

Abstract
During herpes simplex virus (HSV) entry, membrane fusion occurs either on the cell surface or after virus endocytosis. In both cases, binding of glycoprotein D (gD) to a receptor such as nectin-1 or HVEM is required. In this study, we co-cultured cells expressing gD with nectin-1 expressing cells to investigate the effects of gD on nectin-1 at cell contacts. After overnight co-cultures with gD expressing cells, there was a down-regulation of nectin-1 in B78H1-C10, SY5Y, A431 and Hela cells, which HSV enters by endocytosis. In contrast, on Vero cells, which HSV enters at the plasma membrane, nectin-1 was not down-regulated. Further analysis of B78H1-derived cells showed that nectin-1 down-regulation corresponds to the ability of gD to bind nectin-1 and is achieved by internalization and low-pH dependent degradation of nectin-1. Moreover, gD is necessary for virion internalization in B78H1 cells expressing nectin-1. These data suggest that the determinants of gD-mediated internalization of nectin-1 may direct HSV to an endocytic pathway during entry.
Keywords: herpes simplex virus, HSV, entry, nectin-1, glycoprotein, down-regulation, endocytosis, receptor, glycoprotein
INTRODUCTION
Herpes simplex virus (HSV) can infect many cell types in culture, but in its human host it spreads predominantly between epithelial and neuronal cells (Whitley, 2001). This cell tropism is likely determined, at least in part, by the specific interactions between virus and cell that lead to virus entry (reviewed in Krummenacher et al, 2007; Reske et al, 2007). Four envelope glycoproteins are required for HSV entry. Binding of glycoprotein D (gD) to a receptor is an essential step of entry and cell to cell spread but is not sufficient by itself to promote fusion of the viral envelope with membranes of the target cell (Ligas and Johnson, 1988). Rather, receptor binding induces conformational changes in gD that trigger the viral fusion machinery which comprises gB and gH/gL (Fusco et al, 2005; Krummenacher et al., 2005; Subramanian and Geraghty, 2007; Whitbeck et al., 2006; Zago et al, 2004). The gD receptors nectin-1, HVEM (herpesvirus entry mediator) and heparan sulfate modified by 3-O-sulfotransferases are structurally unrelated molecules, but each can mediate HSV entry independently (Geraghty et al., 1998; Krummenacher et al., 1998; Montgomery et al., 1996; Shukla et al., 1999; Whitbeck et al., 1997).
In this study, we focus on one of the gD receptors, nectin-1, which acts as the main receptor on neurons and epithelial cells (Hung et al., 2002; Mata et al., 2001; Simpson et al., 2005). Nectin-1 is a cell adhesion molecule that accumulates at adherens junctions of epithelia, at contacts between cultured cells and at synapses, varicosities and puncta adherentia of neurons (De Regge et al., 2006; Inagaki et al., 2003; Krummenacher et al., 2003; Sakisaka and Takai, 2004; Takai et al, 2003). Ligand binding is required for nectin-1 to accumulate at cell contacts. For example, homophilic trans-interaction of nectin-1 molecules on adjacent cell surfaces occurs on cultured cells (i.e. B78H1-C10, L-cells) (Krummenacher et al., 2002; Takahashi et al., 1999) and heterophilic interaction with nectin-3 has been described at neuronal synapses and in the eye (Inagaki et al., 2005; Mizoguchi et al., 2002). Several splice isoforms are produced from the human nectin-1 gene, PVRL1 (Cocchi et al., 1998; Geraghty et al., 1998). The splice isoform nectin-1α, used in this study, recruits cytoplasmic PDZ-containing proteins such as afadin, to cell contacts to establish adherens junctions (Takahashi et al., 1999; Takai and Nakanishi, 2003).
HSV infects cells as free particles and then can spread to adjacent cells through cell contacts. During the initial infection, HSV enters cells either by direct fusion at the plasma membrane or by fusing its envelope with an endosomal membrane after endocytosis of the virion (Nicola et al, 2003; Wittels and Spear, 1990). The pathway used by virions during entry is cell type dependent (Nicola et al., 2005) but in all cases, the same four essential glycoproteins (gD, gB, gH/gL) and a gD receptor are required (Nicola and Straus, 2004). In Vero cells (African green monkey kidney), fusion occurs at the cell surface in a low-pH independent manner (Milne et al., 2005; Nicola et al., 2005; Wittels and Spear, 1990). Numerous electron microscopy (EM) studies, summarized by Nicola et al (2003) showed fusion of HSV at the cell surface and/or in intracellular vesicles shortly after inoculation. Notably, fusion of virions with the plasma membrane has been observed by EM in Hep-2 cells (Fuller and Spear, 1987) and in explanted neurons from human dorsal root ganglia (Lycke et al., 1988). In other cell lines, however, HSV entry occurs by endocytosis and the viral envelope fuses with an endosomal membrane. In cells of epithelial origin, fusion is more likely to require endosomal acidification, while in neuroblastoma cells it is independent of low pH (Nicola et al., 2005). The cell specific basis for low pH requirement for fusion is unknown and there is no evidence that a particular HSV receptor (nectin-1 or HVEM) is exclusively linked with low pH-dependence (Delboy et al., 2006; Milne et al., 2005; Nicola et al., 2005; Whitbeck et al., 2006). One should note that in their studies, Nicola et al consider only the requirement for low pH for fusion to define endocytic entry (Delboy et al., 2006; Nicola et al., 2005). However, the lack of dependence on low pH for fusion is also compatible with endocytosis because, in cells such as B78H1-C10, pH-independent entry is observed together with virion internalization (Milne et al., 2005). In the present study, we consider a virus to be endocytosed when exposed virion components (e.g. gB) become protected from extracellular protease digestion. Mouse melanoma B78H1 cells, which are devoid of HSV gD receptors, are resistant to HSV entry (Miller et al., 2001) and do not endocytose HSV (Milne et al., 2005). Expression of human nectin-1 or HVEM in these cells (B78H1-C10 and B78H1-A10 respectively) allows virus endocytosis and productive infection (Miller et al., 2001; Milne et al., 2005). In these cells, neutralizing anti-gD antibodies blocked virion internalization (Milne et al., 2005).
Interaction of gD with nectin-1 is not limited to the early steps of infection. We showed previously that expression of gD during the later phases of HSV infection correlates with down-regulation of nectin-1 within the infected cells (effect in cis) (Krummenacher et al., 2003). Newly synthesized gD then accumulates at sites of contact between the infected cell and an adjacent non-infected cell but not between two infected cells. This accumulation of gD requires expression of nectin-1 on the non-infected cells but does not depend on other viral components (Krummenacher et al., 2003). From these studies, we hypothesized that there is a direct trans-interaction between newly synthesized gD and nectin-1 at cell contacts.
To study the interaction of gD with nectin-1 between cells in the absence of infection, we engineered B78H1 (nectin-1 negative) cell lines to constitutively express various forms of gD, as substitutes for infected cells. These effector cells were co-cultivated with target cells that express nectin-1. In most cases, trans-interaction of gD with nectin-1 at contacts between the two cell populations led to down-regulation of nectin-1. This response was specific to gD and consisted in internalization of the receptor from the target cell surface followed by low-pH dependent degradation. We postulate that down-regulation of nectin-1 by gD reflects a form of ligand-induced endocytosis. Interestingly, the cell specificity of nectin-1 down-regulation correlated with the cell specificity of endocytic HSV entry, suggesting that gD-mediated internalization of nectin-1 may direct HSV to an endocytic pathway during entry.
RESULTS
Characterization of cell lines expressing various forms of HSV gD
The goal of this work was to ask how gD binding affects nectin-1 at cell-cell contacts. To separate the effects of gD from those of HSV infection, we developed a cellular co-culture system. A panel of nectin-1 expressing target cells was already available (Krummenacher et al., 2003; Krummenacher et al., 2002). To establish effector cells, we engineered nectin-1-negative mouse melanoma cells (B78H1) that stably express various forms of gD on their surface. In addition to creating a cell line expressing wild type (wt) gD from strain KOS, we made cell lines that express two mutant forms of gD based on their nectin-1 binding properties (Connolly et al., 2005; Krummenacher et al., 2005): gD(W294A) binds nectin-1 with higher affinity than wt gD but cannot complement a gD-null virus, and gD(A3C-Y38C) which does not bind nectin-1 but binds HVEM. The resulting cell lines were named B78-gDwt, B78-gD(W294A) and B78-gD(A3C-Y38C).
Clones were selected based on the level of surface expression of gD as detected by FACS (Fig. 1A) and CELISA (Fig. 1B). The nectin-1 binding activity of the cell lines was tested by CELISA (Fig. 1C). Soluble nectin-1 bound to B78-gDwt cells and showed increased binding to B78-gD(W294A) cells due to the higher affinity of this form of gD (Krummenacher et al., 2005). The negative control cells, B78-gD(A3C-Y38C), did not bind soluble nectin-1 (Fig. 1C) despite higher levels of gD surface expression (Fig. 1B). In contrast, B78-gD(A3C-Y38C) cells did bind soluble HVEM (not shown), indicating that they express a functional mutant of gD with the expected selective receptor usage (Connolly et al., 2005). These results are consistent with the properties of these forms of gD in transient transfection experiments (Connolly et al., 2005; Connolly et al., 2003). The three effector cell lines were next used to study trans-interactions between membrane-bound gD and nectin-1 at cell contacts. Since these cells are not polarized we will refer to the surface areas where cells make contact as “cell contacts” rather than cell junctions.
Figure 1.

