

Abstract
Objective
Frontotemporal lobar degeneration (FTLD) is characterized by impairments in social, behavioral, and/or language function, but postmortem studies indicate that multiple neuropathological entities lead to FTLD. This study assessed whether specific clinical features predict the underlying pathology.
Methods
A clinicopathological correlation was performed on 90 consecutive patients with a pathological diagnosis of frontotemporal dementia and was compared with an additional 24 cases accrued during the same time period with a clinical diagnosis of FTLD, but with pathology not typically associated with frontotemporal dementia.
Results
Postmortem examination showed multiple pathologies including tauopathies (46%), FTLD with ubiquitin-positive inclusions (29%), and Alzheimer’s disease (17%). The pathological groups manifested some distinct demographic, clinical, and neuropsychological features, although these attributes showed only a statistical association with the underlying pathology. FTLD with ubiquitin-positive inclusions was more likely to present with both social and language dysfunction, and motor neuron disease was more likely to emerge in these patients. Tauopathies were more commonly associated with an extrapyramidal disorder. Alzheimer’s disease was associated with relatively greater deficits in memory and executive function.
Interpretation
Clinical and neuropsychological features contribute to delineating the spectrum of pathology underlying a patient diagnosed with FTLD, but biomarkers are needed that, together with the clinical phenotype, can predict the underlying neuropathology.
Frontotemporal lobar degeneration (FTLD) is characterized clinically by progressive changes in social, behavioral, and/or language function.1-3 In the frontal or social/dysexecutive variant of FTLD, there is an early change in social comportment and personality often associated with disinhibition, apathy, and lack of insight. In contrast, the aphasic variant of FTLD is further classified as either progressive nonfluent aphasia, characterized by effortful speech and phonemic errors,4 or semantic dementia, manifested by severe problems with naming and understanding word and object meaning.5 In both the social/dysexecutive and aphasic variants, there is relative preservation of memory, especially in early stages. In addition, some patients with FTLD manifest a motor disorder such as parkinsonism or motor neuron disease (MND).6,7
Multiple neuropathological abnormalities are associated with FTLD,3,8 and this pathological heterogeneity is reflected in a recent classification scheme from an frontotemporal dementia (FTD) work group that was the first to incorporate both immunohistochemical and biochemical data in the diagnostic algorithm (Fig 1).3 Two broad pathological subdivisions of FTD are recognized: brains with tau-positive pathology (tauopathies), and brains free of tau pathology (see Fig 1). The tauopathies are subdivided into several distinct entities, including Pick’s disease (PiD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain disease, tangle-predominant senile dementia, and patients with MAPT gene mutations collectively referred to as frontotemporal dementia with parkinsonism linked to chromosome 17, or FTDP-17. In contrast, brains from the remaining patients lack tau-positive inclusions and are further categorized by the presence or absence of ubiquitin-positive inclusions, designated frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U)9-11 and dementia lacking distinctive histopathology (DLDH), respectively.12-14 A new pathological entity recently was described in FTLD-U patients, designated neuronal intermediate filament inclusion disease, that is characterized by neuronal intermediate filament-positive aggregates in neurons of affected brain regions.15-18 It also should be emphasized that both Alzheimer’s disease (AD) pathology and cortical Lewy bodies formed by filamentous α-synuclein aggregates (dementia with Lewy bodies) may present with clinical features indistinguishable from the FTLD syndromes described earlier (see Fig 1).12,19,20
Fig 1.
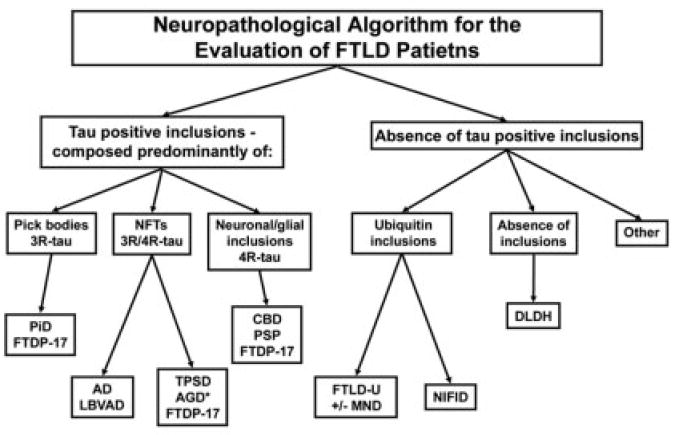
Algorithm for the neuropathological diagnosis of patients with frontotemporal lobar degeneration (FTLD). This algorithm represents an oversimplification of the scheme used to diagnose FTLD patients and does not reflect the complexity of the cases or the commonly observed occurrence of multiple pathologies. 3R-tau, 4R-tau, and 3R/4R-tau refer to the specific subsets of insoluble tau isoforms observed biochemically and characterized by either three or four microtubule binding repeats. AD = Alzheimer’ disease; AGD = argyrophilic grain disease; CBD = corticobasal degeneration; DLDH = dementia lacking distinctive histopathology; FTDP-17 = frontotemporal dementia with parkinsonism linked to chromosome 17; FTLD-U = frontotemporal lobar degeneration with ubiquitin-positive inclusions; LBVAD = Lewy body variant of Alzheimer’s disease; MND = motor neuron disease; NFT = neurofibrillary tangle; NIFID = neuronal intermediate filament inclusion dementia; PiD 3 Pick’s disease; PSP = progressive supranuclear palsy; TPSD = tangle-predominant senile dementia.
FTLD clinical syndromes have been associated with specific neuropathological disorders by some researchers,7 although this association is controversial.3 With the development of therapies that target the underlying pathology and cell biology of specific diseases, classification of patients with various pathological entities will become increasingly important. The clinical and pathological features have been described, but few studies have performed detailed clinicopathological correlations of large series of FTLD patients. In this study, we sought to determine the clinical and/or neuropsychological features that correlate with the underlying pathology in a large series of subjects with a neuropathological diagnosis of FTD who were evaluated over a 10-year period.
