of potentially harmful viral antigens. In one study, vector-mediated cytokine responses were drastically decreased (Mok et al., 2005), but the results were not confirmed by a subsequent publication (Lyons et al., 2006). Others studies aiming at improving systemic delivery have led to coating Ad vectors with polyethyleneglycol, which can help the vector escape from both antibody-mediated (Eto et al., 2005) and innate immune responses (Croyle et al., 2005). Immune responses against AAV vectors can shorten or even prevent transgene expression and, as such, has been a major concern since the first clinical trials. This immunity includes pre-existing immune responses against the wild-type virus or immune responses against the vector owing to repeated administration. Switching serotypes or even inducing immunosuppression are being utilized to overcome such problems (Mingozzi and High, 2007; Zaiss and Muruve, 2008).
Viral vectors can be inactivated by complement attack after administration. One interesting property of retroviral vectors is their ability to be enveloped by different glycoproteins. For example, the G glycoprotein of vesicular stomatitis virus (VSV-G) can be used to create VSV-Gpseudotyped lentiviral vectors that are particularly resistant to centrifugation processes, as compared with other pseudo-types. However, broad tropism and inactivation by human serum might impose a limitation on the use of VSV-G as a glycoprotein to pseudotype vectors for in vivo gene delivery. Similarly, lentiviral vectors pseudotyped with RD114 (feline endogenous retrovirus) glycoproteins are efficiently concentrated, yet, unlike VSV-G pseudotypes, are stable in human and macaque serum, implying that these vectors should retain high infectivity in primate serum after in vivo delivery (Sandrin et al., 2002).
Retroviral vectors made in murine cell lines demonstrated high sensitivity to serum complement (Pensiero et al., 1996). In contrast, retroviral vectors derived from human packaging cell lines are considerably more resistant to inactivation by primate serum complement (Takeuchi et al., 1994; DePolo et al., 1999). Nevertheless, glycoprotein-dependent differences remain, that is VSV-G is very sensitive (DePolo et al., 2000; Guibinga et al., 2004) in contrast to RD114 and its derivatives (Sandrin et al., 2002; Sandrin et al., 2003).
Several other solutions for bypassing the attack by complement have been investigated. Expression of anti-complement decay-accelerating factor molecules on retroviral vector surfaces (Guibinga and Friedmann, 2005; Schauber-Plewa et al., 2005) is one possibility as is PEGylation of VSV-G (Croyle et al., 2004).
The cell targeting issue: tropism and re-targeting
For ex vivo gene therapy, using viral vectors that transduce a large panel of cells (broad tropism) is not a problem, because target cells are first isolated from the organism and then transduced. Thus, the risk of vector dissemination and off-target infection is low. In contrast, restricting infection to the target cells, known as ‘cell targeting’ or ‘transductional targeting’, is a critical issue for efficient and safe in vivo gene delivery. Efficient targeting is a key to enhancing therapeutic effect, reducing side effects and lowering the amounts of vectors required. To achieve this goal, two methods may be used: one is to take advantage of the natural properties of existing viral proteins and the another is to use genetic engineering to retain, abolish or extend the original tropism of vectors (Figure 4, Table 2).
Figure 4.
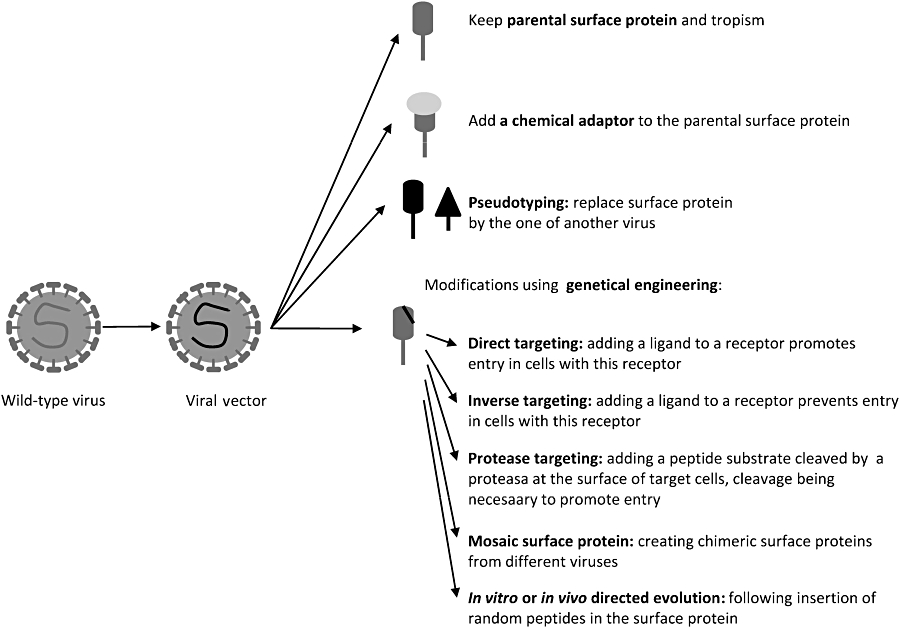
A few targeting and re-targeting options for viral vectors. Retargeting is necessary to alter the tropism of the viral vectors to reach more efficiently the desired target cells and/or prevent transduction of non-target cells.
Table 2.
Overview of properties of lentivectors
| Genus /Glycoprotein | Target cells/organ | Remarks |
|---|---|---|
| Vesiculovirus (VSV-G) | Broad tropism | Stability (complement resistant, physicochemical stability) |
| Arenavirus (LCMV, Lassa) | Liver, cancer cells, pancreatic cells | Stability. Non-toxic |
| Filovirus (Ebola) | Airway epithelium | Apical surface of the epithelium |
| Lyssavirus (Mokola, Rabies) | CNS, myocytes, retina, neuronal progenitors | |
| Murine retrovirus (MLV) | Broad tropism | Efficient re-targetting |
| Feline endogenous retrovirus (RD114) | Haematopoetic cells | Efficient. Non-toxic |
| Human retrovirus (HIV) | T4 cells | Lacks stability |
| Gibbon retrovirus (GALV) | Haematopoetic cells | Efficient re-targetting |
| Hepacivirus (HCV) | Liver cells |
Cell targeting using existing molecules
The most simple, but also most limited method of transductional targeting of viral vectors is to use the natural tropism of the parent virus. For example, amplicons derived from HSV-1 are used to transduce neural cells, taking advantage of the ability of HSV-1 to invade and establish lifelong latent infections in neurons (Cuchet et al., 2007). The viral vector thus inherits the qualities and defaults of the corresponding parental wild-type virus. Tropisms that are more selective can be attained by taking advantage of the natural tropisms of other more or less closely related viruses through pseudotyping. Pseudotyping allows altering of the cell-binding specificity of a vector by changing either its capsid (for non-enveloped vectors) or its surface glycoproteins (for enveloped vectors). This procedure is particularly well established for retroviruses (Verhoeyen and Cosset, 2004). It has also been used to create chimeric Ad vectors derived from a type-5 capsid-46 incorporating type-35 fibre protein (Shayakhmetov et al., 2000). In the case of rAAV, it is possible to package the prototypic AAV-2-derived vector genome in capsids from different AAV serotypes, increasing the diversity of reachable target tissues and transduction efficiency (Rabinowitz et al., 2002; Wu et al., 2006a b).