Cell lines expressing gD. Clonal cell lines derived from B78H1 were engineered to express various forms of HSV-1 gD (gDwt, gD(W294A) and gD(A3C-Y38C). (A) gD expression on cell surface detected by FACS using anti-gD polyclonal serum R7 followed by anti-rabbit Ig coupled with Alexa488. The white histograms represent staining in the absence of R7 and serve as negative controls. (B) Detection of gD on cell surface by CELISA. Cells were stained with anti-gD polyclonal serum R8 and anti-rabbit secondary Ab coupled with peroxidase and substrate. The absorbance detected for untransfected B78H1 cells was subtracted as background. (C) Binding of soluble nectin-1 to cells expressing gD by CELISA. Cells were incubated with increasing concentrations of nectin-1 ectodomain (HveC(346t)). Cell-bound nectin-1 was detected with a mixture of anti-nectin-1 polyclonal rabbit sera R165 and R154. The absorbance detected for untransfected B78H1 cells was subtracted as background.
Down-regulation of nectin-1 from B78H1-C10 cells
To study the interaction of nectin-1 and gD at cell contacts, we co-cultured our gD effector cell lines with target cells that express nectin-1. When B78H1-C10 target cells (Krummenacher et al., 2002) were mixed with B78-gD(A3C-Y38C) effector cells, nectin-1 was clearly detected in the target cells by immunofluorescence (Fig. 2D), while gD was detected in the effector cells (Fig. 2G). Since gD(A3C-Y38C) does not bind to nectin-1, we did not observe co-localization of gD and nectin-1 at effector-target cell contacts (Fig. 2J). To our surprise, when B78H1-C10 target cells were co-cultured with B78-gDwt or B78-gD(W294A) effectors, the detection of nectin-1 was severely decreased in the target cells. Only a few of them still stained positive for nectin-1, but most of these were not in direct contact with effector cells expressing gD (Fig. 2K, L). Indeed, we observed a dose dependent loss of the nectin-1 signal from target cells in the co-cultures when the effector:target ratio was increased (data not shown). Neither gDwt nor gD(W294A) was detected preferentially at contacts with the few target cells where nectin-1 was still detected (Fig. 2K, L). Since the cells were permeabilized prior to staining, the remaining nectin-1 may be within the target cells and not available for gD binding. Nectin-1 fluorescence was consistently dimmer when B78H1-C10 target cells were in contact with effector cells expressing gD(W294A) as compared to cells expressing wt gD. Although not quantified here, we believe it may be a consequence of the higher affinity of gD(W294A) for nectin-1 (Krummenacher et al., 2005).
Figure 2.

Nectin-1 expression on B78H1-C10 cells after co-cultivation with gD expressing cells. Nectin-1 positive B78H1-C10 cells were mixed at an equal ratio with B78-gD(A3C-Y38C) cells (left), B78-gDwt (center) or B78-gD(W294A) cells (right). After 16 h of co-culture, cultures were fixed, permeabilized and stained. Nectin-1 on target B78H1-C10 cells was detected with mouse monoclonal antibody CK41 followed by anti-mouse Ig coupled to Alexa488 shown in green (D,E,F). gD on effector cells was detected with rabbit polyclonal serum R7 followed by anti-rabbit Ig coupled with Alexa594 shown in red (G,H,I). A phase contrast image was taken to show all cells in the co-cultures (A,B,C) and DAPI was used to stain all nuclei (blue) in merged images (J,K,L). Arrows point to target cells not directly in contact with effector cells, thus retaining nectin-1 staining. A representative experiment is shown. (M) Histograms showing that gD expression on effectors cells is not affected after 16 hr co-culture with B78H1-C10 (gray) compared to B78H1 (black line) target cells. gD on effector cells (Qdot655 negative) was detected using mAb MC5 followed by anti-mouse Ig coupled to Alexa488.
One possible explanation for our inability to detect nectin-1 by IFA when target and effector cells were in contact was that the epitope of mAb CK41 used to detect nectin-1 was masked by gD (Krummenacher et al., 2000). To address this and to obtain quantitative data, we developed a FACS-based approach. This approach enabled us to detect nectin-1 expression with mAb CK41 on single target cells in suspension after they had been separated from effector cells. In this assay, B78H1-C10 target cells were labeled with QTracker655 prior to co-cultivation with the effector cells. We used this fluorescent marker to select target cells during FACS analysis and eliminate the background of effector cells (QTracker655-negative, nectin-1-negative, gD-positive). Figure 3 shows nectin-1 expression on the Qtracker655-positive B78H1-C10 target cells after co-culture with the various effector cell lines. When B78H1-C10 target cells were co-cultured with B78H1 cells (no gD), they retained a high level of cell surface nectin-1 (Fig. 3, black profile). This control serves as a reference for the wild-type level of nectin-1 on target B78H1-C10 cells. Similar levels of nectin-1 were detected on B78H1-C10 target cells after co-cultivation with B78-gD(A3C-Y38C) effectors which do not bind nectin-1. In contrast, there was a significant loss of nectin-1 from the surface of B78H1-C10 target cells when they were mixed with effector cells bearing either wt gD or gD(W294A) (Fig. 3). We obtained similar results with another anti-nectin-1 mAb (CK6), which recognizes a different epitope from that seen by CK41 (data not shown).
Figure 3.
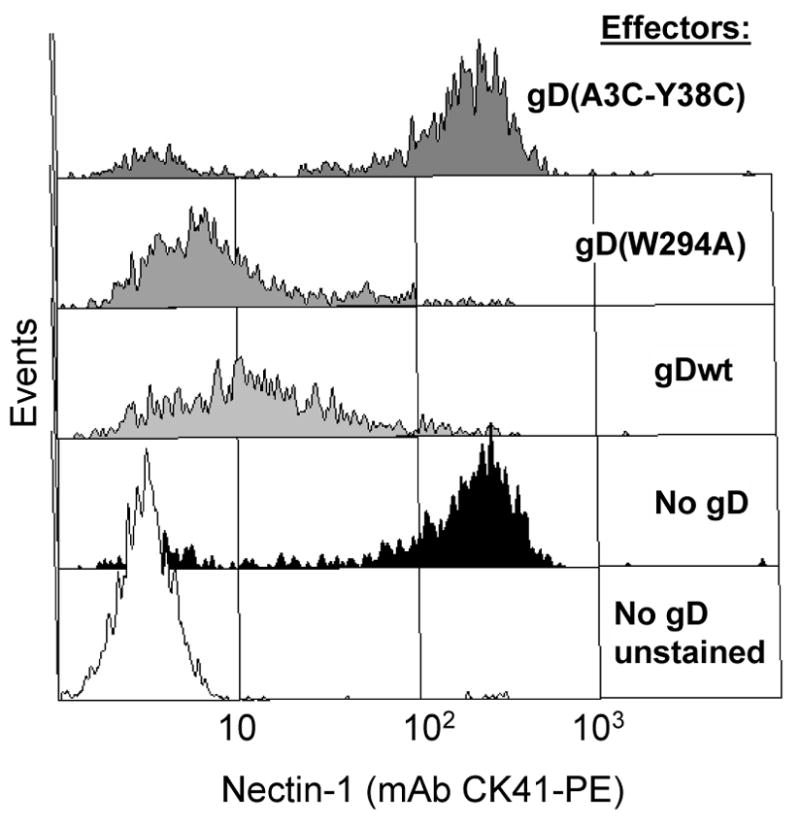
Down-regulation of nectin-1 from cell surface. Target B78H1-C10 cells expressing nectin-1 were labeled with Qtracker655 and mixed with effector cells expressing the indicated forms of gD or with B78 cells (no gD) at a target:effector ratio of 1:2. After overnight co-culture, cells were stained with CK41-PE to detect nectin-1 by FACS. Target B78H1-C10 cells were positively selected based on Qtracker655 fluorescence to be shown on FACS profiles while unlabelled effector cells were excluded. Histograms represent PE fluorescence of Qtracker655-positive target B78H1-C10 cells. The white histogram represents background fluorescence of B78H1-C10 cells (no CK41-PE) co-cultured with B78 cells. A representative experiment is shown.
We also tested whether gD expression on the surface of effector cells was affected by co-culture with nectin-1 expressing cells. By immunofluorescence, B78-gDwt cells in co-cultures are often less intensely stained for gD expression than B78-gD(W294A) and B78(A3C-Y38C) cells (Fig 2H, vs G, I). To determine whether this was an apparent artifact of the method or a genuine effect resulting from exposure of wt gD to nectin-1, we used FACS to quantify gD expression. We measured the amount of gD on the surface of the various effector cells co-cultured with C10 cells expressing nectin-1 or with the receptor-negative B78H1 cells (Fig. 2M). In this case, we found that the surface expression of each form of gD was similar in the presence or absence of nectin-1 on the target cells. Thus nectin-1 does not induce internalization of gD. The FACS results also show that nectin-1 and gD are not internalized as a complex.
These experiments showed that down-regulation of nectin-1 in the target cell is directly related to the ability of gD on the effector cell to bind the receptor in trans. Thus, down-regulation of nectin-1 occurs as a result of a transient heterophilic trans-interaction with gD at cell contacts.
Down-regulated nectin-1 does not accumulate within the cell
Since the IFA experiments examined total amounts of nectin-1 in the target cells, its disappearance suggested that it was not accumulating within the cells after down-regulation from the surface (Fig. 2E, F). We used two approaches to measure the total amount of nectin-1 in target cells. First, we carried out co-culture experiments with B78H1-NGC12 cells, which express a form of nectin-1 with GFP fused to its N-terminus (GFP-nectin-1). This chimeric nectin-1 is functional as a HSV receptor and localizes at cell contacts (Krummenacher et al., 2003). In these cells, we used GFP fluorescence to measure the total amount of GFP-nectin-1 by FACS (Fig. 4A). The level of GFP fluorescence was considered to be maximal (100%) when the target B78H1-NGC12 cells were co-cultured with B78H1 cells (no gD). When the target cells were co-cultured with effectors bearing gD(A3C-Y38C), the amount of GFP remained comparable to the level seen with B78H1 effectors. The level of GFP fluorescence decreased when B78H1-NGC12 cells were exposed to effector cells expressing either wt gD or gD(W294A) (Fig. 4A). We also tested a form of nectin-1 with the GFP tag at the C-terminus of the nectin-1 cytoplasmic tail and obtained similar results (data not shown). The same pattern of down-regulation was observed for total GFP-nectin-1 and for cell surface nectin-1 (Fig. 3 and 4A). Thus, the total amount of GFP-nectin-1 in target cells decreases when these cells are exposed to effector cells expressing a form of gD that binds nectin-1.