Materials and Methods
Subjects
Cases were derived from 510 consecutive patients neuropathologically evaluated from 1995 to 2005 at the University of Pennsylvania (UPenn) as part of a multidisciplinary research program investigating neurodegenerative diseases. Inclusion criteria included all dementia patients with a neuropathological diagnosis consistent with FTD (see Fig 1; n = 90).3 Subjects with PSP or CBD pathology were included only if there was documentation of dementia. In addition, for purposes of comparison, 24 additional subjects were included who met clinical criteria for FTLD,1,8 but with neuropathology not typically associated with FTD. The remaining 396 subjects included patients with non-FTLD dementias (n = 183; ie, AD, dementia with Lewy bodies, cerebrovascular disease), patients with movement disorders without dementia (n = 157; ie, Parkinson’s disease, multiple system atrophy, PSP without documentation of dementia, amyotrophic lateral sclerosis), and control subjects without dementia or a movement disorder (n = 56). Patients were clinically evaluated either at UPenn or at the University of California, San Francisco (UCSF), with the exception of eight FTLD patients who were referred to UPenn for neuropathological evaluation. Consent for autopsy was obtained from the patient or legal next of kin in accordance with Pennsylvania and California state law, as well as protocols approved by UPenn and UCSF Institutional Review Boards.
Neuropathological Classification of Frontotemporal Dementia Patients
To establish a neuropathological diagnosis, two board-certified neuropathologists (M.S.F., J.Q.T.) reviewed all cases, and we established diagnoses according to consensus criteria including the recent Workgroup on FTD and PiD (see Fig 1).3,21-25 Immunohistochemistry was performed using standard and previously published protocols with antibodies that detect phosphorylated tau (PHF126 [generously provided by Dr P. Davies]; β-amyloid [ie, 4G8; Senetek, Maryland Heights, MO]; α-synuclein [Syn30327]; ubiquitin [Chemicon International, Temecula, CA, and Dako Cytomation, Glostrup, Denmark], phosphorylated NF subunits [RMO2428]; and α-internexin [Zymed Laboratories, San Francisco, CA]). Semiquantitative methodologies similar to those described for senile plaques in AD29 (ie, absent, low, moderate, and high) were used to assess the density of senile plaques, as well as tau-, α-synuclein–, and ubiquitin-positive lesions. In instances where multiple distinct pathologies were present, a primary diagnosis was assigned based on the density and distribution of the observed pathology in relation to the clinical phenotype of the patient. Although a small segment of cervical spinal cord was available from most subjects, the entire spinal cord was available only in five cases including four of the six subjects with clinical MND. The availability of the entire spinal cord in only five subjects limited our ability to detect pathological evidence of MND. However, all subjects with clinical MND demonstrated motor neuron degeneration consistent with the pathological diagnosis of FTD-MND.3
Clinical Characterization of Frontotemporal Lobar Degeneration Subjects
Clinical diagnoses at UPenn or UCSF were based on published criteria1,3,8 and established using informant interview, neurological examinations, medical history, neuropsychological evaluation, laboratory screening, and brain imaging when available. Medical records were always obtained, and detailed chart reviews and data collection were performed blinded to neuropathological diagnosis. Accuracy of the clinical diagnosis was bolstered by regular (approximately annually) follow-up evaluations with longitudinal analysis of clinical data. However, due to the long-term nature of this study, more than half of the subjects initially were evaluated before 1998 using the Lund and Manchester clinical criteria8; in these cases, definitive FTLD subtypes as defined by Neary and colleagues1 were not available for analysis.
Symptoms at presentation and signs at first and last clinical evaluation were defined for purposes of data analysis. Presenting symptoms were categorized as follows: social and/or behavioral changes (eg, disinhibition, change in personality, apathy, decreased motivation, and psychiatric illness diagnosed later in life near the onset of symptoms); language dysfunction (eg, naming or word-finding difficulty, impaired comprehension, decline in speech fluency or grammar); other cognitive deficits (eg, memory loss, decline in activities of daily living, attention deficits, executive dysfunction or visuospatial complaints); movement disorders (eg, abnormal gait, falling, rigidity, apraxia, or dysarthria); and focal weakness. Specific clinical features from the neurological examination that were extracted and coded at two time points (onset and before death) included: social dysfunction (behavioral, personality, and psychiatric disorders), aphasia, extrapyramidal features, and pyramidal signs. A limited battery of cognitive tests that was available across referring sites and across the length of patient collection included: Mini-Mental Status Examination (MMSE), a survey measure of dementia severity (maximum score, 30); Boston Naming Test, a measure of confrontational naming for line drawings (percentage correctly named)30; Verbal Fluency, a measure of speech generation based on category naming in a defined time period (number of unique names produced in 60 seconds)31,32; Word List Recall, a measure of episodic memory that requires the recall of a series of words after a brief delay after 3 to 4 presentation trials (percentage correct of 10 words)33; and Digit Span Forward, a measure of selective attention and short-term memory that requires the repetition of a sequence of digits (length of correctly repeated digit span).34
Statistical Analysis
All statistical analyses were performed using SPSS (SPSS, Chicago, IL). Descriptive statistics were used to characterize the entire cohort, as well as each pathological subgroup. t tests were used to investigate differences in demographics and neuropsychological measures among the pathological subgroups. χ2 analyses were used to study the differences in clinical symptoms and signs.
Results
Frontotemporal Dementia Cohort
Among the entire cohort of 114 cases, a clinical diagnosis of FTLD was rendered in 86 (75.4%) patients. The clinical diagnoses in the other subjects with a neuropathological diagnosis of FTD included corticobasal syndrome (n = 16; 14.0%) and PSP (n = 3; 2.6%), which often are considered part of the FTLD spectrum of disorders.7,35 In addition, all subjects in our cohort who were diagnosed with corticobasal syndrome or PSP manifested dementia. The remaining patients were clinically diagnosed with AD (n = 6; 5.3%) or an unclassified dementia (n = 3; 2.6%). In 15 patients, more than 1 clinical diagnosis was made, including 6 who met clinical criteria for both FTLD and amyotrophic lateral sclerosis and 6 who were diagnosed with both FTLD and severe parkinsonism. The mean age at onset of symptoms for the cohort was 60.4 (range, 30–82) years, and the mean duration of disease was 89.1 (range, 17–300) months (Table 1). In contrast, the mean age at onset and disease duration of a well-characterized cohort of patients with a clinical and pathological diagnosis of AD (n = 80) observed at the UPenn AD Center during the same time period was 71.4 years and 108.0 months, respectively [t(189) = 7.99; p < 0.0001; and t(187) = 2.52; p < 0.01]. In addition, 50.9% of the patients were male, and the average years of education was 15.3 (range, 8 –23). A definite or probable family history of a neurodegenerative disease was identified in 17.0% of patients. The group with a definite family history includes five patients from four unrelated families with documented genetic mutations in the MAPT gene, and one patient with a mutation in the valosin-containing protein gene that is associated with the recently described disease “Inclusion body myopathy, Paget’s disease, and frontotemporal dementia.”36 Eleven subjects (9.6%) had a probable family history, indicating multiple first-degree relatives affected in multiple generations. An additional 30 patients (30.0%) had a possible family history defined as at least one affected first-degree relative. In summary, the demographics of our FTD cohort are similar to other large studies that used similar clinical criteria.7,37-39
Table 1.