rAAV offers a wide range of pseudotyping possibilities with more than 50 serotypes described to date, with different origin and tropism. The choice is also extensive for retroviruses. The envelope glycoproteins derived from the majority of the retroviruses, as well as from various families of enveloped viruses, are able to pseudotype onco-retroviral and/or lentiviral cores. Among these non-retroviral glycoproteins, we find glycoproteins derived from the following viruses/families: vesiculovirus (Burns et al., 1993; Naldini et al., 1996), lyssavirus (Mochizuki et al., 1998; Desmaris et al., 2001), arenaviruses (Miletic et al., 1999), hepadnaviridae (Sung and Lai, 2002), paramyxoviridae (Spiegel et al., 1998; Kobinger et al., 2001), orthomyxovirus (Dong et al., 1992; Hatziioannou et al., 1998; Sandrin et al., 2002), filovirus (Wool-Lewis and Bates, 1998; Kobinger et al., 2001) and alphavirus (Suomalainen and Garoff, 1994; Morizono et al., 2001; Sharkey et al., 2001). Excluding some possible exceptions (Desmaris et al., 2001), pseudotyped vectors usually retain the tropism of the parent virus from which the envelope (for enveloped vectors) or capsid (for non enveloped vectors) protein is derived. However, this technique is limited to the number of viral attachment proteins for which a characterized receptor is selectively expressed on target cells of interest.
However, finding the best candidate glycoproteins for pseudotyping retrovirus is not easy. For example, pseudo-typing retroviral vectors with VSV-G glycoprotein allows efficient transduction of most cell types but is not appropriate for in vivo gene transfer to a particular cell type, as the tropism is too large. In contrast, when using oncoretroviral vectors, the RD114 glycoprotein was shown to induce efficient gene transfer into the primary haematopoietic stem cells and lymphocytes peripheral blood lymphocytes (PBL) (Kelly et al., 2000; Sandrin et al., 2002). The RD114 gp does not pseudotype lentiviral vectors very efficiently, as it requires modification of the cytoplasmic tail to increase its incorporation on viral particles (Sandrin et al., 2002 2004; Bouard et al., 2007).
Adaptors can be used to alter the targeting of vectors without changing the surface molecule (for review see Waehler et al., 2007). Adaptors are bi-specific agents that recognize the vector on one side and the target cells on the other. The interactions between the adaptor molecule and the vector can be of different types: receptor–ligand, covalent chemical conjugation, avidin–biotin or antigen– antibody interactions. Adaptors have been used mainly for adenoviral vectors, but they have also been successfully used for rAAV, lentiviral and retroviral vectors. A few proof-ofprinciple studies using this strategy have already been performed in vivo, but this kind of strategy seems to be cumbersome for clinical exploitation. The possible deleterious in vivo instability of the vector–adaptor complex and difficulties in large-scale production of these two-component systems are major concerns that will certainly hinder general use of these re-targeted vectors.
Cell re-targeting using genetic engineering
The modification of surface molecules (capsids or glycoproteins) by genetic engineering is an alternative to the above-mentioned approaches. Direct targeting strategies are based on the simple idea that a ligand inserted at the surface of a viral vector will promote endocytosis or trigger membrane fusion activation and entry of the vector genome when interacting with its specific receptor on target cells. Such strategies were applied to the primary vectors with limited success (for review see Waehler et al., 2007). The inserted peptides can be rationally designed or be the result of direct evolution approaches or library selection approaches (for a review see Yu and Schaffer, 2005). For example, sequences conferring the ability to bind a particular receptor can be inserted in the vicinity of the AAV2 protein. Frequently, this disrupts its ability to be recognized by its wild-type receptor and confers new cell tropism (Perabo et al., 2006). Capsid genes of the Ad can also be genetically engineered to include a small peptide ligand for alternative receptor binding and the abolition of the original receptor binding (Girod et al., 1999; Medina-Kauwe, 2003; Rentsendorj et al., 2006; Campos and Barry, 2007). This approach is limited because the introduction of a peptide can alter the structure and function of the capsid or envelope molecule of the vector and/or the inserted peptide itself. This is especially true not only for the small but highly structured rAAV Cap but also for retroviruses. For the latter, binding and entry are highly regulated by viral surface glycoproteins, which are difficult to modify without creating deleterious effects on infectivity at the stage of membrane fusion. This could explain why direct targeting using specific ligand-target cell receptor binding met with limited success for retroviruses. Instead of targeting the vector to cells bearing the receptor to the introduced ligand, this addition turned out to inhibit the entry of vectors into these cells. Nevertheless, this inability to induce membrane fusion and the sequestration of the targeted receptor-bound retroviral particles could be exploited in inverse targeting strategies. Direct targeting using pH-dependent glycoproteins (such as, influenza virus haemagglutinin, HA) provides an alternative basis for targeting strategies. Retroviruses generated with epidermal growth factor–HA (EGF–HA) (a surface glycoprotein chimera consisting of a fusion protein between HA and EGF) were as infectious as particles carrying only HA for EGF receptor (EFGR)-positive target cells, indicating that the fusion properties of this chimeric glycoprotein were not impaired in this case (Hatziioannou et al., 1999).
Indirect/inverse targeting is a strategy that exploits receptor-mediated virus neutralization. As an example, retroviral vectors displaying EGF fused to retroviral murine leukaemia virus amphotropic strain (MLV-A) glycoproteins could efficiently bind EGFR-positive target cells, but they could not infect them, in contrarst to EGFR-negative target cells. This inverse targeting provides a useful way to de-target the liver (EGFR-positive) (Peng et al., 2001) or stem cells (c-KIT-positive) (Fielding et al., 1998). Thus, it could be used to ameliorate specificity of the vectors but may lead to decreased bioavailability by the mechanism of sequestration. Although potential applications are limited, it may be used to minimize the risk of inadvertent transduction of contaminating cancer cells that express abundant EGFRs. In addition, the re-targeting could be useful for systemic application in the case of rAAV Caps as in Ad serotype 5 vectors (Nicklin et al., 2005; Denby et al., 2007)
Protease targeting, requiring the modification of glycoproteins, is a type of indirect targeting that may be able to exploit and capitalize inverse targeting. Peptide substrates cleaved by cell-surface-specific proteases have been inserted between the displayed virion sequestration ligand and the viral glycoprotein. In a first step, the vector attaches to the cells through the displayed binding domain and then the protease-sensitive linker is cleaved by a specific protease. The underlying glycoprotein is thus exposed and can interact with its receptor and promote virus entry (Peng et al., 1997 1998 1999, 2001; Szecsi et al., 2006).
For some vectors, it is also possible to combine different targeting strategies, for example, direct targeting and pseudotyping. In general, lentiviral vectors are able to deliver effectively their transgene in non-proliferative cells, provided the latter cells are metabolically active. Much research is aimed at finding the conditions for minimal activation of blood cells and their precursors so that they preserve their phenotypical characteristics. Currently, these cells are activated by soluble cytokines. A vector particle carrying the activating molecules would allow the bypassing of this phase of activation by the soluble compounds and would ideally allow vector injections in vivo directly in bone marrow, for example. Several studies showed that it is possible to pseudotype lentivectors with entry glycoproteins (RD114 or VSV-G) and incorporate activators of T lymphocytes (anti-CD3 or interleukin-7) or haematopoietic stem cells (stem cell factor, and thrombopoietin) on vector particles by grafting them with a modified viral glycoprotein derived from murine leukaemia virus or HA (Verhoeyen et al., 2005). The stem cells or T cells are effectively activated and transduced by these vector particles (Maurice et al., 1999 2002; Verhoeyen et al., 2003). The activation of the T cells is minimal, thus making it possible to preserve the phenotype of these cells.