Figure 4.

Detection of total amounts of GFP-nectin-1 and nectin-1 in B78H1-derived cells. (A) Down-regulation of GFP-nectin-1 analyzed by FACS. Target cells expressing GFP-nectin-1 (B78H1-NGC12) were labeled with Qtracker655 and mixed with effector cells expressing the indicated forms of gD or with B78H1 cells (no gD). After overnight co-culture, GFP fluorescence was detected by FACS. Histogram profiles show GFP fluorescence in target cells selected for Qtracker655 labeling. The geometric mean fluorescence is indicated on the right. For comparison background fluorescence of parental B78H1 cells is 3.9. (B) Detection of total amounts of nectin-1 and GFP-nectin-1 by western blot in cell lysates. Target cells (B78H1-C10 or B78H1-NGC12) were mixed with the indicated effector cells. After an overnight co-culture, nectin-1 was detected by mAb CK8 in total cell lysates. Major non-specific bands indicated by asterisks were also observed in B78H1 extracts (not shown). Positions of nectin-1 (N1) and GFP-nectin-1 (GFP-N1) are indicated with arrows. Molecular weight markers are indicated on the left in kilodaltons. (C) Detection of nectin-1 and GFP-nectin-1 by western blot in cell co-culture supernatant. Target B78H1-C10 cells were mixed with effector cells B78-gD(W294A) or B78H1 (no gD). After an overnight co-culture, cell culture supernatant was collected and cells were lysed. 5% of total cell lysate was loaded in lanes 1 and 2 and 5% of supernatant was loaded in lanes 3, 4 and 5 to compare amounts of nectin-1 detected by mAb CK8. The position of full-length nectin-1 (N1) is indicated by an arrow. Lanes 6–9 contain the indicated amount of soluble nectin-1(346t) diluted in fresh culture medium as sensitivity control. The position of nectin-1(346t) is indicated by an arrowhead. The blot is intentionally over-exposed (3.5 min, Supersignal West Dura, Pierce) and non-specific bands detected by mAb CK8 are unlabeled. (D) Effect of bafilomycin A (BFLA). NGC12 target cells expressing GFP-nectin-1 (GFP-N1) were pretreated (+) or not (-) with BFLA for 1 h before and 5 h during co-culture with B78H1 or B78-gD(W294A) effector cells. Cell lysis and analysis were performed as in B.
As a second approach, we carried out western blot analysis of total cell lysates after co-culture (Fig. 4B). Nectin-1 from B78H1-C10 target cells and GFP-nectin-1 from B78H1-NGC12 target cells were each detected when these cells were co-cultured with gD-negative effector B78H1 cells (lanes 1 and 5 respectively) or with cells expressing gD(A3C-Y38C) (lanes 4 and 8). In contrast, neither form of nectin-1 was found after co-culture with effector cells expressing wt gD (lanes 2 and 6) or gD(W294A) (lanes 3 and 7). A crossreacting band of approximately 100 kDa specific to B78-NGC12 cells is also present but its origin has not been defined. This band was also seen with an anti-nectin-1 polyclonal serum (data not shown). Because it is present only in GFP-nectin-1 cells, it is likely to be an intracellular form of nectin-1 related product which is unaffected by gD. Alternatively, it might be a truncated nectin-1, which lacks part of the gD binding domain and therefore cannot be internalized or degraded in response to exposure to gD.
Since nectin-1 can potentially be proteolytically cleaved in its trans-membrane domain (Kim et al, 2002), we looked for the nectin-1 ectodomain in co-culture supernatants by western blot (Fig. 4C). After co-culture with B78-gD(W294A), nectin-1 was absent from the cell lysate (Fig. 4C, lane 1) but it was present in the lysate from the control co-culture (lane 2). Because we overexposed the film to increase the sensitivity of detection, several non-specific bands were observed but no nectin-1 degradation products specific to the B78H1-C10 + B78-gD(W294A) co-culture could be identified (lane 1 vs 2). We also attempted to detect nectin-1 degradation products using a polyclonal serum or a pool of several mAbs against different regions of nectin-1. These antibodies did not detect specific cleavage products and also showed higher non-specific background (data not shown). Furthermore, no additional band was detected by mAb CK8 in supernatant from the B78H1-C10 and B78-gD(W294A) co-culture compared to controls (lane 3 vs lanes 4, 5). We used soluble nectin-1 (Krummenacher et al., 1998) diluted in fresh medium (lanes 6–9) as a sensitivity control to show that very low amounts of nectin-1 could be detected in the culture supernatant. These results show that gD does not induce release of nectin-1 from the target cell surface.
Consequently, we tested whether the loss of nectin-1 involved intracellular degradation after internalization. We used bafilomycin A1 (BFLA) to prevent endosome/lysosome acidification, thereby preventing activation of lysosomal proteases. NGC12 target cells were treated with BFLA before and during co-culture with effector cells (Fig. 4D). Under these conditions, accumulation of nectin-1 in total cell lysate would indicate that nectin-1 degradation occurs in lysosomes. In the absence of BFLA, the amount of GFP-nectin-1 was decreased in co-cultures with B78-gD(W294A) cells compared to co-cultures with B78H1 cells (Fig. 4D, lanes 1 and 2). After BFLA treatment, the level of GFP-nectin-1 did not change, even in the presence of B78-gD(W294A) cells (lane 4). Thus, endosome/lysosome acidification is required for nectin-1 degradation. We also observed that gD-induced nectin-1 degradation was prevented in target cells treated with the cathepsin/calpain inhibitor E-64d (Chandran et al., 2005)(data not shown). These results indicate that nectin-1 is degraded in lysosomes by low-pH dependent cysteine-proteases.
Homophilic trans-interaction does not induce down-regulation of nectin-1
HSV gD binding to the V-domain of nectin-1 disrupts its homophilic trans-interactions (Krummenacher et al., 2002; Sakisaka et al., 2001). This suggests that gD and nectin-1 share a similar binding site on nectin-1. Therefore, we tested whether nectin-1 induces the same response as gD on nectin-1 in target cells during co-culture. To differentiate nectin-1 on target cells from that on effector cells we used B78H1-NGC12 cells as targets. We previously showed that GFP-nectin-1 in these cells was functionally similar to wt nectin-1 as a receptor for HSV and as a cell adhesion molecule (Krummenacher et al., 2003). This indicates that GFP does not significantly affect homophilic trans-interaction and gD binding. Since these cells express GFP-nectin-1, we could detect surface GFP-nectin-1 on B78H1-NGC12 cells with an anti-GFP pAb. Furthermore, the use of an anti-GFP Ab, which binds to the GFP portion fused to nectin-1, well away from the gD binding site, should confirm that the lack of detection of nectin-1 is not due to epitope masking by gD. As anticipated, GFP-nectin-1 was down-regulated from the B78H1-NGC12 cell surface after co-culture with effectors expressing gD(W294A) (Fig. 5A). In contrast, nectin-1 on effector B78H1-C10 cells had little effect on cell surface GFP-nectin-1 (Fig. 5A). This is in accordance with the presence of nectin-1 at cell contacts between nectin-1 expressing cells (Krummenacher et al., 2003). In the same co-cultures, we used GFP fluorescence to look at the total amount of GFP-nectin-1 in target cells (Fig. 5B). Exposure of B78H1-NGC12 target cells to nectin-1 effector B78H1-C10 cells had no effect on the total amount of GFP-nectin-1 present within target cells (Fig. 5B). Moreover, effector B78H1 cells did not cause down-regulation of nectin-1 on target B78H1-C10 cells (Fig. 3) or GFP-nectin-1 on B78H1-NGC12 cells (Fig. 4, 5). These data suggest that internalization and degradation of nectin-1 is a specific response to gD binding and that gD actively affects nectin-1 in ways that differ from the homophilic trans-interaction.
Figure 5.
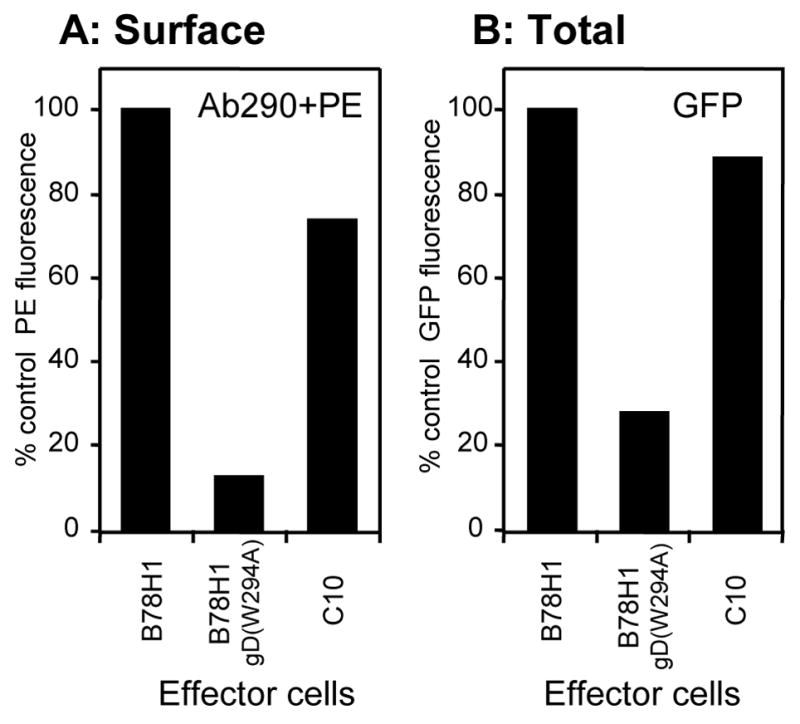
FACS analysis comparing effects of nectin-1 versus gD as ligands for nectin-1. Target B78H1-NGC12 cells (expressing GFP-nectin-1) were stained with Qtracker655 and co-cultivated with B78H1-C10 effector cells expressing nectin-1, B78H1 cells expressing gD(W294A) or untransfected B78H1 cells. GFP-nectin-1 was detected on Qtracker655-positive target B78H1-NGC12 cells in each co-culture. (A) Surface expression was detected with anti-GFP antibody Ab290 followed by PE-coupled secondary antibody. (B) Total expression was measured by GFP fluorescence. Results are reported as percentages of fluorescence after co-culture with B78H1 cells (no gD) used as control effectors. A representative experiment is shown.