Demographics of Frontotemporal Dementia Patients
| Demographics | Total | Tauopathy | FTLD-Ua | DLDH | AD | Other |
|---|---|---|---|---|---|---|
| Age at onset (range), yr | 60.4 (30-82), n = 111b |
61.0 (30-77), n = 51 |
60.3 (46-79), n = 33 |
54.8 (43-80), n = 4 |
60.3 (34-82), n = 19 |
59.8 (50-70), n = 4 |
| Sex, M:F (% male) | 58:56 (50.9%) | 28:25 (53%) | 14:19 (42%) | 2:2 (50%) | 10:9 (53%) | 4:1 (80%) |
| Education (range), yr | 15.3 (8-23), n = 85 |
15.3 (10-21), n = 36 |
15.2 (10-23), n = 25 |
16.0 (15-17), n = 3 |
15.8 (12-19), n = 18 |
12.0 (8-16), n = 3 |
| Disease duration, (range), mo | 89.1 (17-300), n = 109b |
90.8 (24-300), n = 49 |
81.5 (20-264), n = 32 |
82.8 (36-120), n = 4 |
107.0 (24-204), n = 19 |
57.0 (17-151), n = 5 |
| Family historyc | Probable: 17.0% Possible: 30.0% |
Probable: 22.4% Possible: 22.4% |
Probable: 19.2% Possible: 30.8% |
Possible: 75.0% | Probable: 6.3% Possible: 43.8% |
Possible: 20.0% |
Six patients with frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) pathology were diagnosed clinically with motor neuron disease (MND) at some point after presentation. Mean age of onset = 51.3 years; mean education = 17.3 years; mean disease duration = 49.3 months.
Information regarding age at onset of dementia was not available in three patients.
Family history was coded according to the following criteria: definite = family history consistent with autosomal dominant pattern of inheritance (ie, at least three affected first-degree relatives in multiple generations) and/or identification of mutations in MAPT or valosin-containing protein genes; probable = two first-degree relatives with an FTLD diagnosis or related dementia; possible = one first-degree relative or two second-degree relatives with a diagnosis of FTLD or related dementia; none = no family history of FTLD or related condition. Information regarding family history was available in only 100 of the 114 patients.
DLDH = dementia lacking distinctive histopathology; AD = Alzheimer’s disease.
Neuropathological Classification
The final neuropathological diagnosis for the subjects in this study is summarized in Figure 2. The majority of patients were classified as tauopathies (n = 53; 46.5%) based on the presence of tau-positive inclusions. The tauopathy group was further classified as CBD (n = 22; 19.3%), PiD (n = 9; 7.9%), PSP (n =7; 6.1%), argyrophilic grain disease/tangle-predominant senile dementia (n = 6; 5.3%), FTDP-17 (n = 5; 4.4%), and tauopathy, not otherwise specified (n = 4; 3.5%) (see Fig 2B). Other patients had pathologies without tau inclusions, including FTLD-U (n = 33; 28.9%) based on the presence of ubiquitin-positive, tau-negative inclusions. Four patients (3.5%) were diagnosed with DLDH characterized by the relative absence of inclusion pathology. In 19 patients (16.7%), a primary neuropathological diagnosis of AD was made despite these patients meeting strict clinical criteria for FTLD. On neuropathological examination, these patients demonstrated both robust senile plaques and tau pathology including abundant neurofibrillary tangles (mean Braak stage, 5.8), and four patients also showed neocortical Lewy bodies that were diagnosed as Lewy body variant of AD. The topographic distribution of inclusion pathology in FTLD-U, tauopathy, and AD patients at the time of autopsy was widespread (Fig 3), which is consistent with the clinical progression of these patients to a common status of global impairment. Lastly, the remaining 5 patients were diagnosed with Lewy body disease (n = 2), Creutzfeldt–Jakob disease (n = 1), cerebrovascular disease (n = 1), and normal adult brain (n = 1). In summary, the clinical diagnosis of FTLD is associated with a wide range of pathologies, including a large group of patients meeting neuropathological criteria for the diagnosis of AD.
Fig 2.
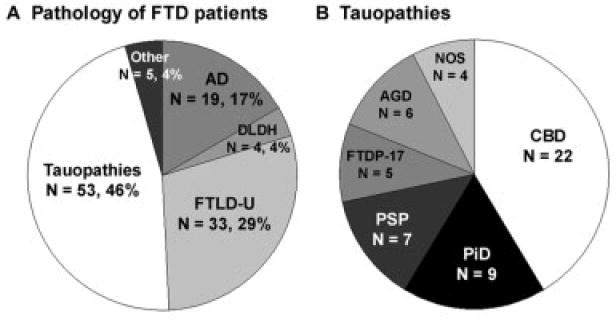
Neuropathology of frontotemporal dementia (FTD). (A) Primary neuropathological diagnosis of patients who presented clinically with frontotemporal lobar degeneration (FTLD). The number of patients and percentage of total are indicated. The “other” category includes patients with Lewy body disease (n = 2), Creutzfeldt–Jakob disease (n = 1), cerebrovascular disease (n = 1), and normal adult brain (n = 1). Additional, less robust pathology was present in 20.4% of patients and consisted of either a second, less robust, neurodegenerative disorder present in 13.8% of patients or cerebrovascular disease present in 6.6% of patients. (B) Neuropathological subtypes of tauopathies. The number of patients in each category is indicated. AD = Alzheimer’ disease; AGD = argyrophilic grain disease; CBD = corticobasal degeneration; DLDH = dementia lacking distinctive histopathology; FTDP-17 = frontotemporal dementia with parkinsonism linked to chromosome 17; FTLD-U = frontotemporal lobar degeneration with ubiquitin-positive inclusions; NOS = not otherwise specified; PiD = Pick’s disease; PSP = progressive supranuclear palsy.
Fig 3.