Transgene expression and maintenance – transcriptional targeting
Choosing the appropriate promoter
Once a gene is transferred to the desired target cells, to exert efficient therapeutic effect, it should be expressed at an appropriate level and time. The use of different types of promoters can be envisioned to regulate transgene expression: (i) high-level and constitutive viral promoters, (ii) inducible promoters, (iii) tissue-specific promoters or (iv) endogenous promoters, including the complete genomic locus and other regulatory sequences.
High-level expression from constitutive viral promoters is often used when there is no need for tightly regulated expression, as is the case in most vaccine approaches where the goal is a strong expression of the transgene. This is also the case for proof-of-principle preliminary studies. A first level of transgene expression regulation is possible using regulation expression systems (for a review see Goverdhana et al., 2005). An ability to switch transgene expression on or off on demand would not only be desirable for tight control of the regimen but may also limit side effects.
The tetracycline-based transcription regulation system is the most commonly used, and has already been tested in retroviral, lentiviral vectors, rAAV and HSV vectors, yielding successful gene regulation (for review see Goverdhana et al., 2005). This system may be preferred to others for clinical gene therapy, as its regulation is well characterized and that the inducer is a well-used and well-characterized antibiotic. Other interesting regulation strategies are envisioned, such as X-ray radiation-induced promoters to breast cancer, hypoxia-driven promoters against myocardial ischaemia or glucose-responsive elements in the case of diabetes.
In contrast, one way to permit only switch-off transgene expression is to use an excisable transgene. Recombination systems, such as the Cre–loxP nuclease design, have already been attempted in lentiviral vectors (Delenda, 2004).
The use of tissue-specific promoters is also a very attractive strategy because it allows an additional level of cell targeting known as transcriptional targeting (Beck et al., 2004; Saukkonen and Hemminki, 2004). This can limit possible side effects if the transgene should be transferred to neighbouring cells by the bystander effect. Designing this type of promoter requires detailed knowledge of transcription regulatory sequences, which is becoming available for an increasing number of genes. When gene therapy is used to correct a genetic defect, the best regulatory sequences for the replacing transgene would of course be the native promoter and upstream and downstream flanking sequence. When compared with vectors expressing complementary DNA using heterologous promoters, this should result in improvements in expression at physiological levels, expression of multiple splice variants and correct tissue specificity. Such a strategy can, however, only be envisioned using high-capacity vectors, such as helper-dependent Ad (37 kb maximum) or HSV amplicons (100 kb maximum). Such amplicons for example showed good physiological expression of the human low-density lipoprotein receptor gene in proliferating cells (Wade-Martins et al., 2003). In contrast to adenovirus-and HSV-1-derived vectors, a major shortcoming of rAAV vector is its limited 5 kb packaging capacity, which prohibits its use for such an approach. The increasing knowledge available for gene transcription regulatory sequences and the refinement of inducible techniques are currently opening a wealth of possibilities. Nevertheless, one solution to the low delivery of rAAV vectors could be overcome using transpliced and overlapping vectors (Ghosh and Duan, 2007; Ghosh et al., 2007 2008).
Stability of transgene expression
Adenovirus-and herpes virus-derived vectors are typically short-term expression vectors, whereas rAAV and retroviral vectors allow for long-term expression. Short-lived expression from the former vectors is due to their non-integrative nature and due to the immune response that they induce, which ultimately leads to the loss of transgene expressing cells. Improving production techniques to eliminate all contamination with highly immunostimulatory helper viruses has been demonstrated to increase the length of expression (Cuchet et al., 2007). The recent emergence of adenoviral vectors for stable correction of genetic disorders is discussed by Jager and Ehrhardt (2007). As prolonged expression is not always essential to obtain a therapeutic effect (for example, in some anti-cancer or vaccine strategies), these vectors may, nevertheless, be an important component of the therapeutic arsenal. Integration-competent vectors are, in contrast, the tools of choice when genetic correction needs to be achieved in cells that differentiate from precursor cells after active proliferation. The ability of wild-type AAV to achieve site-specific integration raised considerable interest in this virus as a vector for long-term gene correction. Unfortunately, this ability is lost in vectors due to the elimination of Rep proteins (Ponnazhagan et al., 1997). rAAV persist in cells mainly in the episomal form and were demonstrated to be able to induce life-long expression of a transgene in different tissues despite their generally non-integrative nature (Duan et al., 1998; Nakai et al., 2001). rAAVs are, nevertheless, capable of low levels of integration in the genome and possible random integration is a concern, despite the high number of studies demonstrating their highly safe profile (Porteus et al., 2006; Kay, 2007).
Retrovirus-derived vectors are therefore the choice for long-term expression in dividing cells, because of their ability to integrate the transgene. However, integration per se is not a guarantee of stable expression, as integrated transgenes can be silenced over time (Ellis, 2005; Ellis and Yao, 2005). Moreover, integration can occur in unwanted locations. Integrations that disrupt some essential genes can be distinguished from those that lead to oncogenic misregulations (Baum et al., 2006). The dramatic potential of first-generation oncoretroviral vectors to activate protooncogenes was unfortunately demonstrated in both the French and the British X-linked severe combined immunodeficiency gene therapy trials (ESGT, 2003; Gansbacher, 2003; Thrasher et al., 2006; Thrasher and Gaspar, 2007). Improvement of vector design to limit these severe adverse effects is an area of intense research. Firstly, in self-inactivated vectors, already routinely used in the lab and evaluated for clinical use (Thornhill et al., 2008), the intrinsic promoter activity of the long terminal repeat was suppressed to prevent unwanted activation of neighbouring genes (Montini et al., 2006; Schambach et al., 2007). self-inactivated vectors, in addition, cannot be further mobilized and this modification decreased transactivation of transgene on surrounding sequences (Yu et al., 1986; Modlich et al., 2006). Secondly, the so-called ‘insulator’ sequences can be used to isolate further the transgene from the influence of neighbouring sequences. β-LCR sequences discovered far upstream of the human β-globin cluster have been reported to confer position-independent and copy number-dependent expression (Chung et al., 1993). This region, originally 20 kb long, has been reduced to forms of only a few hundred base pairs that still confer the silencing modulation activity (Chung et al., 1997). Several chromatin insulator boundary elements have also been described to protect expression cassettes from position effects (Rivella et al., 2000). These insulators were added in retroviral vectors and seem to be efficient at isolating the transgene cassette from the genomic surrounding sequences and vice versa. This not only protects the genome from unwanted gene expression alteration but also the transgene from silencing, which is often observed over time (Rivella et al., 2000). This silencing has been attributed to de novo cytosine methylation and histone deacetylation, and ultimately leads to chromatin condensation. Nevertheless, these insulators cannot circumvent mutational insertions. They may protect the genome from the insertional activation of proto-oncogenes, but they will not protect against the insertional inactivation of tumour suppressors genes. Although it may not be possible to completely eliminate risk, an additional strategy to limit the consequences of oncogenesis is to engineer suicide genes into the vector backbone to provide an inducible self-destruction mechanism (Uchiyama et al., 2006).