HSV gD induces down-regulation of nectin-1 in non-transfected cell lines
As a model, we have studied the effects of cell-bound gD on human nectin-1 expressed in transfected B78H1 cells. We next asked whether endogenous nectin-1 was down-regulated by gD in natural HSV permissive cell lines. Human neuroblastoma SY5Y, epidermoid A431 and Hela cells as well as African green monkey Vero cells were selected because they express enough nectin-1 for it to be detected by FACS (Krummenacher et al., 2004). Each of these cell lines was pre-labeled with QTracker655 and co-cultured overnight with gD expressing effector cells. FACS analysis showed that nectin-1 was significantly down-regulated from the surface of SY5Y, A431 and Hela cells after co-culture with cells expressing gDwt or gD(W294A) (Fig. 6, white and gray bars respectively). Co-culture with effector cells expressing the non-binding gD(A3C-Y38C) (Fig. 6, black bars) had no significant effect as compared to the control co-culture with gD-negative B78H1 cells (Fig. 6, dotted line). Thus, nectin-1 expressed on the surface of several human cell lines is down-regulated upon trans-interaction with gD. In contrast, there was no evidence of nectin-1 down-regulation from the surface of Vero cells (Fig. 6). Vero nectin-1 functions as a gD receptor when transfected into the receptor-negative B78H1 cells (Milne et al., 2003). Thus, the absence of down-regulation of nectin-1 in Vero cells is not due to a lack of interaction with gD. Entry into human Hep-2 cells has been shown to occur at the plasma membrane (Wittels and Spear, 1990), however in these cells, nectin-1 expression was not detectable by FACS (data not shown) and thus, Hep-2 cells could not be included in the down-regulation experiment. We did however detect HVEM on these cells (data not shown). These observations do not rule out the use of nectin-1 as a receptor for HSV on these cells since we found previously that even levels of nectin-1 that were below the threshold of detection could be efficiently used for entry (Krummenacher et al., 2004).
Figure 6.
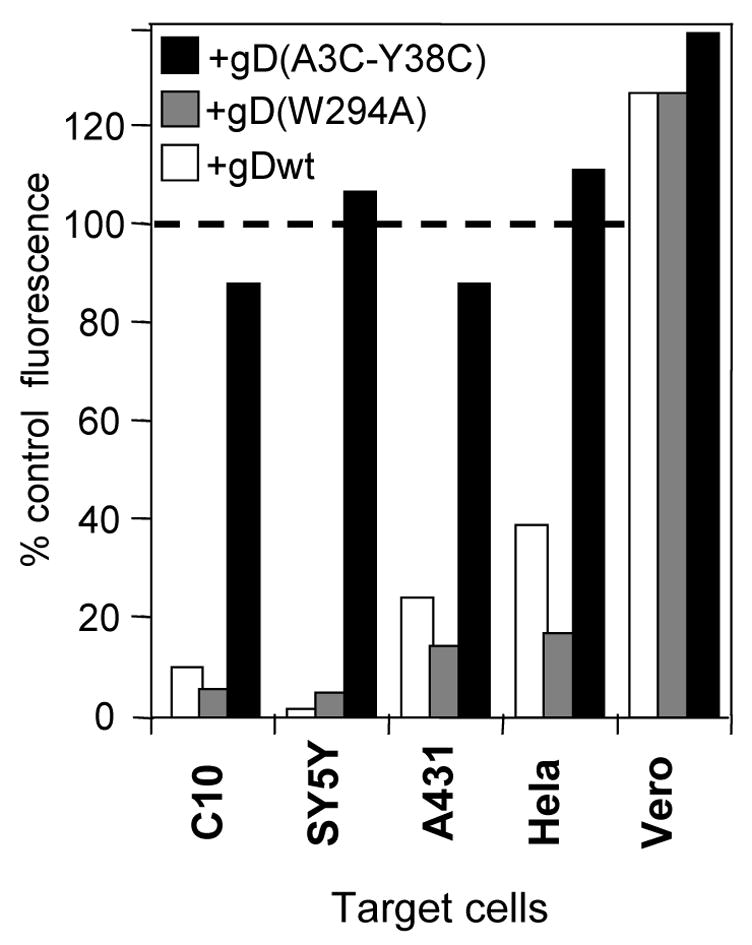
Down-regulation of receptors on non-transfected cell lines. Target cells were labeled with Qtracker655 and mixed with effector cells expressing various forms of gD. After an overnight co-culture, cells were stained with the appropriate antibody: CK41-PE (anti-nectin-1) for B78H1-C10, A431, SY5Y and Hela cells, and CK6 (anti-nectin-1) followed by anti-mouse Ig coupled with Alexa488 in the case of primate Vero cells. Antibody CK6 is used for Vero cells because CK41 does not recognize African green monkey nectin-1. CK6, like CK41, detects down-regulation of human nectin-1 (not shown). Geometric mean fluorescence was measured from Qtracker655-positive target cells after each co-culture. For comparison between cell lines, fluorescence on target cells is reported as the percentage of fluorescence after co-culture with control B78H1 (no gD) effector cells. A representative experiment is shown.
HSV enters A431 and SY5Y by endocytosis
Our data show that HSV gD induces down-regulation of nectin-1 from the surface of B78H1-C10 and Hela cells but not Vero cells. Interestingly, HSV enters B78H1-C10 and Hela cells by endocytosis, whereas it enters Vero cells by direct fusion with the plasma membrane (Milne et al., 2005; Nicola et al, 2003). Because down-regulation of nectin-1 likely reflects ligand-induced endocytosis, we hypothesized that gD-mediated internalization of nectin-1 is part of the mechanism involved in endocytosis of HSV. If so, virus entry into SY5Y and A431 cells, where nectin-1 is down-regulated by gD (Fig. 6), should be by endocytosis. We used a protease protection assay (Milne et al., 2005) to determine whether virions became endocytosed during HSV entry into A431 and SY5Y cells. Purified virus was added to cells at 4°C for attachment before entry was allowed to proceed for 15 min at 37°C. Then, the virus cell mixture was treated with proteinase K (PK) or mock digested. Glycoproteins that remain on the cell surface after direct fusion of the envelope with the plasma membrane or on attached virions are degraded by PK. In contrast, glycoproteins are protected from PK if the virus has been endocytosed (Milne et al., 2005). Total cell lysates were analyzed by Western blot to detect gB. We detected the same amount of virion gB bound to each cell type in the absence of PK treatment (Fig. 7A). As a control for protease activity, cells were left at 4°C (no entry) and PK completely degraded all the gB as all the virions remained attached to the cell surface (Fig. 7B). When the virus-cell mixture was incubated for 15 min at 37°C and then treated with PK, full-length gB was detected in A431 and SY5Y cells (Fig. 7C). As controls for endocytic entry, we used nectin-1 bearing B78H1-C10 cells (lane 1, positive control) and receptor negative B78H1 cells (lane 2, negative control) (Milne et al., 2005). The amount of protected gB represents only a fraction of the total input (Fig 7, panel C vs A) because internalization is not complete after 15 min (Milne et al., 2005). This assay does not rule out the possibility that some fusion takes place at the plasma membrane of A431 or SY5Y cells. However, in B78H1-C10 cells, fusion does not occur at the plasma membrane since blocking endocytosis abolishes virus entry into these cells (Milne et al., 2005).
Figure 7.
