

Abstract
Objective
To teach doctor of pharmacy (PharmD) students how to apply organ clearance concepts in a clinical setting in order to optimize dose management, select the right drug product, and promote better patient-centered care practices.
Design
A student-focused 5-hour topic entitled "Organ Clearance Concepts: Modeling and Clinical Applications" was developed and delivered to second-year PharmD students. Active-learning techniques, such as reading assignments and thought-provoking questions, and collaborative learning techniques, such as small groups, were used. Student learning was assessed using application cards and a minute paper.
Assessment
Overall student responses to topic presentation were overwhelmingly positive. The teaching strategies here discussed allowed students to play an active role in their own learning process and provided the necessary connection to keep them motivated, as mentioned in the application cards and minute paper assessments. Students scored an average of 88% on the examination given at the end of the course.
Conclusion
By incorporating active-learning and collaborative-learning techniques in presenting material on organ clearance concept, students gained a more thorough knowledge of dose management and drug-drug interactions than if the concepts had been presented using a traditional lecture format. This knowledge will help students in solving critical patient situations in a real-world context.
INTRODUCTION
Teaching strategies to ensure students' adeptness in critical thinking and problem solving need to be an integral part of the PharmD curriculum. A topic entitled “Organ Clearance Concepts: Modeling and Clinical Applications,” offered in the PharmD program of the University of Puerto Rico School of Pharmacy (UPR-SP) in the last 2 academic years is described and its outcomes discussed. This curricular effort followed a smooth transition from dependent to independent learning and involved PharmD students as active, self-directed learners in solving problems related to clinical pharmacokinetics that are commonly encountered in the practice of pharmacy.
Clearance, the parameter which relates rate of elimination to drug concentration, is important because it defines the rate of administration required to maintain a plateau drug concentration. Together with the extent of distribution outside of plasma, clearance also determines the speed at which a drug is eliminated from the body.1-2 The sensitivity of organ clearance of a drug to changes in binding within blood depends on its unbound clearance. If unbound clearance is low, relative to organ blood flow, the extraction ratio (and clearance) will always be low and dependent on plasma binding.1-3 If the extraction ratio is high, elimination becomes perfusion rate-limited and clearance will be relatively insensitive to changes in binding, but oral bioavailability may exhibit dependence on binding if the liver is the major eliminating organ.1-4 A full insight into the implications of altered binding on pharmacokinetics requires a sound understanding of the physiology both of the eliminating organs and the distribution of drug within the body.1, 5
This physiological approach to hepatic drug clearance recognizes that hepatic blood flow, the activity of the overall elimination process (intrinsic clearance), drug binding in the blood, and the anatomical arrangement of the hepatic circulation are the major biological determinants of hepatic drug clearance.6 This approach permits quantitative prediction of both the unbound and total drug concentration over time relationships in the blood after intravenous and oral administration, and any changes that may occur as a result of alterations in the above biological parameters. These considerations have led to a classification of drug metabolism based on the hepatic extraction ratio. The proposed classification allows prediction and interpretation of the effects of individual variations in drug-metabolizing activity, route of administration, pharmacokinetic interactions, and disease states on hepatic drug elimination and consequently on body clearance and blood drug concentrations. Not surprisingly, these alterations might also be associated to the inter-subject variability in dose-response relationship.
An understanding of organ clearance concepts, linked to basic principles of clinical pharmacokinetics and therapeutic drug monitoring, will facilitate interpretation of blood drug concentrations in individual patients and consequently help individualize drug dosages to achieve steady-state concentrations within a range of values that correlate well with desired patient response.2-4 The physiological approach to the hepatic clearance of drugs has been discussed by Zimmerman.7
The significance of this curricular effort from the perspective of the Accreditation Council on Pharmaceutical Education (ACPE) competencies and Center for Advancement of Pharmacy Education (CAPE) educational outcomes is based on providing patient-centered pharmaceutical care through sound therapeutic principles and evidence-based data, taking into account relevant professional issues and evolving pharmaceutical and clinical sciences that may impact therapeutic outcomes.
DESIGN
After the completion of all the learning activities and assignments for this topic, students will be able to:
(1) Explain the role of organ clearance concepts for analyzing many clinical situations;
(2) Know the major biological (physiological and biochemical) determinants of organ drug clearance;
(3) Apply organ clearance model expressions for both the corresponding calculation of relevant parameters and interpretation of individual variations. Interpretation of such variations allow students to individualize drug dosages in order to achieve steady-state concentrations within a range of values that correlate well with desired patient response;
(4) Explain the effects of individual variations in drug-metabolizing activity, route of administration, pharmacokinetic drug interactions, disease-states, age and habits on hepatic drug elimination and consequently on total body clearance and blood drug concentrations;
(5) Know when a drug is showing a restrictive clearance;
(6) Determine whether an altered drug-protein binding is clinically significant.
The specific learning objectives of the topic are for students to accomplish the following:
(1) Predict and interpret the clinical significance of individual variations in drug-metabolizing activity, route of administration, pharmacokinetic drug interactions, disease-states, age and habits.
(2) Derive specific clearance models and relationships following basic organ clearance concepts and principles;
(3) Calculate the individual patient physiologically based pharmacokinetic parameters from organ clearance relationships.
(4) Differentiate a drug showing a restrictive clearance from one having a non-restrictive clearance.
(5) Evaluate the effect of altered plasma protein binding on drug concentrations and response by using the organ clearance concepts and models.