The ultimate strategy to limit integration side effects would be to control the sites of integration. Hybrid HSV-1/AAV amplicon vectors, for example, have been developed to try to combine the large transgene capacity of HSV-1-derived amplicons, which provides Rep activity to the rAVV vectors with the potential for site-specific genomic integration and stable transgene expression of AAV. These chimeric vectors have been shown to support transgene expression for significantly longer periods than standard HSV-1 amplicons (Cortes et al., 2008). Moreover, HSV/AAV hybrid vectors may mediate site-specific integration at the AAV integration site on human chromosome 19 at a relatively high rate, although random integration has also been observed (see Glauser et al., 2006). Concerning retroviruses, considerable attention was suddenly focused on integration sites of lenti-and onco-retroviral vectors after the repeated development of leukaemia in X-linked severe combined immunodeficiency clinical trials, as we mentioned earlier. The dogma that integration of these vectors was a random process has now been disproved. It has been possible to define a weak consensus target sequence for human immunodeficiency virus and avian leukosis virus (Weidhaas et al., 2000; Bushman, 2002a b). Recently, integration sites of retroviral and lentiviral vectors were mapped in CD34+ cells (Cattoglio et al., 2007). These investigations are very important for the estimation of integrative vector genotoxicity. To date, it is believed that lentiviruses (human immunodeficiency virus) integrate preferentially within the active transcription units, whereas γ-retroviruses (murine leukaemia virus) favour transcription start sites, and that integrase is the principle viral determinant of integration specificity.
Currently, two methods for targeting transgene integration of retroviral vectors are envisioned, they are: the use of (i) homologous recombination and (ii) the specific integration properties of C31 phage (Ginsburg and Calos, 2005).
The spontaneous rate of homologous recombination in mammalian cells is currently too low to be considered for gene therapy purposes. It would therefore be necessary to increase the rate of homologous recombination to levels that might be of therapeutic interest. Interesting results have already been obtained with rAAV (Vasileva and Jessberger, 2005; Miller et al., 2006). Some sequence-specific endonucleases (meganucleases) could also be used to induce site-specific recombination and thus the integration of transgene (non-integrative lentiviral vectors) (Paques and Duchateau, 2007). The sequence specificity of these endonucleases could be redesigned (Ashworth et al., 2006). The phage C31 integrase catalyses efficient, site-specific recombination between two relatively short sequences. Some encouraging studies showed efficient and stable genomic integration using vectors encoding for this integrase (Held et al., 2005; Ishikawa et al., 2006). Unfortunately, studies examining the integration specificity activity of C31 integrase for the human genome estimated between 100 and 1000 pseudoattP sites. There is concern that C31 could catalyse recombination between these pseudo-attP sites, leading to genomic instability (Chalberg et al., 2006). It might be possible to restrict integration into a smaller subpopulation of these sites by directed mutagenesis of the integrase (Sclimenti et al., 2001); this approach has been exploited in non-viral delivery systems. Incorporating the C31 integrase system into a viral vector is a possible next step. Last but not least, targeting lentiviral integration by fusion integrase (IN) and sequence-specific DNA-binding protein has already been reported (Tan et al., 2006).
Conclusions and perspectives
The choice of the appropriate vector for a specific gene transfer application requires the careful consideration of several parameters, including, as discussed in this review, production processes and stability constraints, the eventual need for long-term or transient expression and for the regulation of transgene expression. Delivery procedures and availability of target cells upon delivery are also important decision factors and will influence the spread of vector particles. Similar to HSV-based vectors, rAAV vectors have the ability to efficiently transduce neurons, yet delivery procedures are very different for either vector. Particularly, HSV-derived vectors can reach the CNS via vector delivery to peripheral neurons. Similarly, although most viral vectors can be directly injected in blood, tumours or muscle, Ad and rAAV vectors can also be delivered by inhalation.
Since the early 1980s, the field of gene transfer has experienced rapid development and continuous progress. Nevertheless, considerable research effort is still needed to achieve clinical applications of gene transfer vectors. Despite undeniable recent successes and important clinical benefits, such as correction of X-linked and ADA deficiencies by γ-retroviral vectors and in haemophilia B treatment by rAAV2, some fatal adverse events have been observed with most vectors (Ad, rAAV and retroviral vectors). Thus, improving the design and biosafety of the vectors is a critical challenge for the future. Similarly, careful investigation of vector properties will require a greater understanding of basic virology, as well as immunological and genetic interaction to rule out or eliminate unpredicted events. We should remember that the initial conceptual problem associated with the use of retroviral vectors as therapeutic agents of gene therapy was the isolation of replication-competent retroviruses. Now, we have to understand and deal with the events associated with insertional mutagenesis induced by replication-defective vectors.
 |
Acknowledgments
We apologise to all our colleagues whose work could not be cited due to the brevity of this review. We thank Anna Salvetti for critical reading of this manuscript. Our work is supported by INSERM, ENS Lyon and UCB Lyon-I, and by grants from the Ligue Nationale Contre le Cancer, the European Community (contract LSHB-CT-2004-005242 ‘CONSERT’) and the Agence Nationale de Recherches sur le SIDA et les Hépatites Virales (ANRS). DB was supported by a fellowship from the Association Française contre les Myopathies (AFM).
Glossary
Abbreviations
- AAV
adeno-associated virus
- Ad
adenovirus
- Cap
AAV capsid protein
- EGF
epidermal growth factor
- EGFR
EGF receptor
- HA
haemagglutinin (glycoprotein of influenza virus)
- HSV-1
herpes simplex virus-1
- rAAV
recombinant adeno-associated virus
- RD114
RD114 gp
- Rep
AAV replication protein
- VSV-G
G glycoprotein of vesicular stomatitis virus
Conflict of interest
The authors state no conflict of interest.