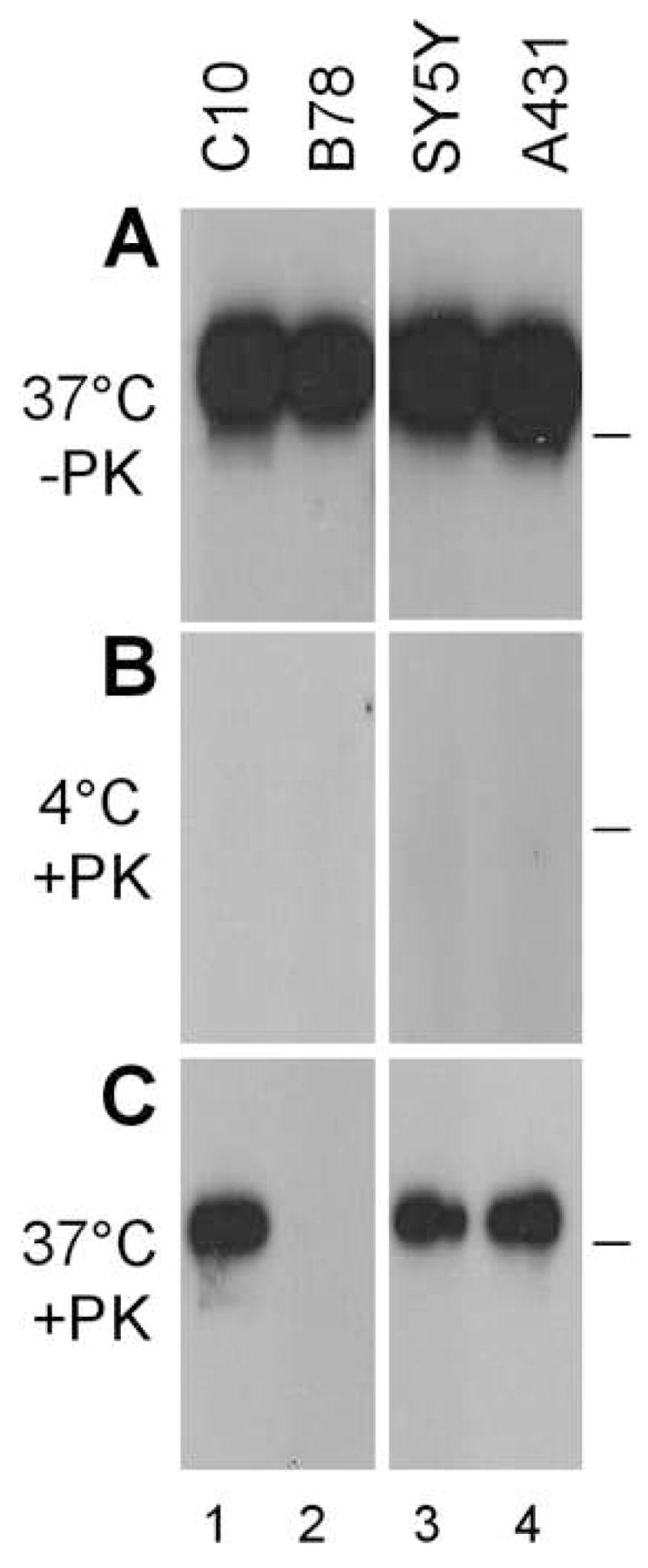
Virus protection during entry in B78H1-C10, B78, SY5Y and A431 cells. After attachment on the surface of the indicated cells at 4°C, HSV-1 KOS infection was allowed to proceed at 37°C for 15 min (A, C) or left at 4°C (B). Cells were then chilled and treated with proteinase K (B, C) or mock digested (A). After lysis in the presence of protease inhibitors, full-length gB was detected by immunoprecipitation (mAb DL16) and western blot (pAb R69). (A) Total amount of gB bound to cells without proteinase K digestion. (B) Control for total digestion of surface gB by proteinase K in the absence of entry. (C) Protected gB indicative of virus internalization at 37°C prior to protease treatment. PK means proteinase K and the bar on the right indicates the position of the 114 kDa molecular weight marker.
We previously showed that virion gB was protected from PK treatment during entry into Hela cells but not into Vero cells (Milne et al., 2005). Thus, the same cells that respond to gD binding by internalizing nectin-1 (i.e. B78H1-C10, SY5Y, A431 and Hela) are also permissive for HSV entry by endocytosis (Fig. 7 and (Milne et al., 2005; Nicola et al, 2003)). Altogether, this suggests that the two events might be related.
gD is required for HSV endocytosis in B78H1-C10 cells
HSV gD is necessary for fusion during endocytic entry (Nicola and Straus, 2004). Here, we analyzed the role of gD in the initial internalization of virions from the cell surface. We examined whether virion gB was protected from protease digestion using preparations of the gD-null virus KOSgDβ that had been either grown in VD60 cells to provide wt gD (complemented) or grown in Vero cells (non complemented). The presence or absence of gD was confirmed for each virus preparation by Western blot (WB) (data not shown). Since non-complemented gD-null virus could not be directly titered, the amount of each virus added to B78H1-C10 cells was based on equal amounts of capsid protein VP5 as detected by Western blotting (Handler et al, 1996). When complemented and non-complemented HSV KOSgDβ were attached to cells at 4°C and then allowed to enter at 37°C for 15 min, we detected similar amounts of gB (Fig. 8, lanes 1 and 2, no PK treatment). When cells were left at 4°C to prevent entry, all gB from both virus preparations was digested by PK (Fig. 8, lanes 3 and 4). When the two virus preparations were incubated for 15 min at 37°C and treated with PK, a significant fraction of gB from the gD-complemented virus was protected (Fig. 8, lane 5). No protected gB was detected when the non-complemented gD-null virus was used (Fig. 8, lane 6). This shows that gD is necessary for HSV endocytosis during entry. Since nectin-1 is also required for HSV endocytosis in B78H1-C10 cells (Fig. 6 and (Milne et al., 2005)), these data support the role for the gD-nectin-1 interaction in this process.
Figure 8.

Virus protection from protease digestion requires envelope gD. KOSgDβ virions devoid of envelope gD (Non-complemented virus labeled N) or complemented with wt gD KOS (labeled C) were attached to B78H1-C10 cells at 4°C. After 45 min temperature was shifted to 37°C for 15 min or the cells were left at 4°C as indicated. Cells were chilled on ice and extracellular proteins were digested with proteinase K where indicated (+PK). After lysis in the presence of protease inhibitors, gB was detected by immunoprecipitation with mAb DL16 and western blot (pAb R69). The bar on the right indicates the position of the 114 kDa molecular weight marker.
DISCUSSION
Our data show that the trans-interaction of gD with nectin-1 on the cell surface is sufficient to induce internalization of the receptor on adjacent cells in the absence of other viral components. This response was observed in cells that HSV infects by endocytosis but not in Vero cells where infection occurs at the plasma membrane. We also showed that gD is necessary for HSV endocytosis into cells expressing nectin-1. Taken together, these results suggest that gD binding to nectin-1 during entry actively directs HSV to an endocytic pathway in cell types where this pathway is preferred.
gD actively induces nectin-1 down-regulation in trans
The loss of nectin-1 occurs when target cells were co-cultivated with effector cells expressing forms of gD capable of binding to nectin-1. There is no effect from gD(A3C-Y38C), a form of gD which cannot bind nectin-1. We therefore infer that nectin-1 internalization is a consequence of an interaction with gD. The consequences of gD binding to nectin-1 differ dramatically from those that follow homophilic nectin-1 trans-interaction; binding of gD results in internalization and degradation of nectin-1 whereas homophilic binding of nectin-1 leads to accumulation at cell contacts (Krummenacher et al., 2002; Miyahara et al., 2000).
We previously showed that gD co-localized with nectin-1 at junctions between infected and non-infected B78H1 expressing nectin-1-GFP cells, notably at the edge of viral plaques (Krummenacher et al., 2003). Such co-localization was also observed, albeit rarely, in cultures of B78H1-C10 cells infected at high MOI with HSV. However, we did not see any co-localization of gD with nectin-1 in overnight co-cultures between gD and nectin-1 expressing cells (Fig. 2). We observed extensive co-localization of gD and nectin-1 between target and effector cells after short co-cultures (up to 1h) and a decrease in their occurrence at later times (data not shown). This is consistent with a transient interaction between gD and nectin-1 which is followed by down-regulation of the receptor in the target cells. Such temporal co-localization would be expected at the edge of viral plaques, where infected cells continuously interact with new uninfected cells as the plaque expands.
Nectin-1 does not accumulate at contacts with cells that do not express a functional ligand (e.g. B78H1 cells, or B78-gD(A3C-Y38C)) however, it remains on the cell surface. The internalization of nectin-1 only occurred in the presence of gD, suggesting that the viral glycoprotein actively induces this specific response of its cellular receptor.
Internalization of nectin-1 in response to gD in trans leads to an apparent paradox. We have shown that surface nectin-1 is reduced on cells that have been co-cultures with gD expressing cells. A similar situation may occur with infected cells since gD is known to occur before progeny virions are released. If gD on the surface of infected cells causes internalization of nectin-1 on adjacent non-infected cells, does this make the uninfected cell resistant to infection? Overall the availability of nectin-1 may be decreased. We hypothesize that uninfected cells continuously produce the receptor on their surface that is rapidly endocytosed upon binding to gD. In that case, newly made receptor appearing on the surface of uninfected cells may encounter either cell-bound gD and be down-regulated, or gD on egressing virions and lead to virus endocytosis. It is not yet clear whether down-regulation of nectin-1 plays a role in slowing down or shaping virus spread.
Furthermore, it appears that gD expressed on the cell surface is sufficient to trigger nectin-1 internalization. Soluble gD blocks HSV entry by binding to the receptor. It is possible that, if it can lead to nectin-1 internalization, this may partially contribute to the inhibition of HSV entry (Nicola et al, 1997). Interestingly, gD does not need to be functional in membrane fusion to trigger nectin-1 internalization. We previously showed that gD(W294A) was functionally impaired in complementing a gD-null virus despite a higher affinity for nectin-1 and HVEM (Krummenacher et al., 2005). We also identified the defect in gD(W294A) as the inability to trigger the viral fusion machinery after receptor binding (Krummenacher et al., 2005). Here, nectin-1 internalization relies on the ability of gD to bind, even if it is unable to induce fusion. One could anticipate that virions carrying gD(W294A) could be internalized but unable to fuse with the endosome in B78H1-C10 cells. Thus, there appears to be a temporal separation between gD binding to its receptor on the cell surface and fusion initiation after internalization during endocytic entry.
Nectin-1 internalization leads to low-pH dependent degradation
The inability to detect nectin-1 in the supernatant of co-cultured cells indicates that nectin-1 was not released from the cell surface upon gD binding but was internalized. The efficient internalization of nectin-1 induced by gD that is anchored on another cell implies either dissociation of the two molecules before nectin-1 internalization or co-internalization of both gD (or a portion of it) and nectin-1. In the case of dissociation, the ability of the high-affinity gD(W294A) to achieve nectin-1 down-regulation as efficiently as wt gD may seem surprising, but in fact, both forms of gD dissociate from nectin-1 with the same kinetics in vitro (Krummenacher et al., 2005). It is not known if virion gD dissociates from its receptor in the process leading to membrane fusion during entry. Alternatively, it is equally possible that gD remains bound and that all or part of it is transferred to the target cell. We have failed to detect the transfer of gD to the surface of or inside target cells as a result of binding to nectin-1 (Fig. 2 and FACS data not shown). The effect of BFLA on nectin-1 degradation shows, albeit indirectly, that the receptor is internalized during down-regulation. Internalization of nectin-1 is followed by low-pH dependent intracellular degradation. The internalization pathway followed by nectin-1 has not yet been defined, but the effect of BFLA indicates that it leads to an acidic compartment. The inhibition of nectin-1 degradation by BFLA and by cysteine-protease inhibitor E-64d suggests that lysosomal low-pH dependent cathepsins are involved in this process.
gD-mediated internalization of nectin-1 correlates with HSV endocytic entry
The fact that wt gD anchored in a plasma membrane can induce internalization of its receptor by binding in trans suggests that the same response can be triggered by gD exposed on the viral envelope. In most cell types, virions are endocytosed prior to fusion of the envelope with a vesicular membrane in a low pH-dependent or independent manner (Milne et al., 2005; Nicola et al, 2003). In transfected B78H1 cells, a gD receptor, nectin-1 or HVEM, is required for internalization of HSV (Fig. 7 and Milne et al., 2005). Here, we show that gD on the viral envelope is necessary for virus endocytosis in B78H1-C10 cells expressing nectin-1 (Fig. 8). Thus, in these cells the interaction between gD and nectin-1 appears to be necessary to direct virions to an endocytic pathway.