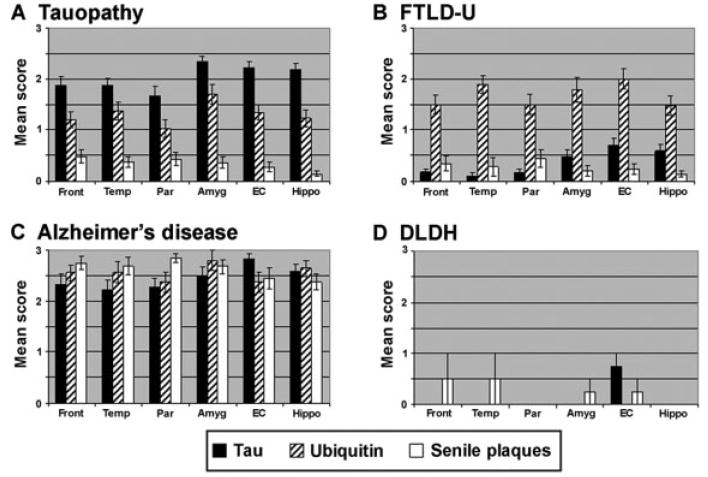
Topographical distribution of pathological inclusions in frontotemporal dementia (FTD) subgroups. Semiquantitative distribution of tau (black bars), ubiquitin (striped bars), and senile plaque (white bars) inclusion pathology in FTD subgroups. The tau pathology in the Alzheimer’s disease (AD) patients was largely Thioflavine S–positive, whereas in the tauopathies, the pathology was largely Thioflavine S–negative. This is reflected in the Braak stage for each of the subgroups: (A) tau = 1.7 ± 1.1; (B) frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) = 0.8 ± 0.7; (C) AD = 5.8 ± 0.8; (D) dementia lacking distinctive histopathology (DLDH) = 0.8 ± 0.5. Amyg = amygdala; EC = entorhinal cortex; Front = middle frontal gyrus; Hippo = hippocampus, CA1/subiculum; Par = inferior parietal lobule; Temp = superior temporal gyrus.
Demographics of Frontotemporal Dementia Patients
The demographics of the FTD patients are presented in Table 1. When age at onset, education, and sex were compared across the different pathological diagnoses, there were no significant differences among the subgroups. In contrast, when the individual tauopathies were compared, the cohort of PSP patients had a significantly later age at disease onset (71.4 years) relative to each of the other tauopathies, FTLD-U, and AD, whereas the FTDP-17 patients had an earlier age at onset (46.0 years) (Table 2). However, the number of subjects with each of these neuropathological diagnoses was small. It is noteworthy that 13 of the 19 AD patients presenting with FTLD manifested disease symptomatology before the age of 65 years, which is in contrast with patients with both a clinical and pathological diagnosis of AD (n = 80) observed at the UPenn AD Center during the same 10-year period [χ2(1) = 20.1; p < 0.0001]; the mean age at onset of this cohort was 71.4 (±8.4) years, and only 17.5% of patients with both a clinical and a neuropathological diagnosis of AD presented before the age of 65 years. There was a trend toward shorter disease duration in patients with FTLD-U pathology (81.5 months) relative to the subjects with FTLD and AD pathology [107 months; t(49) = 1.74; p < 0.09] or tauopathies (90.8 months), although this difference did not reach statistical significance. Moreover, when the FTLD-U patients who met clinical criteria for MND40 (n = 6) were removed from this group, the disease duration in FTLD-U was increased to 88.9 months, further reducing differences among pathological subgroups. Consistent with previous work,41 the age at onset of the FTLD-U patients with clinically evident MND (n = 6) was significantly earlier (mean, 51.3 years) than patients with a tauopathy and patients with FTLD-U but without clinical MND [t(55) = 2.44; p < 0.05; and t(31) = 3.02; p < 0.01, respectively]. There was also a shorter duration of disease in the patients with FTLD-U and clinically evident MND (mean, 49 months) relative to patients in the AD cohort [t(23) = 2.43; p < 0.05], as well as patients with tauopathies and patients with FTLD-U without clinical MND, although these latter two subgroups did not reach statistical significance. Thus, the demographics of pathological FTD subgroups are indistinguishable from each other and from AD patients presenting with FTLD. However, the presence of clinical MND associated with FTLD-U was associated with an earlier age at onset and shorter disease duration.
Table 2.
Demographics of the Tauopathy Cohort
| Demographics | CBD | PiD | PSP | FTDP-17 | AGD/TPSD |
|---|---|---|---|---|---|
| Age at onset (range), yr | 61.3 (51-74), n = 22 |
60.9 (47-73), n = 9 |
71.4 (66-77), n = 7a |
46.0 (30-55), n = 5b |
61.4 (46-69), n = 5 |
| Sex, M:F (% male) | 8:14 (36%) | 5:4 (56%) | 7:0 (100%) | 2:3 (40%) | 4:2 (67%) |
| Education (range), yr | 15.0 (10-20), n = 17 |
16.5 (12-20), n = 6 |
15.8 (12-21), n = 6 |
NA | 14.8 (12-18), n = 4 |
| Disease duration (range), mo | 68.6 (24-96),c n = 22 |
99.6 (36-164), n = 9 |
88.3 (60-120), n = 7 |
124.0 (48-240), n = 3 |
134.4 (24-300), n = 5 |
Age at onset in progressive supranuclear palsy (PSP) was significantly greater than all other tau subgroups, frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U), and dementia lacking distinctive histopathology (DLDH) (p < 0.05).
Age at onset in frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) was significantly younger than all other tau subgroups, FTLD-U, and Alzheimer’s disease (AD) (p < 0.05).
Disease duration in corticobasal degeneration (CBD) was significantly shorter than all other tau subgroups and AD (p < 0.05).
PiD = Pick’s disease; AGD = argyrophilic grain disease; TPSD = tangle-predominant senile dementia; NA = not available.
Clinical Features of Frontotemporal Dementia Pathological Subgroups
Patients and/or informants reported a variety of symptoms at presentation, and greater than 50% of patients reported multiple categories of symptoms. Behavioral/ social or language problems were reported in 75% of patients. Cognitive complaints that included memory problems, decline in activities of daily living, attention deficits, or executive dysfunction also were reported in 51% of patients at presentation, whereas movement disorders and weakness were reported at presentation in only 16 and 0% of patients, respectively. In view of the small numbers of patients with DLDH or a specific tau-positive pathology, we evaluated clinical features in this cohort by comparing groups of patients with tau pathology, those without tau pathology (FTLD-U and DLDH), and patients with a FTLD clinical presentation and AD pathology. When stratified by these pathological categories, the presenting symptoms showed patterns that distinguished these pathological subgroups [χ2(6) = 14.98; p < 0.02] (Fig 4A). Although there was no difference in prevalence of social/ behavioral or language complaints associated with these pathologies, patients with tau-positive or FTLD-U/ DLDH pathology were less likely to present with cognitive complaints when compared with the subgroup with AD pathology [FTLD-U/DLDH: χ2(1) = 3.79; p < 0.05; tau-positive: χ2(1) =7.96; p < 0.01]. In addition, there was a higher frequency of movement disorder complaints in the tauopathy group compared with FTLD-U/DLDH [χ2(1) = 9.00; p < 0.01]. Notably, weakness or clinically evident MND was never reported as a presenting symptom, including the six patients who later met clinical criteria for MND. Thus, the presenting symptoms are helpful in predicting the underlying pathology. Specifically, behavioral/social and language complaints are common to all pathological subgroups, but patients with a tauopathy are more likely to present with a movement disorder, and individuals with AD pathology are more likely to present with cognitive complaints other than language and social difficulties.