References
- Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- Andreadis S, Roth C, Doux JL, Morgan J, Yarmush M. Large-scale processing of recombinant retroviruses for gene therapy. Biotechnol Prog. 1999;15:1–11. doi: 10.1021/bp980106m. [DOI] [PubMed] [Google Scholar]
- Ashworth J, Havranek JJ, Duarte CM, Sussman D, Monnat RJ, Jr, Stoddard BL, et al. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17:253–263. doi: 10.1089/hum.2006.17.253. [DOI] [PubMed] [Google Scholar]
- Beck C, Uramoto H, Boren J, Akyurek LM. Tissue-specific targeting for cardiovascular gene transfer. Potential vectors and future challenges. Curr Gene Ther. 2004;4:457–467. doi: 10.2174/1566523043346138. [DOI] [PubMed] [Google Scholar]
- Blouin V, Brument N, Toublanc E, Raimbaud I, Moullier P, Salvetti A. Improving rAAV production and purification: towards the definition of a scaleable process. J Gene Med. 2004;6(Suppl)(1):S223–S228. doi: 10.1002/jgm.505. [DOI] [PubMed] [Google Scholar]
- Bouard D, Sandrin V, Boson B, Negre D, Thomas G, Granier C, et al. An acidic cluster of the cytoplasmic tail of the RD114 virus glycoprotein controls assembly of retroviral envelopes. Traffic. 2007;8:835–847. doi: 10.1111/j.1600-0854.2007.00581.x. [DOI] [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman F. Constructing the vertebrate genome: evidence from eels that LINEs mobilize SINEs. Mol Cell. 2002a;10:961–962. doi: 10.1016/s1097-2765(02)00747-5. [DOI] [PubMed] [Google Scholar]
- Bushman FD. Integration site selection by lentiviruses: biology and possible control. Curr Top Microbiol Immunol. 2002b;261:165–177. doi: 10.1007/978-3-642-56114-6_8. [DOI] [PubMed] [Google Scholar]
- Campos SK, Barry MA. Current advances and future challenges in adenoviral vector biology and targeting. Curr Gene Ther. 2007;7:189–204. doi: 10.2174/156652307780859062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, et al. Hot spots of retroviral integration in human CD34 + hematopoietic cells. Blood. 2007;110:1770–1778. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- Chalberg TW, Portlock JL, Olivares EC, Thyagarajan B, Kirby PJ, Hillman RT, et al. Integration specificity of phage phiC31 integrase in the human genome. J Mol Biol. 2006;357:28–48. doi: 10.1016/j.jmb.2005.11.098. [DOI] [PubMed] [Google Scholar]
- Chung JH, Bell AC, Felsenfeld G. Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci USA. 1997;94:575–580. doi: 10.1073/pnas.94.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JH, Whiteley M, Felsenfeld G. A 50 element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993;74:505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- Cortes ML, Oehmig A, Saydam O, Sanford JD, Perry KF, Fraefel C, et al. Targeted integration of functional human ATM cDNA into genome mediated by HSV/AAV hybrid amplicon vector. Mol Ther. 2008;16:81–88. doi: 10.1038/sj.mt.6300338. [DOI] [PubMed] [Google Scholar]
- Cosset FL, Takeuchi Y, Battini JL, Weiss RA, Collins MK. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J Virol. 1995;69:7430–7436. doi: 10.1128/jvi.69.12.7430-7436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croyle MA, Callahan SM, Auricchio A, Schumer G, Linse KD, Wilson JM, et al. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J Virol. 2004;78:912–921. doi: 10.1128/JVI.78.2.912-921.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croyle MA, Le HT, Linse KD, Cerullo V, Toietta G, Beaudet A, et al. PEGylated helper-dependent adenoviral vectors: highly efficient vectors with an enhanced safety profile. Gene Ther. 2005;12:579–587. doi: 10.1038/sj.gt.3302441. [DOI] [PubMed] [Google Scholar]
- Cuchet D, Potel C, Thomas J, Epstein AL. HSV-1 amplicon vectors: a promising and versatile tool for gene delivery. Expert Opin Biol Ther. 2007;7:975–995. doi: 10.1517/14712598.7.7.975. [DOI] [PubMed] [Google Scholar]
- Delenda C. Lentiviral vectors: optimization of packaging, transduction and gene expression. J Gene Med. 2004;6(Suppl)(1):S125–S138. doi: 10.1002/jgm.501. [DOI] [PubMed] [Google Scholar]
- Denby L, Work LM, Seggern DJ, Wu E, McVey JH, Nicklin SA, et al. Development of renal-targeted vectors through combined in vivo phage display and capsid engineering of adenoviral fibers from serotype 19p. Mol Ther. 2007;15:1647–1654. doi: 10.1038/sj.mt.6300214. [DOI] [PubMed] [Google Scholar]
- DePolo NJ, Harkleroad CE, Bodner M, Watt AT, Anderson CG, Greengard JS, et al. The resistance of retroviral vectors produced from human cells to serum inactivation in vivo and in vitro is primate species dependent. J Virol. 1999;73:6708–6714. doi: 10.1128/jvi.73.8.6708-6714.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePolo NJ, Reed JD, Sheridan PL, Townsend K, Sauter SL, Jolly DJ, et al. VSV-G pseudotyped lentiviral vector particles produced in human cells are inactivated by human serum. Mol Ther. 2000;2:218–222. doi: 10.1006/mthe.2000.0116. [DOI] [PubMed] [Google Scholar]
- Desmaris N, Bosch A, Salaun C, Petit C, Prevost MC, Tordo N, et al. Production and neurotropism of lentivirus vectors pseudo-typed with lyssavirus envelope glycoproteins. Mol Ther. 2001;4:149–156. doi: 10.1006/mthe.2001.0431. [DOI] [PubMed] [Google Scholar]
- Dong J, Roth MG, Hunter E. A chimeric avian retrovirus containing the influenza virus hemagglutinin gene has an expanded host range. J Virol. 1992;66:7374–7382. doi: 10.1128/jvi.66.12.7374-7382.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, et al. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther. 2005;16:1241–1246. doi: 10.1089/hum.2005.16.1241. [DOI] [PubMed] [Google Scholar]
- Ellis J, Yao S. Retrovirus silencing and vector design: relevance to normal and cancer stem cells? Curr Gene Ther. 2005;5:367–373. doi: 10.2174/1566523054546233. [DOI] [PubMed] [Google Scholar]
- ESGT, Board of Directors of (BoDo) French gene therapy group reports on the adverse event in a clinical trial of gene therapy for X-linked severe combined immune deficiency (X-SCID). Position statement from the European Society of Gene Therapy. J Gene Med. 2003;5:82–84. doi: 10.1002/jgm.364. [DOI] [PubMed] [Google Scholar]
- Eto Y, Gao JQ, Sekiguchi F, Kurachi S, Katayama K, Maeda M, et al. PEGylated adenovirus vectors containing RGD peptides on the tip of PEG show high transduction efficiency and antibody evasion ability. J Gene Med. 2005;7:604–612. doi: 10.1002/jgm.699. [DOI] [PubMed] [Google Scholar]
- Fielding AK, Maurice M, Morling FJ, Cosset FL, Russell SJ. Inverse targeting of retroviral vectors: selective gene transfer in a mixed population of hematopoietic and nonhematopoietic cells. Blood. 1998;91:1802–1809. [PubMed] [Google Scholar]