In other cell lines where virus entry follows an endocytic pathway, such as Hela (Nicola and Straus, 2004), SY5Y and A431 cells (Fig. 7), gD also induced nectin-1 down-regulation. In contrast, virions are not endocytosed during entry in Vero cells (Milne et al., 2005; Nicola et al, 2003), and similarly, nectin-1 is not down-regulated upon exposure to gD expressing cells. The fact that gD-mediated down-regulation of nectin-1 and HSV endocytosis appear to have the same cell type specificity further supports the idea that the gD-receptor interaction is involved in selection of the entry pathway. Identification of additional human cell types that support fusion at the plasma membrane will be instrumental in reinforcing the correlation between nectin-1 internalization and virus endocytosis. Unfortunately, the low level of nectin-1 expression in Hep-2 cells, which, like Vero cells, are infected at the plasma membrane (Wittels and Spear, 1990) did not allow the use of these cells to further support this correlation. Since the gD-nectin-1 interaction is not sufficient for virus endocytosis or for nectin-1 internalization in Vero cells, other cell characteristics allowing fusion at the plasma membrane and hindering nectin-1 down-regulation likely affect the route of HSV entry.
Other determinants of HSV endocytosis
Currently, Vero cells provide the only well-characterized model for HSV induced fusion at the plasma membrane and for the absence of down-regulation of nectin-1. It is not yet clear whether Vero cells are an exception or if they are representative of a category of cells with similar characteristics. In primary sensory neurons, virions fusing at the plasma membrane and naked cytoplasmic capsids were observed 2 hours post-infection (Lycke et al., 1988). This contrasts with data from our virus protection assay showing that virions are rapidly endocytosed in neuroblastoma SY5Y cells. Virus endocytosis occurs within the first 30 min with kinetics that are consistent with the rapid rate of virus entry in synchronized infections (Milne et al., 2005; Nicola et al., 2005). Thus, it is not clear if fusion at the plasma membrane occurring 2h p.i. would lead to a productive infection. At that late time, it is also possible that capsids in the cytoplasm resulted from fusion with an endosome. However, it is conceivable that cellular differences between primary sensory neurons and passaged neuroblastoma cell lines affect the HSV entry pathway.
HSV can enter cells in a low pH independent fashion, at the cell surface (e.g. on Vero cells) and after endocytosis (e.g. in B78H1-C10 cells)(Milne et al., 2005; Nicola and Straus, 2004). We previously showed that HSV entry into B78H1-C10 cells was prevented when endocytosis was inhibited (Milne et al., 2005). This suggested that the virus cannot fuse on the surface of these cells when its route of entry (endocytosis) is blocked. HSV entry by low pH independent endocytosis was also observed for SY5Y and A431 cells, however, further studies are required to determine if, like for the model B78H1-C10 cells, entry into cells using this pathway is also prevented by inhibiting endocytosis. Ultimately, it is likely that the cell specific determinants that allow endocytosis differ from those that prevent fusion on the cell surface.
We have shown that A431 and Hela cells, in addition to nectin-1, have detectable levels of HVEM on their surface (Krummenacher et al., 2004). It is possible that HVEM can be used by HSV for entry into these cells. HVEM is a functional receptor in oral epithelial cells (Hung et al., 2002) and in corneal fibroblasts and trabecular meshwork cells in the eye (Tiwari et al., 2005; Tiwari et al., 2007). The entry pathway into these cells has not yet been defined. Since HVEM, like nectin-1, can mediate endocytic entry into CHO cells (Nicola and Straus, 2004), the entry pathway does not appear to be specified only by the nature of the receptor. Based on our results with nectin-1, we speculate that gD and HVEM may be involved in internalization of virions during endocytic entry into cells expressing HVEM as the only receptor, such as CHO-HVEM cells (Nicola et al, 2003) or B78H1-A10 (Milne et al., 2005). It is important to note that in CHO cells lacking a receptor, HSV is endocytosed and degraded since a gD receptor is necessary for productive entry (Nicola et al, 2003). It is unclear if CHO cells with and without gD-receptor endocytose HSV in the same manner.
Recently, Delboy et al. (2006) analyzed entry of the mutant strain HSV-1 ANG path into CHO cells expressing nectin-1 or nectin-2 as the gD receptor. This study showed that the route of entry of ANG path in CHO cells is affected by gD determinants influencing receptor usage and by cellular factors. Our data using HSV-1 KOS and various human cell lines are in accordance with these notions and lead to the conclusion that the selection of an entry pathway involves gD binding to its receptor and depends on cell characteristics that vary from one cell type to the other.
Down-regulation of receptors by other neurotropic viruses
Many viruses are endocytosed upon attaching to a cell surface receptor (Smith and Helenius, 2004). In most cases, it is difficult to distinguish between active triggering of internalization and passive endocytosis whereby virions bind to shuttling receptors. For HSV, our data on nectin-1 down-regulation suggest an active induction of receptor internalization by gD leading to virion endocytosis. Constitutive nectin-1 internalization might occur naturally but there is no reported indication of shuttling of nectin-1 to and from the cell surface. Should it happen, our data would indicate that gD binding would accelerate internalization of nectin-1 from the cell surface and redirect the internalized nectin-1 to a degradation pathway.
In the nectin family, the poliovirus receptor (necl-5) mediates internalization of poliovirus and further allows endosomal movement by interacting with the dynein light chain (Ohka et al., 2004). Another neurotropic virus, Measles virus (MV) can use two unrelated receptors for entry, the homophilic adhesion molecule SLAM and CD46 (Dorig et al., 1993; Tatsuo et al., 2000). Both receptors are down-regulated from the cell surface upon trans-interacting with wt MV hemagglutinin in co-culture assays (Tanaka et al., 2002; Welstead et al., 2004). Although virus endocytosis is not limited to neurotropic viruses, the down-regulation of receptor as part of virus entry that we observed with HSV gD appears to be shared by viruses in different families using receptors with related structures (Ig-like) and functions (cell adhesion).
MATERIALS AND METHODS
Viruses, antibodies and proteins
i) Viruses
HSV-1 KOS was grown and titered on Vero cells and purified as described (Handler et al, 1996). KOS gDβ was obtained from Dr. P. G. Spear (Dean et al., 1994). Non-complemented KOSgDβ was produced in Vero cells and complemented viruses were produced on VD60 cells (Dean et al., 1994; Ligas and Johnson, 1988). Titration of KOSgDβ was performed on VD60 cells.
ii) Antibodies
Anti-nectin-1 monoclonal antibodies (mAbs) CK6, CK8 and CK41 as well as rabbit polyclonal sera (pAb) R154 and R165 were described previously (Krummenacher et al., 1998; Shukla et al., 2000). For FACS, CK41 was directly coupled with phycoerythrin (PE) at Molecular Probes/Invitrogen. Anti-HSV glycoproteins antibodies were as follows: Against gD, rabbit polyclonal sera, pAbs R7 and R8 (Isola et al., 1989), mouse monoclonal Ab MC5 (Atanasiu et al., 2007); against gB, pAb R69 (Eisenberg et al., 1987) and mAb DL16 (Bender et al., 2005). Rabbit anti-GFP pAb Ab290 was purchased from Abcam. Secondary anti-IgG coupled with PE, Alexa594 or Alexa488 were purchased from Invitrogen.
iii) Cell lines
SY5Y, A431 and Hep-2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum (FCS) and antibiotics (penicillin and streptomycin). Vero, Hela and murine melanoma B78H1 cells were grown in DMEM with 5% FCS and antibiotics. Previously described B78H1 transfected cell lines B78H1-C10, B78H1-NGC12 and B78H1-control-16 (abbreviated here B78) were grown in the same medium supplemented with 500 μg/ml G418 (Krummenacher et al., 2003).