Fig 4.
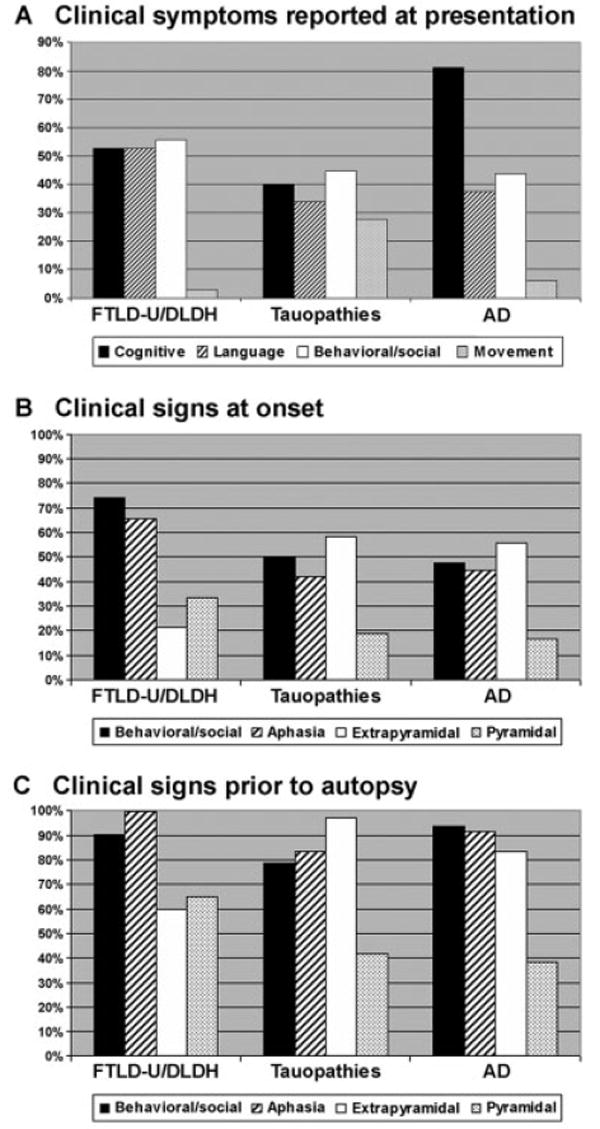
Clinical symptoms and signs distinguish pathological subgroups of frontotemporal dementia (FTD). (A) The percentage of patients and/or informants reporting the presence of the clinical features indicated. When stratified by pathological diagnosis, the presenting symptoms showed patterns that distinguish the various pathological subgroups. Behavioral/social (white bars) = disinhibition, change in personality, apathy, decreased motivation, and psychiatric illness diagnosed later in life near the onset of symptoms; language disorders (striped bars) = naming or word-finding difficulty, decline in speech or grammar; cognitive deficits (black bars) = memory loss, decline in activities of daily living, attention deficits, executive dysfunction, or visuospatial complaints; movement problems (gray bars) = abnormal gait, falling, rigidity, apraxia, or dysarthria. (B, C) The percentage of patients diagnosed at first (B) and last (C) clinical examination in the major pathological subgroups with language (striped bars), behavioral/social (black bars), extrapyramidal (white bars), and/or pyramidal dysfunction (gray bars) is indicated. Although the pattern of clinical signs did not significantly distinguish the pathological subgroups, the presence of both social dysfunction and aphasia at first examination was specific for frontotemporal lobar degeneration with ubiquitin-positive inclusions/dementia lacking distinctive histopathology (FTLD-U/DLDH) compared with Alzheimer’s disease (AD) and tauopathy groups. Extrapyramidal symptoms were more common in the tauopathy subgroups (58%) relative to the FTLD-U/DLDH subgroups (21%), and pyramidal symptoms were frequently identified in FTLD-U/ DLDH patients, although this difference did not reach statistical significance. With disease progression, the incidence of social dysfunction, aphasia, extrapyramidal signs, and pyramidal signs increased in all subgroups to a common status of global impairment in all pathology subgroups.
At the time of first neurological examination, social/ behavioral dysfunction and aphasia were documented in 57 and 51% of patients, respectively, whereas extrapyramidal and pyramidal signs were identified in 46 and 23% of patients, respectively. Moreover, in greater than 55% of patients, multiple clinical features were manifest, including 30% of patients with both social/ behavioral and language dysfunction at the initial clinical evaluation. The profile of clinical signs at first examination also showed differences among the pathologically defined subgroups (see Fig 4B). Specifically, the presence of both a behavioral/social disorder and aphasia at onset was more evident in FTLD-U/DLDH compared with AD and tauopathy groups [χ2(1) = 6.11; p < 0.01, χ2(1) = 8.72; p < 0.01], respectively, although the presence of either a behavioral/social disorder or aphasia was not significantly different among the pathological groups. Notably, 46 and 44% of FTLD subjects with AD pathology manifest signs of a behavioral/social disorder and/or aphasia, respectively, and these individuals did not manifest significant impairment in memory function early in the course of disease. Furthermore, extrapyramidal signs were more common in the tauopathy subgroup (58%) relative to FTLD-U/DLDH (21%) [χ2(1) = 10.98; p < 0.001]. Conversely, there was a trend toward an increased frequency of pyramidal signs in FTLD-U/DLDH patients (33 vs 19% in the tauopathy subgroup and 17% in the AD subgroup), although these differences were not significant. With disease progression, the incidence of social/behavioral dysfunction, aphasia, extrapyramidal signs, and pyramidal signs at last examination increased in all subgroups to 85, 90, 82, and 48% of patients, respectively (see Fig 4C), with no significant differences in the incidence of language or behavioral/social dysfunction. Extrapyramidal signs in the FTLD-U/ DLDH subgroup were less prevalent compared with the tauopathy group [χ2(1) = 13.6; p < 0.0001], whereas there was a trend toward an increased frequency of pyramidal features in patients with FTLD-U/DLDH pathology. Thus, at clinical presentation, the presence of both behavioral/social and language dysfunction and a lack of extrapyramidal signs are associated with FTLD-U or DLDH pathology. In contrast, extrapyramidal signs are more commonly identified both early and late in the disease course in patients with an underlying tauopathy. With disease progression, however, there is convergence to a common status of global impairment.