- Gansbacher B. Report of a second serious adverse event in a clinical trial of gene therapy for X-linked severe combined immune deficiency (X-SCID). Position of the European Society of Gene Therapy (ESGT) J Gene Med. 2003;5:261–262. doi: 10.1002/jgm.390. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Duan D. Expanding adeno-associated viral vector capacity: a tale of two vectors. Biotechnol Genet Eng Rev. 2007;24:165–177. doi: 10.1080/02648725.2007.10648098. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Lai Y, Duan D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol Ther. 2008;16:124–130. doi: 10.1038/sj.mt.6300322. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Long C, Bostick B, Duan D. Efficient whole-body transduction with trans-splicing adeno-associated viral vectors. Mol Ther. 2007;15:750–755. doi: 10.1038/sj.mt.6300081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg DS, Calos MP. Site-specific integration with phiC31 integrase for prolonged expression of therapeutic genes. Adv Genet. 2005;54:179–187. doi: 10.1016/S0065-2660(05)54008-2. [DOI] [PubMed] [Google Scholar]
- Girod A, Ried M, Wobus C, Lahm H, Leike K, Kleinschmidt J, et al. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat Med. 1999;5:1438. doi: 10.1038/71021. [DOI] [PubMed] [Google Scholar]
- Glauser DL, Ackermann M, Saydam O, Fraefel C. Chimeric herpes simplex virus/adeno-associated virus amplicon vectors. Curr Gene Ther. 2006;6:315–324. doi: 10.2174/156652306777592090. [DOI] [PubMed] [Google Scholar]
- Goverdhana S, Puntel M, Xiong W, Zirger JM, Barcia C, Curtin JF, et al. Regulatable gene expression systems for gene therapy applications: progress and future challenges. Mol Ther. 2005;12:189–211. doi: 10.1016/j.ymthe.2005.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Kleinschmidt JA. Progress in adeno-associated virus type 2 vector production: promises and prospects for clinical use. Hum Gene Ther. 1999;10:2445–2450. doi: 10.1089/10430349950016799. [DOI] [PubMed] [Google Scholar]
- Guibinga GH, Friedmann T. Baculovirus GP64-pseudotyped HIV-based lentivirus vectors are stabilized against complement inactivation by codisplay of decay accelerating factor (DAF) or of a GP64-DAF fusion protein. Mol Ther. 2005;11:645–651. doi: 10.1016/j.ymthe.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Guibinga GH, Hall FL, Gordon EM, Ruoslahti E, Friedmann T. Ligand-modified vesicular stomatitis virus glycoprotein displays a temperature-sensitive intracellular trafficking and virus assembly phenotype. Mol Ther. 2004;9:76–84. doi: 10.1016/j.ymthe.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- Hatziioannou T, Delahaye E, Martin F, Russell SJ, Cosset F-L. Retroviral display of functional binding domains fused to the amino-terminus of influenza haemagglutinin. Hum Gene Ther. 1999;10:1533–1544. doi: 10.1089/10430349950017860. [DOI] [PubMed] [Google Scholar]
- Hatziioannou T, Valsesia-Wittmann S, Russell S, Cosset F-L. Incorporation of fowl plague virus hemagglutinin into murine leukemia virus particles and analysis of the infectivity of the pseudotyped retroviruses. J Virol. 1998;72:5313–5317. doi: 10.1128/jvi.72.6.5313-5317.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Huang KS, Lo WH, Chung YC, Lai YK, Chen CY, Chou ST, et al. Combination of baculovirus-mediated gene delivery and packed-bed reactor for scalable production of adeno-associated virus. Hum Gene Ther. 2007;18:1161–1170. doi: 10.1089/hum.2007.107. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Tanaka N, Murakami K, Uchiyama T, Kumaki S, Tsuchiya S, et al. Phage phiC31 integrase-mediated genomic integration of the common cytokine receptor gamma chain in human T-cell lines. J Gene Med. 2006;8:646–653. doi: 10.1002/jgm.891. [DOI] [PubMed] [Google Scholar]
- Jager L, Ehrhardt A. Emerging adenoviral vectors for stable correction of genetic disorders. Curr Gene Ther. 2007;7:272–283. doi: 10.2174/156652307781369074. [DOI] [PubMed] [Google Scholar]
- Kaptein LC, Greijer AE, Valerio D, van Beusechem VW. Optimized conditions for the production of recombinant amphotropic retroviral vector preparations. Gene Ther. 1997;4:172–176. doi: 10.1038/sj.gt.3300373. [DOI] [PubMed] [Google Scholar]
- Kay MA. AAV vectors and tumorigenicity. Nat Biotechnol. 2007;25:1111–1113. doi: 10.1038/nbt1007-1111. [DOI] [PubMed] [Google Scholar]
- Kelly PF, Vandergriff J, Nathwani A, Nienhuis AW, Vanin EF. Highly efficient gene transfer into cord blood nonobese diabetic/severe combined immunodeficiency repopulating cells by oncoretroviral vector particles pseudotyped with the feline endogenous retrovirus (RD114) envelope protein. Blood. 2000;96:1206–1214. [PubMed] [Google Scholar]
- Kobinger GP, Weiner DJ, Yu QC, Wilson JM. Filoviruspseudotyped lentiviral vector can efficiently and stably transduce airway epithelia in vivo. Nat Biotechnol. 2001;19:225–230. doi: 10.1038/85664. [DOI] [PubMed] [Google Scholar]
- Kotani H, Newton PB, III, Zhang S, Chiang YL, Otto E, Weaver L, et al. Improved methods of retroviral vector transduction and production for gene therapy. Hum Gene Ther. 1994;5:19–28. doi: 10.1089/hum.1994.5.1-19. [DOI] [PubMed] [Google Scholar]
- Lee SG, Kim S, Robbins PD, Kim BG. Optimization of environmental factors for the production and handling of recombinant retrovirus. Appl Microbiol Biotechnol. 1996;45:477–483. doi: 10.1007/BF00578459. [DOI] [PubMed] [Google Scholar]
- Loewen N, Barraza R, Whitwam T, Saenz DT, Kemler I, Poeschla EM. FIV vectors. Methods Mol Biol. 2003a;229:251–271. doi: 10.1385/1-59259-393-3:251. [DOI] [PubMed] [Google Scholar]
- Loewen N, Leske DA, Chen Y, Teo WL, Saenz DT, Peretz M, et al. Comparison of wild-type and class I integrase mutant-FIV vectors in retina demonstrates sustained expression of integrated transgenes in retinal pigment epithelium. J Gene Med. 2003b;5:1009–1017. doi: 10.1002/jgm.447. [DOI] [PubMed] [Google Scholar]
- Lyons M, Onion D, Green NK, Aslan K, Rajaratnam R, Bazan-Peregrino M, et al. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol Ther. 2006;14:118–128. doi: 10.1016/j.ymthe.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Maurice M, Mazur S, Bullough FJ, Salvetti A, Collins MKL, Russell SJ, et al. Efficient gene delivery to quiescent IL2-dependent cells by murine leukemia virus-derived vectors harboring IL2 chimeric envelopes glycoproteins. Blood. 1999;94:401–410. [PubMed] [Google Scholar]
- Maurice M, Verhoeyen E, Salmon P, Trono D, Russell SJ, Cosset F-L. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood. 2002;99:2342–2350. doi: 10.1182/blood.v99.7.2342. [DOI] [PubMed] [Google Scholar]
- McTaggart S, Al-Rubeai M. Retroviral vectors for human gene delivery. Biotechnol Adv. 2002;20:1–31. doi: 10.1016/s0734-9750(01)00087-8. [DOI] [PubMed] [Google Scholar]
- Medina-Kauwe LK. Endocytosis of adenovirus and adenovirus capsid proteins. Adv Drug Deliv Rev. 2003;55:1485–1496. doi: 10.1016/j.addr.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Merten OW. State-of-the-art of the production of retroviral vectors. J Gene Med. 2004;6(Suppl)(1):S105–S124. doi: 10.1002/jgm.499. [DOI] [PubMed] [Google Scholar]
- Miletic H, Bruns M, Tsiakas K, Vogt B, Rezal R, Baum C, et al. Retroviral vectors pseudotyped with lymphocytic choriomeningitis virus. J Virol. 1999;73:6114–6116. doi: 10.1128/jvi.73.7.6114-6116.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DG, Wang PR, Petek LM, Hirata RK, Sands MS, Russell DW. Gene targeting in vivo by adeno-associated virus vectors. Nat Biotechnol. 2006;24:1022–1026. doi: 10.1038/nbt1231. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2007;7:316–324. doi: 10.2174/156652307782151425. [DOI] [PubMed] [Google Scholar]