Cell lines expressing HSV gD
i) Selection
B78H1 cells were transfected with plasmids pSC390 to express gDwt (Connolly et al., 2003), pDL473 for gD(W294A) (Krummenacher et al., 2005) or pDL490 for gD(A3C-Y38C) (Connolly et al., 2005) as described for nectin-1-GFP constructs (Krummenacher et al., 2003). Clones were selected by limiting dilution in the presence of 1 mg/ml G418 and further maintained in DMEM with 5% FCS, antibiotics and 500 μg/ml G418. Clones to be used in this study were selected by immunofluorescence, CELISA and FACS for their level of gD expression. Clonal cell lines used in this study are: B78-gDwt clone #13, B78-gD(W294A) clone #23 and B78-gD(A3C-Y38C) clone #25.
ii) gD expression
For flow cytometry (FACS) experiments, cells were detached with 0.02% di-sodium EDTA (w/v) in PBS (Versene; Gibco-BRL) and resuspended in PBS containing 3% FCS, 0.01% sodium azide (PBS-FCS). Anti-gD serum R7 was diluted 100x in cold PBS-FCS, 50μl was added to 3×105 cells. Cells were incubated for 30 min on ice and then washed with 1 ml cold PBS-FCS before being incubated in 50ul PBS-FCS containing anti-rabbit IgG coupled with Alexa488 (Mol. Probes) for 30 min on ice. Cells were washed as previously and fixed in 300 μl 3% paraformaldehyde in PBS-FCS. Cell surface expression was also tested by Cellular ELISA (CELISA) using anti-gD rabbit pAb R8, as described by Connolly et al. (Connolly et al., 2005).
iii) Binding of soluble nectin-1 by CELISA
Soluble nectin-1 ectodomain (HveC(346t)) was produced in a baculovirus expression system (Krummenacher et al., 1998) and the purified protein was diluted in DMEM, 5% FCS, added to 5×104 cells in 96-well plates and incubated for 30 min at 37°C. Cells were washed 3 times with PBS and fixed with 100 μl 3% paraformaldehyde in PBS at room temperature (RT) for 30 min. A mixture of anti-nectin-1 rabbit sera R154 and R165, each one diluted 1000x in PBS containing 5% FCS, were added to cells and incubated for 90 min at RT. Cells were washed as before and incubated with anti-rabbit IgG coupled to peroxidase (2 μg/ml) for 1h followed by substrate (ABTS, Moss Inc.).
Cell co-cultures
i) Immunofluorescence analysis
Target and effector cells were detached with trypsin, mixed at a 1:1 ratio and co-cultured for 16 h on glass coverslips (1–2×105 cells per coverslip). The culture medium was DMEM supplemented with 5% FCS and antibiotics. Cells were fixed with 3% paraformaldehyde in PBS for 30 min at RT. Quenching, permeabilization and staining were performed as previously described (Krummenacher et al., 2003; Sodeik et al, 1997). Anti-nectin-1 mAb CK41 and anti-gD pAb R7 were added first, then followed by goat anti-mouse IgG coupled with Alexa488 (for CK41) and goat anti-rabbit IgG coupled with Alexa594 (for R7). No crossreaction was observed. Images were collected on a Nikon Eclipse E600 microscope equipped with a 40X objective and analyzed using Image-Pro Plus software (Media Cybernetics, Inc).
ii) FACS analysis
Target cells expressing nectin-1 were detached with trypsin, then counted and divided in 0.8 ml aliquots at 5×106 cells/ml for labeling with Qdots using the Qtracker ™ 655 labeling kit (Quantum Dot Corp/Invitrogen, Hayward CA). Cells in suspension were exposed to the dye for 1h at 37°C in DMEM supplemented with 5% FCS following the manufacturer’s instructions. This labeling was done to distinguish and gate target cells during FACS analysis. These target cells were washed 3 times with culture medium and mixed with twice as many unlabeled effector cells that had been previously detached and counted. A total of 1.5×106 cells were plated in each well of a 6-well plate and cultivated overnight in DMEM supplemented with 5% FCS and antibiotics. Target and effector cells were also cultivated separately as controls. Cells were detached with Versene and resuspended in cold PBS-FCS. Labeling with anti-nectin-1 mAb CK41-PE (5 μg/ml) was performed on 0.5×106 cells (50 μl) for 30 min on ice. Mock stained cells were used as negative controls. Alternatively cells were stained with anti-GFP pAb Ab290 (diluted 400x in PBS-FCS) followed by PE-labeled secondary antibody. Cells were washed with cold PBS-FCS and fixed with 3% paraformaldehyde in PBS-FCS. In order to gate target cells during FACS analysis, Qtracker655 was detected by excitation at 630 nm and reading at 660+/−15 nm. Qtracker655-positive target cells were positively selected for measurement of PE and/or GFP fluorescence.
iii) Western blot analysis
1×106 target cells were mixed with 1.5×106 effector cells in 12-well plates and co-cultured overnight in 2 ml DMEM supplemented with 5% FCS and antibiotics. Cells were washed with PBS, detached with Versene, pelleted and resuspended in 200 μl cold lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100) with Complete protease inhibitor cocktail (Roche). After lysis on ice for 30 min, lysates were cleared by centrifugation. Proteins were separated by SDS-PAGE under reducing and denaturing conditions on 10% acrylamide gels and transferred to nitrocellulose for detection with anti-nectin-1 mAb CK8. In specific experiments, bafilomycin A1 (BFLA, Sigma-Aldrich Co.) diluted in DMSO was added to the medium of target cells to a final concentration of 10 nM. As a control, DMSO only was used. After 1 h incubation, effector cells were added and the same concentration of BFLA or DMSO was maintained during the 5 h of co-culture. Cells were washed and lysed as above. For detection of nectin-1 in co-culture supernatant, cells were mixed as above and co-cultivated in 0.5 ml growth medium (DMEM supplemented with 5% FCS, 500 μg/ml G418, and penicillin/streptomycin). The supernatant was collected after overnight co-culture, 10 microliters of 50x complete protease inhibitor cocktail (Roche) was added. Cell debris were eliminated by centrifugation (20 min, 12000 rpm, 4°C). Cells were washed three times with cold PBS and lysed as above. For western blot analysis, an equivalent amount of cell lysate and co-culture supernatant (5% of total for each sample) were loaded for direct quantitative comparison. As a control we electrophoresed known amounts of recombinant nectin-1 ectodomain (nectin-1(346t)) (Krummenacher et al., 1998) diluted in fresh culture medium.
Proteinase K protection assay
To detect virion internalization, proteinase K protection assays were carried out largely according to a published method (Milne et al., 2005). Briefly, confluent monolayers of cells in 25 cm2 flasks (3×106 cells/flask) were chilled on ice. Three flasks of each cell type were inoculated with sucrose gradient purified HSV-1 KOS (input multiplicity 30 pfu/cell in 10% DMEM containing 30mM HEPES). After 45 min adsorption at 4°C, two flasks were transferred to a 37°C water bath to initiate virus entry, while the third remained at 4°C. After 15 min, all three flasks were placed on ice. Cells were washed once with Hank’s balanced salt solution containing 30 mM HEPES (HBSS-HEPES), then treated or mock treated for 1 h with proteinase K (50 μg/ml in HBSS-HEPES, 1 mM CaCl2). Cells were pelleted in a 2 ml tube and lysed on ice with Tris buffered saline (TBS) containing 1% NP40 and 1mM PMSF. After 20 minutes, lysates were cleared by microcentrifugation. gB was immunoprecipitated with mAb DL16, resolved on SDS-PAGE and detected by western blotting using rabbit serum R69. When gD-null virus was used, the input multiplicity was based on the titer (on VD60 cells) of the phenotypically complemented virus preparation.
Propagation and partial purification of KOS gDβ
The gD negative virus KOSgDβ was propagated on VD60 cells, which supply gD upon infection (Dean et al., 1994; Ligas and Johnson, 1988) for complementation. To produce paired stocks of phenotypically null and gD complemented virions, duplicate confluent 225 cm2 flasks of Vero or VD60 cells were inoculated with complemented virus stocks at an input multiplicity of 5 pfu/cell. When the cytopathic effects had progressed to completion (about 48 h), cells and medium were collected, subjected to two cycles of freezing at −70°C and thawing at 37°C. Cell debris was removed by centrifugation at 3500 rpm (2850x g), then each of the cleared virus preparations was layered onto 20% sucrose (in PBS) and the virions pelleted at 25000 rpm in a Beckman SW28 rotor. Virions were resuspended overnight in PBS at 4°C, then pelleted again using the same conditions. Virus pellets were finally resuspended in PBS, aliquoted and stored at −70°C. Preparations were analyzed for virion proteins by western blotting and for infectivity by titration on Vero and VD60 cells.
Acknowledgments
This investigation was supported by Public Health Service grant AI-056045 to R.J.E. from the National Institute of Allergy and Infectious Diseases as well as AI-18289 to G.H.C. from NIAID and NS-36731 to R.J.E. from the National Institute of Neurological Disorders and Stroke. C.K. was supported by an award from the University of Pennsylvania Research Foundation. K.M.S. was supported by NIH training grant T32-GM07229.