Neuropsychological Testing in Frontotemporal Dementia Patients
The neuropsychological tests performed at the initial examination at both centers also demonstrated findings that stratified the various pathological subgroups of FTD (Table 3). The mean Mini-Mental Status Examination for the entire cohort of patients was in the mildly demented range at presentation, and there were no significant differences among the pathological subgroups. In contrast, the neuropsychological profiles showed statistical association with the underlying pathology. Thus, the tauopathy patients were distinguished from AD by greater impairment on a measure of verbal fluency [t(43) = 2.04; p < 0.05], whereas the AD cohort performed significantly worse on the word list recall task compared with the tauopathy and FTLD-U/DLDH subgroups [t(29) = 2.05; p < 0.05; and t(42) = 2.95; p < 0.01, respectively]. Further-more, the AD subgroup performed significantly worse than the FTLD-U/DLDH patients on digit span forward [t(19) = 2.43; p < 0.05]. FTLD-U/DLDH patients also showed greater deficits than the tauopathy cohort on the Boston Naming Test [t(47) = 2.28; p < 0.05]. In summary, tauopathy patients were impaired on the executive measure of category naming fluency, tau-negative patients were significantly compromised on a measure of naming, and patients with AD pathology had difficulty on measures of episodic and short-term memory.
Table 3.
Neuropsychological Profile of Frontotemporal Dementia Patients
| Cognitive Tests | Total | Tauopathy | FTLD-U/DLDH | AD |
|---|---|---|---|---|
| MMSE score | 22.0 (2-30), n = 83 |
22.6 (4-30), n = 38 |
22.4 (8-30), n = 26 |
20.1 (2-29), n = 17 |
| Boston Naming (range), % correct | 75.3 (0-100), n = 64 |
81.2 (0-100), n = 33 |
64.1 (20-100),a n = 16 |
73.4 (7-100), n = 14 |
| Animal naming fluency (range), number per minute | 7.9 (0-18), n = 67 |
6.8 (0-18),b n = 32 |
7.9 (1-17), n = 20 |
9.9 (3-16), n = 13 |
| Word list recall (range), % correct | 33.9 (0-100), n = 63 |
41.7 (0-100), n = 31 |
33.3 (0-70), n = 18 |
15.9 (0-56),c n = 13 |
| Digit span forward (range), number of digits produced | 5. 7 (0-8), n = 43 |
5.8 (4-8), n = 21 |
6.4 (5-8), n = 12 |
4.6 (0-8),d n = 9 |
Naming was significantly impaired in frontotemporal lobar degeneration with ubiquitin-positive inclusions/dementia lacking distinctive histopathology (FTLD-U/DLDH) compared with tauopathy (p < 0.05).
Animal naming fluency was significantly impaired relative to frontal variant Alzheimer’s disease (AD) (p < 0.05).
Word list recall was significantly worse in frontal variant AD compared with tauopathy and FTLD-U/DLDH (p < 0.05).
Digit span forward was significantly worse in frontal variant AD than FTLD-U/DLDH.
MMSE = Mini-Mental State Examination.
Discussion
We performed a detailed analysis of clinicopathological features in 90 consecutive patients with a neuropathological diagnosis of FTD and compared these features with an additional 24 patients clinically diagnosed with FTLD but with pathology not typically associated with FTD. We found a broad spectrum of distinct neuropathological findings, including tauopathies (46.5%) and FTLD-U (28.9%). Moreover, we observed clinical and neuropsychological differences among the pathological subgroups that provide hints at the underlying pathology. These clinicopathological correlations presumably reflect differences in the pathogenesis of disorders that affect a similar regional topography in the brain. Together with other biomarkers, these clinical data will contribute to a more accurate antemortem diagnosis of FTD patients.
The spectrum of pathologies in our study was similar to several recently published series.7,37,42,43 In all but one of these studies, FTLD-U was the single most common neuropathological diagnosis. However, in our series, the tauopathies as a group were more abundant than FTLD-U. In addition, similar to Lipton and colleagues’ results,42 we found that DLDH represents only a small fraction of our neuropathological diagnoses, accounting for less than 4% of our FTD cohort. These data stand in contrast with older reports that up to 60% of FTD patients have DLDH pathology.1,12,13 There were also some notable differences in the relative proportions of the tauopathies PiD, PSP, and CBD that may reflect differences in the clinic populations evaluated at UPenn and UCSF, as well as the institutions at which these prior studies were performed. For instance, Hodges and colleagues7 reported PiD as the most common pathology, present in almost one third of their FTLD patient population, whereas both Lipton and colleagues’42 and Knopman and coworkers’37 reports, PSP was the most common tauopathy, present in 29% and 24% of their respective patient cohorts. In our analysis, CBD was the most common tauopathy, diagnosed in 19.3% of patients, similar to Kertesz and colleagues’ study43 in which CBD accounted for 20% of their FTLD cohort. In contrast with these recent studies, we also found AD pathology in 16.7% of the patients with the clinical diagnosis of FTLD.7,42 The basis for this discrepancy between our study and previous reports is unclear. One possibility is the variability in inclusion criteria and/or subject selection. For instance, patients with AD and other unrelated pathologies were specifically excluded from analysis in some previous studies. Alternatively, AD may be challenging to distinguish from progressive aphasia because verbal memory is difficult to ascertain in these circumstances. For example, two recent studies describing the pathology associated with semantic dementia and progressive aphasia reported AD pathology in 11 and 38% of subjects, respectively.44,45 In our cohort, 44% of FTLD patients with AD pathology manifested progressive aphasia. A final possibility relates to the description of a frontal variant of AD that is distinct both clinically and pathologically from typical AD.19 Knopman and coworkers37 also reported AD pathology in 12% of a small, pathologically confirmed, FTD cohort, whereas Kertesz and colleagues43 reported AD or Lewy body variant of AD in 20% of their patients. Thus, the broad spectrum of pathologies presenting clinically with FTLD emphasizes the importance of identifying clinical features and molecular biomarkers that can provide more precise antemortem predictions of the specific underlying pathology.