- Mochizuki H, Schwartz JP, Tanaka K, Brady RO, Reiser J. High-titer human immunodeficiency virus type 1-based vector systems for gene delivery into nondividing cells. J Virol. 1998;72:8873–8883. doi: 10.1128/jvi.72.11.8873-8883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knoss S, Schambach A, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108:2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok H, Palmer DJ, Ng P, Barry MA. Evaluation of polyethylene glycol modification of first-generation and helper-dependent adenoviral vectors to reduce innate immune responses. Mol Ther. 2005;11:66–79. doi: 10.1016/j.ymthe.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- Morizono K, Bristol G, Xie Y-M Kung SK-P, Chen ISC. Antibody-directed targeting of retroviral vectors via cell surface antigens. J Virol. 2001;75:8016–8020. doi: 10.1128/JVI.75.17.8016-8020.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nicklin SA, Wu E, Nemerow GR, Baker AH. The influence of adenovirus fiber structure and function on vector development for gene therapy. Mol Ther. 2005;12:384–393. doi: 10.1016/j.ymthe.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Nunes FA, Furth EE, Wilson JM, Raper SE. Gene transfer into the liver of nonhuman primates with E1-deleted recombinant adenoviral vectors: safety of readministration. Hum Gene Ther. 1999;10:2515–2526. doi: 10.1089/10430349950016852. [DOI] [PubMed] [Google Scholar]
- Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- Parks RJ, Chen L, Anton M, Sankar U, Rudnicki MA, Graham FL. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci USA. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng KW, Morling FJ, Cosset FL, Murphy G, Russell SJ. A gene delivery system activatable by disease-associated matrix metalloproteinases. Hum Gene Ther. 1997;8:729–738. doi: 10.1089/hum.1997.8.6-729. [DOI] [PubMed] [Google Scholar]
- Peng KW, Morling FJ, Cosset F-L, Russell SJ. A retroviral gene delivery system activatable by plasmin. Tumor Target. 1998;3:112–120. [Google Scholar]
- Peng KW, Pham L, Ye H, Zufferey R, Trono D, Cosset FL, et al. Organ distribution of gene expression after intravenous infusion of targeted and untargeted lentiviral vectors. Gene Ther. 2001;8:1456–1463. doi: 10.1038/sj.gt.3301552. [DOI] [PubMed] [Google Scholar]
- Peng KW, Vile R, Cosset FL, Russell S. Selective transduction of protease-rich tumors by matrix-metalloproteinase-targeted retroviral vectors. Gene Ther. 1999;6:1552–1557. doi: 10.1038/sj.gt.3300982. [DOI] [PubMed] [Google Scholar]
- Pensiero MN, Wysocki CA, Nader K, Kikuchi GE. Development of amphotropic murine retrovirus vectors resistant to inactivation by human serum. Hum Gene Ther. 1996;7:1095–1101. doi: 10.1089/hum.1996.7.9-1095. [DOI] [PubMed] [Google Scholar]
- Perabo L, Goldnau D, White K, Endell J, Boucas J, Humme S, et al. Heparan sulfate proteoglycan binding properties of adenoassociated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80:7265–7269. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnazhagan S, Erikson D, Kearns WG, Zhou SZ, Nahreini P, Wang XS, et al. Lack of site-specific integration of the recombinant adeno-associated virus 2 genomes in human cells. Hum Gene Ther. 1997;8:275–284. doi: 10.1089/hum.1997.8.3-275. [DOI] [PubMed] [Google Scholar]
- Porteus MH, Connelly JP, Pruett SM. A look to future directions in gene therapy research for monogenic diseases. PLoS Genet. 2006;2:e133. doi: 10.1371/journal.pgen.0020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Rentsendorj A, Xie J, MacVeigh M, Agadjanian H, Bass S, Kim DH, et al. Typical and atypical trafficking pathways of Ad5 penton base recombinant protein: implications for gene transfer. Gene Ther. 2006;13:821–836. doi: 10.1038/sj.gt.3302729. [DOI] [PubMed] [Google Scholar]
- Rethwilm A. Foamy virus vectors: an awaited alternative to gammaretro-and lentiviral vectors. Curr Gene Ther. 2007;7:261–271. doi: 10.2174/156652307781369092. [DOI] [PubMed] [Google Scholar]
- Rivella S, Callegari JA, May C, Tan CW, Sadelain M. The cHS4 insulator increases the probability of retroviral expression at random chromosomal integration sites. J Virol. 2000;74:4679–4687. doi: 10.1128/jvi.74.10.4679-4687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S, Lowenthal A, Terheggen HG, Columbo JP. Induction of arginase activity with the Shope papilloma virus in tissue culture cells from an argininemic patient. JExp Med. 1973;137:1091–1096. doi: 10.1084/jem.137.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans – immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- Saeki Y, Fraefel C, Ichikawa T, Breakefield XO, Chiocca EA. Improved helper virus-free packaging system for HSV amplicon vectors using an ICP27-deleted, oversized HSV-1 DNA in a bacterial artificial chromosome. Mol Ther. 2001;3:591–601. doi: 10.1006/mthe.2001.0294. [DOI] [PubMed] [Google Scholar]
- Sanders DA. Ebola virus glycoproteins: guidance devices for targeting gene therapy vectors. Expert Opin Biol Ther. 2004;4:329–336. doi: 10.1517/14712598.4.3.329. [DOI] [PubMed] [Google Scholar]
- Sandrin V, Boson B, Salmon P, Gay W, Negre D, Le Grand R, et al. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood. 2002;100:823–832. doi: 10.1182/blood-2001-11-0042. [DOI] [PubMed] [Google Scholar]
- Sandrin V, Muriaux D, Darlix JL, Cosset FL. Intracellular trafficking of Gag and Env proteins and their interactions modulate pseudotyping of retroviruses. J Virol. 2004;78:7153–7164. doi: 10.1128/JVI.78.13.7153-7164.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrin V, Russell SJ, Cosset FL. Targeting retroviral and lentiviral vectors. Curr Top Microbiol Immunol. 2003;281:137–178. doi: 10.1007/978-3-642-19012-4_4. [DOI] [PubMed] [Google Scholar]
- Saukkonen K, Hemminki A. Tissue-specific promoters for cancer gene therapy. Expert Opin Biol Ther. 2004;4:683–696. doi: 10.1517/14712598.4.5.683. [DOI] [PubMed] [Google Scholar]
- Schambach A, Galla M, Maetzig T, Loew R, Baum C. Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol Ther. 2007;15:1167–1173. doi: 10.1038/sj.mt.6300152. [DOI] [PubMed] [Google Scholar]
- Schauber-Plewa C, Simmons A, Tuerk MJ, Pacheco CD, Veres G. Complement regulatory proteins are incorporated into lentiviral vectors and protect particles against complement inactivation. Gene Ther. 2005;12:238–245. doi: 10.1038/sj.gt.3302399. [DOI] [PubMed] [Google Scholar]
- Sclimenti CR, Thyagarajan B, Calos MP. Directed evolution of a recombinase for improved genomic integration at a native human sequence. Nucleic Acids Res. 2001;29:5044–5051. doi: 10.1093/nar/29.24.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey CM, North CL, Kuhn RJ, Sanders DA. Ross River virus glycoprotein-pseudotyped retroviruses and stable cell lines for their production. J Virol. 2001;75:2653–2659. doi: 10.1128/JVI.75.6.2653-2659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A. Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J Virol. 2000;74:2567–2583. doi: 10.1128/jvi.74.6.2567-2583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimotohno K, Temin HM. Formation of infectious progeny virus after insertion of herpes simplex thymidine kinase gene into DNA of an avian retrovirus. Cell. 1981;26:67–77. doi: 10.1016/0092-8674(81)90034-9. [DOI] [PubMed] [Google Scholar]