We are grateful to P. G. Spear for the gD-null KOSgDβ virus and Hep-2 cells and D. C. Johnson for VD60 cells. We thank Ali Zekavat and Bruce Shenker for FACS processing at the Flow Cytometry Facility of the UPENN School of Dental Medicine. We thank Sarah Connolly and Dan Landsburg for plasmids and Isabelle Baribaud for help with fluorescence microscopy. We are grateful to the members of the Cohen and Eisenberg laboratory for advice and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0707452104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol. 2005;79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, Menotti L, Mirandola P, Lopez M, Campadelli-Fiume G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attribute of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J Virol. 1998;72:9992–10002. doi: 10.1128/jvi.72.12.9992-10002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ. Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J Virol. 2005;79:1282–1295. doi: 10.1128/JVI.79.2.1282-1295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD/HveA interface. J Virol. 2003;77:8127–8140. doi: 10.1128/JVI.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Regge N, Nauwynck HJ, Geenen K, Krummenacher C, Cohen GH, Eisenberg RJ, Mettenleiter TC, Favoreel HW. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J Cell Biol. 2006;174:267–275. doi: 10.1083/jcb.200510156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean HJ, Terhune SS, Shieh M, Susmarski N, Spear PG. Single amino acid substitutions in gD of herpes simplex virus 1 confer resistance to gD-mediated interference and cause cell-type-dependent alterations in infectivity. Virology. 1994;199:67–80. doi: 10.1006/viro.1994.1098. [DOI] [PubMed] [Google Scholar]
- Delboy MG, Patterson JL, Hollander AM, Nicola AV. Nectin-2-mediated entry of a syncytial strain of herpes simplex virus via pH-independent fusion with the plasma membrane of Chinese hamster ovary cells. Virol J. 2006;3:105. doi: 10.1186/1743-422X-3-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- Eisenberg RJ, Ponce de Leon M, Friedman HM, Fries LF, Frank MM, Hastings JC, Cohen GH. Complement component C3b binds directly to purified glycoprotein C of herpes simplex virus types 1 and 2. Microb Pathog. 1987;3:423–435. doi: 10.1016/0882-4010(87)90012-x. [DOI] [PubMed] [Google Scholar]
- Fuller AO, Spear PG. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc Natl Acad Sci USA. 1987;84:5454–5458. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Forghieri C, Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D, gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci USA. 2005;102:9323–9328. doi: 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Eisenberg RJ, Cohen GH, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Handler CG, Cohen GH, Eisenberg RJ. Crosslinking of glycoprotein oligomers during herpes simplex virus type 1 entry. J Virol. 1996;70:6076–6082. doi: 10.1128/jvi.70.9.6076-6082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung SL, Cheng YY, Wang YH, Chang KW, Chen YT. Expression and roles of herpesvirus entry mediators A and C in cells of oral origin. Oral Microbiol Immunol. 2002;17:215–223. doi: 10.1034/j.1399-302x.2002.170403.x. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Irie K, Deguchi-Tawarada M, Ikeda W, Ohtsuka T, Takeuchi M, Takai Y. Nectin-dependent localization of ZO-1 at puncta adhaerentia junctions between the mossy fiber terminals and the dendrites of the pyramidal cells in the CA3 area of adult mouse hippocampus. J Comp Neurol. 2003;460:514–524. doi: 10.1002/cne.10653. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Irie K, Ishizaki H, Tanaka-Okamoto M, Morimoto K, Inoue E, Ohtsuka T, Miyoshi J, Takai Y. Roles of cell-adhesion molecules nectin 1 and nectin 3 in ciliary body development. Development. 2005;132:1525–1537. doi: 10.1242/dev.01697. [DOI] [PubMed] [Google Scholar]
- Isola VJ, Eisenberg RJ, Siebert GR, Heilman CJ, Wilcox WC, Cohen GH. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J Virol. 1989;63:2325–2334. doi: 10.1128/jvi.63.5.2325-2334.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Webb M, Wisner TW, Brunetti C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol. 2001;75:821–833. doi: 10.1128/JVI.75.2.821-833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Ingano LA, Kovacs DM. Nectin-1alpha, an immunoglobulin-like receptor involved in the formation of synapses, is a substrate for presenilin/gamma-secretase-like cleavage. J Biol Chem. 2002;277:49976–49981. doi: 10.1074/jbc.M210179200. [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud F, Ponce De Leon M, Baribaud I, Whitbeck JC, Xu R, Cohen GH, Eisenberg RJ. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology. 2004;322:286–299. doi: 10.1016/j.virol.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Eisenberg RJ, Cohen GH. Cellular localization of nectin-1 and glycoprotein D during herpes simplex virus infection. J Virol. 2003;77:8985–8999. doi: 10.1128/JVI.77.16.8985-8999.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Ponce de Leon M, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Localization of a binding site for herpes simplex virus glycoprotein D on the herpesvirus entry mediator C by using anti-receptor monoclonal antibodies. J Virol. 2000;74:10863–10872. doi: 10.1128/jvi.74.23.10863-10872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Sanzo JF, Cohen GH, Eisenberg RJ. Effects of herpes simplex virus on structure and function of nectin- 1/HveC. J Virol. 2002;76:2424–2433. doi: 10.1128/jvi.76.5.2424-2433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Carfi A, Eisenberg RJ, Cohen GH. Pöhlmann S, Simmons G, editors. Herpesvirus entry into cells: The Enigma Variations. Viral entry into host cells Landes Bioscience. 2007 Vol. in press. Online edition http://www.eurekah.com/chapter/3035.
- Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol. 1998;72:7064–7074. doi: 10.1128/jvi.72.9.7064-7074.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005;24:4144–4153. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligas MW, Johnson DC. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycke E, Hamark B, Johansson M, Krotochwil A, Lycke J, Svennerholm B. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch Virol. 1988;101:87–104. doi: 10.1007/BF01314654. [DOI] [PubMed] [Google Scholar]
- Mata M, Zhang M, Hu X, Fink DJ. HveC (nectin-1) is expressed at high levels in sensory neurons, but not in motor neurons, of the rat peripheral nervous system. J Neurovirol. 2001;7:476–480. doi: 10.1080/135502801753170336. [DOI] [PubMed] [Google Scholar]
- Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol Ther. 2001;3:160–168. doi: 10.1006/mthe.2000.0240. [DOI] [PubMed] [Google Scholar]
- Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol. 2005;79:6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RSB, Hanna SL, Rux AH, Willis SH, Cohen GH, Eisenberg RJ. Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J Virol. 2003;77:8962–8972. doi: 10.1128/JVI.77.16.8962-8972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara M, Nakanishi H, Takahashi K, Satoh-Horikawa K, Tachibana K, Takai Y. Interaction of nectin with afadin is necessary for its clustering at cell-cell contact sites but not for its cis dimerization or trans interaction. J Biol Chem. 2000;275:613–618. doi: 10.1074/jbc.275.1.613. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Nakanishi H, Kimura K, Matsubara K, Ozaki-Kuroda K, Katata T, Honda T, Kiyohara Y, Heo K, Higashi M, Tsutsumi T, Sonoda S, Ide C, Takai Y. Nectin: an adhesion molecule involved in formation of synapses. J Cell Biol. 2002;156:555–65. doi: 10.1083/jcb.200103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Nicola AV, Hou J, Major EO, Straus SE. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J Virol. 2005;79:7609–7616. doi: 10.1128/JVI.79.12.7609-7616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Peng C, Lou H, Cohen GH, Eisenberg RJ. Antigenic structure of soluble herpes simplex virus (HSV) glycoprotein D correlates with inhibition of HSV infection. J Virol. 1997;71:2940–2946. doi: 10.1128/jvi.71.4.2940-2946.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Straus SE. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol. 2004;78:7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka S, Matsuda N, Tohyama K, Oda T, Morikawa M, Kuge S, Nomoto A. Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J Virol. 2004;78:7186–7198. doi: 10.1128/JVI.78.13.7186-7198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reske A, Pollara G, Krummenacher C, Chain BM, Katz DR. Understanding HSV-1 entry glycoproteins. Rev Med Virol. 2007;17:205–215. doi: 10.1002/rmv.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakisaka T, Takai Y. Biology and pathology of nectins and nectin-like molecules. Curr Opin Cell Biol. 2004;16:513–521. doi: 10.1016/j.ceb.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Sakisaka T, Taniguchi T, Nakanishi H, Takahashi K, Miyahara M, Ikeda W, Yokoyama S, Peng YF, Yamanishi K, Takai Y. Requirement of interaction of nectin-1alpha/HveC with afadin for efficient cell-cell spread of herpes simplex virus type 1. J Virol. 2001;75:4734–4743. doi: 10.1128/JVI.75.10.4734-4743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, DalCanto MC, Rowe CL, Spear PG. Striking similarity of murine nectin-1α to human nectin-1α (HveC) in sequence and activity as a gD receptor for alphaherpesvirus entry. J Virol. 2000;74:11773–11781. doi: 10.1128/jvi.74.24.11773-11781.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol. 2005;11:208–18. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- Smith AE, Helenius A. How viruses enter animal cells. Science. 2004;304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol. 1997;136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian RP, Geraghty RJ. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci USA. 2007;104:2903–2908. doi: 10.1073/pnas.0608374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, Mizoguchi A, Takai Y. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with afadin, a PDZ domain-containing protein. J Cell Biol. 1999;145:539–549. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Nakanishi H. Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci. 2003;116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- Takai Y, Shimizu K, Ohtsuka T. The roles of cadherins and nectins in interneuronal synapse formation. Curr Opin Neurobiol. 2003;13:520–526. doi: 10.1016/j.conb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Minagawa H, Xie MF, Yanagi Y. The measles virus hemagglutinin downregulates the cellular receptor SLAM (CD150) Arch Virol. 2002;147:195–203. doi: 10.1007/s705-002-8312-0. [DOI] [PubMed] [Google Scholar]
- Tatsuo H, Ono N, Tanaka K, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- Tiwari V, Clement C, Scanlan PM, Kowlessur D, Yue BY, Shukla D. A role for herpesvirus entry mediator as the receptor for herpes simplex virus 1 entry into primary human trabecular meshwork cells. J Virol. 2005;79:13173–13179. doi: 10.1128/JVI.79.20.13173-13179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V, Shukla SY, Yue BY, Shukla D. Herpes simplex virus type 2 entry into cultured human corneal fibroblasts is mediated by herpesvirus entry mediator. J Gen Virol. 2007;88:2106–2110. doi: 10.1099/vir.0.82830-0. [DOI] [PubMed] [Google Scholar]
- Welstead GG, Hsu EC, Iorio C, Bolotin S, Richardson CD. Mechanism of CD150 (SLAM) down regulation from the host cell surface by measles virus hemagglutinin protein. J Virol. 2004;78:9666–9674. doi: 10.1128/JVI.78.18.9666-9674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the TNFR superfamily and a mediator of HSV entry. J Virol. 1997;71:6083–6093. doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitbeck JC, Zuo Y, Milne RSB, Cohen GH, Eisenberg RJ. Stable association of herpes simplex virus with target membrane is triggered by low pH in the presence of the gD receptor HVEM. J Virol. 2006;80:3773–3780. doi: 10.1128/JVI.80.8.3773-3780.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ. Herpes simplex viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott, Williams and Wilkins; Philadelphia: 2001. pp. 2461–2509. [Google Scholar]
- Wittels M, Spear PG. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 1990;18:271–290. doi: 10.1016/0168-1702(91)90024-p. [DOI] [PubMed] [Google Scholar]
- Zago A, Jogger CR, Spear PG. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc Natl Acad Sci USA. 2004;101:17498–17503. doi: 10.1073/pnas.0408186101. [DOI] [PMC free article] [PubMed] [Google Scholar]