The demographic characteristics of our FTD cohort are similar to several large clinical series, and we did not find significant differences in the demographics of tauopathy, FTLD-U/DLDH, and AD pathological subgroups.7,37-39 These data contrast with recent studies that found a significant difference between groups with and without tau pathology in the age at diagnosis and/or the age at death.7,43 These discrepancies may be due in part to the inclusion of patients with clinical MND who have an underlying pathology of FTLD-U, because these individuals manifest both a younger age at onset and shorter disease duration.41,46 In our analysis, differences between tauopathy and FTLD-U/ DLDH groups in age at onset and disease duration disappeared once clinical MND was considered. Notably, the age at onset of FTLD patients with AD pathology was 60.3 years, which is virtually identical to the entire FTD cohort (mean age at onset, 60.4 years). The age of disease onset of this subgroup of AD patients is significantly earlier than patients with both a clinical and pathological diagnosis of AD,47 including those observed at the UPenn AD Center. Although a biological explanation of the early age at onset in the AD patients presenting clinically with FTLD is unknown, this early age at onset might have contributed to the clinical categorization of these patients as FTLD.
The clinical signs and symptoms at presentation showed some distinct patterns in the pathological subgroups that may help to determine the underlying pathology in cases of FTD. Thus, FTLD-U was always the neuropathological diagnosis in cases with a clinical diagnosis of both FTLD and MND. Recent studies also reported a similar association of combined MND and FTLD with FTLD-U pathology in 100% of patients (n = 9).7,48 However, notably, MND was diagnosed in only 18.2% of FTLD-U cases and was never a presenting symptom. This rare but statistically reliable prediction of a specific underlying pathology is in contrast with the associations between other clinical features and an underlying pathology. For instance, the presence of clinically diagnosed social/behavioral and/or language dysfunction was statistically more prevalent in the subgroup with FTLD-U or DLDH compared with patients with tau or AD pathology. The clinical presentation of progressive aphasia has been associated with tau-negative pathologies such as FTLD-U,43,44 whereas other reports have related progressive nonfluent aphasia with PiD7 or CBD.43 We did not find a specific clinical association of a language disorder with any of the pathological subgroups. In contrast, the AD patients clinically diagnosed with FTLD were more likely to manifest deficits in other domains of cognition such as impaired memory and executive function when compared with both the tauopathy and FTLD-U/DLDH subgroups. Furthermore, patients with a tauopathy more frequently manifest extrapyramidal features consistent with the presence of both CBD and PSP in this subgroup. These observations parallel the findings of a smaller series, associating extrapyramidal features such as rigidity with CBD and the pyramidal features of clinical MND with FTLD-U.7 These statistical associations were derived in part from clustering across pathologies sharing an immunohistochemical profile. However, the tau-negative pathologies such as FTLD-U and DLDH, although sharing the absence of tau pathology, differ in many important ways including the absence of ubiquitin-immunoreactive inclusions. Another limitation to a clinically based histopathological diagnosis is that our FTD cohort of patients tended to decline to a common phenotype with global impairment, rendering a clinical diagnosis less transparent at later stages of disease. These clinical findings are supported by the widespread distribution of pathology at the time of autopsy in all pathological subgroups of FTD without a specific predilection for the frontal lobes. This is in contrast with Johnson and colleagues’19 observations, which showed a significantly higher load of tau pathology in the frontal lobe of FTLD subjects with AD pathology. However, in this study, we used a semiquantitative assessment of tau pathology, whereas in Johnson and colleagues’19 report, tau pathology was assessed using area-occupied volumetric measurements. Nonetheless, similar to Kertesz and colleagues’43 description, additional clinical features emerge with disease progression in our study and lead to the finding that the FTD phenotypes, albeit initially distinct, converge over time in a single patient. Thus, clinical features detected at the first clinical examination rather than at later time points may contribute in a statistical sense to distinguishing the pathological subgroups of FTD.
A limited battery of neuropsychological tests showed some statistical associations with pathological subgroups of FTD, thus suggesting that antemortem neuropsychological evaluation also may contribute to identifying the underlying pathology of FTLD cases. However, quantitative neuropsychological findings that discriminate among pathologically defined subgroups must be interpreted with caution because of their modest statistical significance and the limited scope of the assessment available across clinics during the decade when data were collected. Nevertheless, psychometric testing may be helpful in distinguishing the pathological subgroups with particular attention to the domains of memory and language. Thus, AD patients clinically diagnosed with FTLD were impaired in episodic memory compared with both the tauopathy and FTLD-U/DLDH subgroups, as well as a test of attention relative to patients with FTLD-U/DLDH pathology. Conversely, tauopathy and FTLD-U/DLDH patients showed greater deficits on tests of verbal fluency and confrontational naming, respectively. Additional work clearly is necessary with larger panels of psychometric testing to further refine the neuropsychological profile of the pathological subgroups of FTLD patients.
In summary, the relation between FTLD and the pathology underlying this family of clinical syndromes is complex. Clinical and neuropsychological features of FTLD patients early in the course of disease are associated with an underlying pathology. However, these associations are statistical effects found in group analyses, and the particular clinical features observed in any individual patient rarely predict the specific underlying pathology with reliability. This study highlights the need for additional radiological49 and/or biological markers such as cerebrospinal fluid tau50 that, in association with clinical and neuropsychological features, can consistently delineate the specific pathology underlying a patient with the clinical diagnosis of FTLD.
Acknowledgments
This work was supported by the NIH (P01 AG17586, M.S.F., V.V.D., V.M.Y-L., J.Q.T., M.G., P01 AG09215, VMY-L., J.Q.T., P01 AG19724, J.K.J., H.J.R., B.L.M., P30 AG10124, M.S.F., C.M.C., S.E.A., J.H.K., J.Q.T., R01 AG15116, M.G., R01 AG 022538, J.K.J., K08 AG20073, M.S.F., National Institute on Neurological Disorders and Stroke, R01 NS44266, M.G., K08 NS41408, V.V.D.).
We thank the families of the patients participating in our studies for making this research possible.
Footnotes
Published online May 22, 2006 in Wiley InterScience (www.interscience.wiley.com).