- Sinn PL, Hickey MA, Staber PD, Dylla DE, Jeffers SA, Davidson BL, et al. Lentivirus vectors pseudotyped with filoviral envelope glycoproteins transduce airway epithelia from the apical surface independently of folate receptor alpha. J Virol. 2003;77:5902–5910. doi: 10.1128/JVI.77.10.5902-5910.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel M, Bitzer M, Schenk A, Rossmann H, Neubert WJ, Seidler U, et al. Pseudotype formation of moloney murine leukemia virus with sendai virus glycoprotein F. J Virol. 1998;72:5269–5302. doi: 10.1128/jvi.72.6.5296-5302.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung VM, Lai MM. Murine retroviral pseudotype virus containing hepatitis B virus large and small surface antigens confers specific tropism for primary human hepatocytes: a potential liver-specific targeting system. J Virol. 2002;76:912–917. doi: 10.1128/JVI.76.2.912-917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Suomalainen M, Garoff H. Incorporation of homologous and heterologous proteins into the envelope of Moloney murine leukemia virus. J Virol. 1994;68:4879–4889. doi: 10.1128/jvi.68.8.4879-4889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland S. Ebola glycoprotein: the key to successful gene therapy? Drug Discov Today. 2003;8:609–610. doi: 10.1016/s1359-6446(03)02774-0. [DOI] [PubMed] [Google Scholar]
- Szecsi J, Drury R, Josserand V, Grange MP, Boson B, Hartl I, et al. Targeted retroviral vectors displaying a cleavage site-engineered hemagglutinin (HA) through HA-protease interactions. Mol Ther. 2006;14:735–744. doi: 10.1016/j.ymthe.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Tabin CJ, Hoffmann JW, Goff SP, Weinberg RA. Adaptation of a retrovirus as a eucaryotic vector transmitting the herpes simplex virus thymidine kinase gene. Mol Cell Biol. 1982;2:426–436. doi: 10.1128/mcb.2.4.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Cosset FL, Lachmann PJ, Okada H, Weiss RA, Collins MKL. Type C retrovirus inactivation by human complement is determined by both the viral genome and producer cell. J Virol. 1994;68:8001–8007. doi: 10.1128/jvi.68.12.8001-8007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Dong Z, Wilkinson TA, Barbas CF, III, Chow SA. Human immunodeficiency virus type 1 incorporated with fusion proteins consisting of integrase and the designed polydactyl zinc finger protein E2C can bias integration of viral DNA into a predetermined chromosomal region in human cells. J Virol. 2006;80:1939–1948. doi: 10.1128/JVI.80.4.1939-1948.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornhill SI, Schambach A, Howe SJ, Ulaganathan M, Grassman E, Williams D, et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol Ther. 2008;16:590–598. doi: 10.1038/sj.mt.6300393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrasher AJ, Gaspar HB. Severe Adverse Event in Clinical Trial of Gene Therapy for X-SCID. http://www.esgct.org/upload/X-SCID_statement_AT.pdf.
- Thrasher AJ, Gaspar HB, Baum C, Modlich U, Schambach A, Candotti F, et al. Gene therapy: X-SCID transgene leukaemogenicity. Nature 443: E5–E6; discussion E6–E7. [DOI] [PubMed]
- Uchiyama T, Kumaki S, Ishikawa Y, Onodera M, Sato M, Du W, et al. Application of HSVtk suicide gene to X-SCID gene therapy: ganciclovir treatment offsets gene corrected X-SCID B cells. Biochem Biophys Res Commun. 2006;341:391–398. doi: 10.1016/j.bbrc.2005.12.199. [DOI] [PubMed] [Google Scholar]
- Vasileva A, Jessberger R. Precise hit: adeno-associated virus in gene targeting. Nat Rev Microbiol. 2005;3:837–847. doi: 10.1038/nrmicro1266. [DOI] [PubMed] [Google Scholar]
- Verhoeyen E, Cosset FL. Surface-engineering of lentiviral vectors. J Gene Med. 2004;6(Suppl)(1):S83–S94. doi: 10.1002/jgm.494. [DOI] [PubMed] [Google Scholar]
- Verhoeyen E, Dardalhon V, Ducrey-Rundquist O, Trono D, Taylor N, Cosset F-L. IL-7 surface-engineered lentiviral vectors promote survival and efficient gene transfer in resting primary T-lymphocytes. Blood. 2003;101:2167–2174. doi: 10.1182/blood-2002-07-2224. [DOI] [PubMed] [Google Scholar]
- Verhoeyen E, Wiznerowicz M, Olivier D, Izac B, Trono D, Dubart-Kupperschmitt A, et al. Novel lentiviral vectors displaying ‘early-acting cytokines’ selectively promote survival and transduction of NOD/SCID repopulating human hematopoietic stem cells. Blood. 2005;106:3386–3395. doi: 10.1182/blood-2004-12-4736. [DOI] [PubMed] [Google Scholar]
- Wade-Martins R, Saeki Y, Chiocca EA. Infectious delivery of a 135-kb LDLR genomic locus leads to regulated complementation of low-density lipoprotein receptor deficiency in human cells. Mol Ther. 2003;7:604–612. doi: 10.1016/s1525-0016(03)00060-1. [DOI] [PubMed] [Google Scholar]
- Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Gibson M, Spear PG, Scolnick EM. Construction and isolation of a transmissible retrovirus containing the src gene of Harvey murine sarcoma virus and the thymidine kinase gene of herpes simplex virus type 1. J Virol. 1981;39:935–944. doi: 10.1128/jvi.39.3.935-944.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidhaas JB, Angelichio EL, Fenner S, Coffin JM. Relationship between retroviral DNA integration and gene expression. J Virol. 2000;74:8382–8389. doi: 10.1128/jvi.74.18.8382-8389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol. 1998;72:3155–3160. doi: 10.1128/jvi.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Asokan A, Grieger JC, Govindasamy L, Agbandje-McKenna M, Samulski RJ. Single amino acid changes can influence titer, heparin binding, and tissue tropism in different adenoassociated virus serotypes. J Virol. 2006a;80:11393–11397. doi: 10.1128/JVI.01288-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006b;14:316–327. doi: 10.1016/j.ymthe.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Yu JH, Schaffer DV. Advanced targeting strategies for murine retroviral and adeno-associated viral vectors. Adv Biochem Eng Biotechnol. 2005;99:147–167. doi: 10.1007/10_006. [DOI] [PubMed] [Google Scholar]
- Yu SF, von Ruden T, Kantoff PW, Garber C, Seiberg M, Ruther U, et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc Natl Acad Sci USA. 1986;83:3194–3198. doi: 10.1073/pnas.83.10.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss AK, Muruve DA. Immunity to adeno-associated virus vectors in animals and humans: a continued challenge. Gene Ther. 2008;15:808–816. doi: 10.1038/gt.2008.54. [DOI] [PubMed] [Google Scholar]
- Zaupa C, Revol-Guyot V, Epstein AL. Improved packaging system for generation of high-level noncytotoxic HSV-1 amplicon vectors using Cre-loxP site-specific recombination to delete the packaging signals of defective helper genomes. Hum Gene Ther. 2003;14:1049–1063. doi: 10.1089/104303403322124774. [DOI] [PubMed] [Google Scholar]