References
- 1.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Grossman M. Frontotemporal dementia: a review. J Int Neuropsychol Soc. 2002;8:566–583. doi: 10.1017/s1355617702814357. [DOI] [PubMed] [Google Scholar]
- 3.McKhann GM, Albert MS, Grossman M, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 4.Grossman M, Ash S. Primary progressive aphasia: a review. Neurocase. 2004;10:3–18. doi: 10.1080/13554790490960440. [DOI] [PubMed] [Google Scholar]
- 5.Hodges JR, Patterson K, Oxbury S, et al. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115(pt 6):1783–1806. doi: 10.1093/brain/115.6.1783. [DOI] [PubMed] [Google Scholar]
- 6.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 7.Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- 8.Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson M, Lennox G, Lowe J. Motor neurone disease-inclusion dementia. Neurodegeneration. 1996;5:339–350. doi: 10.1006/neur.1996.0046. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto K, Hirai S, Yamazaki T, et al. New ubiquitin-positive intraneuronal inclusions in the extra-motor cortices in patients with amyotrophic lateral sclerosis. Neurosci Lett. 1991;129:233–236. doi: 10.1016/0304-3940(91)90469-a. [DOI] [PubMed] [Google Scholar]
- 11.Wightman G, Anderson VE, Martin J, et al. Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett. 1992;139:269–274. doi: 10.1016/0304-3940(92)90569-s. [DOI] [PubMed] [Google Scholar]
- 12.Brun A. Frontal lobe degeneration of non-Alzheimer type. I. Neuropathology. Arch Gerontol Geriatr. 1987;6:193–208. doi: 10.1016/0167-4943(87)90021-5. [DOI] [PubMed] [Google Scholar]
- 13.Knopman DS, Mastri AR, Frey WH, et al. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–256. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- 14.Neary D, Snowden JS, Northen B, et al. Dementia of frontal lobe type. J Neurol Neurosurg Psychiatry. 1988;51:353–361. doi: 10.1136/jnnp.51.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bigio EH, Lipton AM, White CL, III, et al. Frontotemporal and motor neurone degeneration with neurofilament inclusion bodies: additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol. 2003;29:239–253. doi: 10.1046/j.1365-2990.2003.00466.x. [DOI] [PubMed] [Google Scholar]
- 16.Cairns NJ, Perry RH, Jaros E, et al. Patients with a novel neurofilamentopathy: dementia with neurofilament inclusions. Neurosci Lett. 2003;341:177–180. doi: 10.1016/s0304-3940(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 17.Cairns NJ, Zhukareva V, Uryu K, et al. Alpha-Internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004;164:2153–2161. doi: 10.1016/s0002-9440(10)63773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126:2291–2303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 19.Johnson JK, Head E, Kim R, et al. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999;56:1233–1239. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 20.Downes JJ, Priestley NM, Doran M, et al. Intellectual, mnemonic, and frontal functions in dementia with Lewy bodies: a comparison with early and advanced Parkinson’s disease. Behav Neurol. 1998;11:173–183. [PubMed] [Google Scholar]
- 21.Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 22.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 23.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 24.Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61:935–946. doi: 10.1093/jnen/61.11.935. [DOI] [PubMed] [Google Scholar]
- 25.Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy) Neurology. 1994;44:2015–2019. doi: 10.1212/wnl.44.11.2015. [DOI] [PubMed] [Google Scholar]
- 26.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giasson BI, Duda JE, Murray IV, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 28.Lee VM-Y, Otvos L, Jr, Carden MJ, et al. Identification of the major multiphosphorylation site in mammalian neurofilaments. Proc Natl Acad Sci U S A. 1988;85:1998–2002. doi: 10.1073/pnas.85.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea and Febiger; 1983. [Google Scholar]
- 31.Grossman M. Semantic and perceptual errors in aphasics’ free-hand category drawing. Neuropsychology. 1993;7:27–40. [Google Scholar]
- 32.Delis DC, Kramer JH, Kaplan E. California Verbal Learning Test. 2. San Antonio, TX: The Psychological Corporation; 2000. [Google Scholar]
- 33.Welsh K, Butters N, Hughes J, et al. Detection of abnormal memory decline in mild cases of Alzheimer’s disease using CERAD neuropsychological measures. Arch Neurol. 1991;48:278–281. doi: 10.1001/archneur.1991.00530150046016. [DOI] [PubMed] [Google Scholar]
- 34.Wechsler D. Wechsler Memory Scale—Revised. San Antonio, TX: The Psychological Corporation; 1987. [Google Scholar]
- 35.Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006;66:41–48. doi: 10.1212/01.wnl.0000191307.69661.c3. [DOI] [PubMed] [Google Scholar]
- 36.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 37.Knopman DS, Boeve BF, Parisi JE, et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol. 2005;57:480–488. doi: 10.1002/ana.20425. [DOI] [PubMed] [Google Scholar]
- 38.Stevens M, van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50:1541–1545. doi: 10.1212/wnl.50.6.1541. [DOI] [PubMed] [Google Scholar]
- 39.Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–930. doi: 10.1001/archneur.62.6.925. [DOI] [PubMed] [Google Scholar]
- 40.Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 41.Hodges JR, Davies R, Xuereb J, et al. Survival in frontotemporal dementia. Neurology. 2003;61:349–354. doi: 10.1212/01.wnl.0000078928.20107.52. [DOI] [PubMed] [Google Scholar]
- 42.Lipton AM, White CL, III, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol (Berl) 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 43.Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 44.Davies RR, Hodges JR, Kril JJ, et al. The pathological basis of semantic dementia. Brain. 2005;128:1984–1995. doi: 10.1093/brain/awh582. [DOI] [PubMed] [Google Scholar]
- 45.Knibb JA, Xuereb JH, Patterson K, et al. Clinical and pathological characterization of progressive aphasia. Ann Neurol. 2006;59:156–165. doi: 10.1002/ana.20700. [DOI] [PubMed] [Google Scholar]
- 46.Roberson ED, Hesse JH, Rose KD, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005;65:719–725. doi: 10.1212/01.wnl.0000173837.82820.9f. [DOI] [PubMed] [Google Scholar]
- 47.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology. 2004;24:87–102. doi: 10.1111/j.1440-1789.2003.00544.x. [DOI] [PubMed] [Google Scholar]
- 49.Grossman M, McMillan C, Moore P, et al. What’s in a name: voxel-based morphometric analyses of MRI and naming difficulty in Alzheimer’s disease, frontotemporal dementia and corticobasal degeneration. Brain. 2004;127:628–649. doi: 10.1093/brain/awh075. [DOI] [PubMed] [Google Scholar]
- 50.Grossman M, Farmer J, Leight S, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol. 2005;57:721–729. doi: 10.1002/ana.20477. [DOI] [PubMed] [Google Scholar]