This topic is focused on development of the professional abilities that may create the skills to evaluate and modify or recommend modifications in drug therapy in order to ensure effective and safe patient care; to use clinical data to optimize therapeutic drug regimens; to retrieve, evaluate, and manage professional information and literature; and to identify, assess, and solve medication-related problems and provide a clinical judgment as to the continuing effectiveness of individualized therapeutic plans and intended therapeutic outcomes.
Educational Environment
The UPR-SP PharmD degree program is structured in 4 academic years in order to ensure the achievement of the abilities necessary to become a generalist practitioner who renders pharmaceutical care. The Basic Pharmacokinetics and Biopharmaceutics unit of the Integrated Pharmaceutical Sciences and Therapeutic Agents II course develops the fundamentals of biopharmaceutics and pharmacokinetics in order to reinforce the major concept of the drug. The students are expected to acquire relevant information for better understanding the potential benefit related to the safe and effective use of drug product. It is aimed at enhancing the students' skills in developing and assessing formulations based on the relationship between the drug, the dosage form, and the living system. The unit brings together disciplines like pharmacokinetics, biopharmaceutics, physical pharmacy, compounding, and therapeutics. It strengthens some cardinal concepts related to the optimization of drug product uses, and improves knowledge of the relationship between drug exposure and clinical outcome, with emphasis on supporting the patient-oriented pharmaceutical care goals, to refine drug dosage regimens and identify factors determining untoward responses. Active-learning strategies and methodologies are used across the entire course, as are lectures and case discussions.
The pharmacokinetic unit is particularly designed to provide core scientific knowledge and develop learning skills that will become the basis for highly competent application and practice of pharmacy in clinical courses later in the didactic PharmD curriculum, such as Integrated Sciences, Therapeutics and Patient Care, and Integrative Seminar of Pharmaceutical Care and Human Development, as well as a series of structured experiential learning practicum courses.
Although other relevant topics in pharmacokinetics are also developed (eg, modeling, non-compartmental analysis, and residence time concepts), this unit emphasizes the development of conceptual knowledge in clinical pharmacokinetics, and its importance as a foundation for the practice of pharmacy in different settings. It also incorporates the problem-solving and case-discussion processes to enable students to prevent, identify, and/or solve problems related to clinical pharmacokinetics that are commonly encountered in the practice of pharmacy. Accordingly, this unit fosters the integration of knowledge based on professional practice experience in a systematic ability-based curriculum.
A student-focused 5-hour topic entitled “Organ Clearance Concepts: Modeling and Clinical Applications” was developed and presented to 43 second-year PharmD students. In preceding sessions, and as part of this course unit, students were instructed in the use of critical concepts and principles in clinical pharmacokinetics and nonlinearity. This knowledge helped them understand the material, particularly as it pertains to clinical situations involving dosage adjustments. Classroom and computer center facilities at the UPR-SP, Medical Sciences Campus, are used to cover this topic.
Pedagogy/Andragogy
In this section, description of and rationale for the use of specific teaching and learning methods, use of instructional tools and technology, and the role of students in this topic are discussed. Table 1 depicts the instructional design. Before the introductory session, students were provided with some short readings1,6-8 and at least 3 thought-provoking questions leading to the next session. In doing so, students became invested in and motivated toward participating in the forthcoming activities. The reading assignment was also provided online to students and had to be completed before attending the class session for the discussion of the topic.
Table 1.
Instructional Design for the Topic “Organ Clearance Concepts: Modeling and Clinical Applications” Developed and Delivered to 43 Second-Professional Year PharmD Students

The provided questions guided students' reflection and preparation for the next session by reading these articles. The following are those that have raised more discussion:
What would happen if the drug alters its own metabolism (ie,intrinsic clearance)?
Orally administered medications must pass through the liver before entering the systemic circulation, so if the drug shows a very high hepatic extraction ratio (first-pass effect), how could it be administered orally and still have an acceptable response?
Otherwise, if the drug has a very low hepatic extraction ratio but is primarily metabolized by the liver (ie, renal excretion is negligible), how is the drug supposed to be removed from the body?
In your opinion, why are some drugs affected but most are not influenced by changes in plasma protein binding?
The first portion of the topic was devoted to delivering an active, enthusiastic 2-hour lecture in a supportive classroom environment. A PowerPoint slide presentation that encouraged questions and handouts was the material presented during this introductory session. The slides were also uploaded to the Blackboard Learning System and thus available online, in a Web-enhanced format, to the PharmD students. External links to related material were also provided.9,10 The structure of the lecture consisted of an introduction to the learning objectives; a discussion of the relevant organ clearance concepts and principles (mainly hepatic but also renal); relations to extraction ratio and bioavailability (first-pass effect); identification of major biological determinants of organ drug clearance; modeling and derivations for low/high hepatic extraction ratio; examination of major pharmacokinetic drug interactions; cause and clinical significance; how to differentiate a drug showing a restrictive versus non-restrictive clearance and examples of the use of this knowledge to manage clinical situations. As a foundation material, this introductory lecture covered major aspects of this subject and created a conceptual framework that allowed students to perform better on future problem-solving activities. Students had already been introduced to bioavailability and first-pass effect concepts during an earlier session.
The lecture culminated with an opportunity for students to apply the information they had learned. Students were divided into 10 small groups for the next class period and asked to designate a spokesperson for the group. Each of the 10 groups was provided with a practice problem ahead of the final encounter for this topic. The practice problems (see Appendix 1) were similar to the examples discussed in the classroom. However, in addition to the already discussed predictions, students were also required to recalculate the dosage regimen in the particular patient (individualization), using the measured and desired drug concentrations at steady state, when the dosing interval (τ = 24 hours) is given.23 Students were expected to work together on the problem before attending the next class, consulting the reading notes and the reference literature as a team (each member having a well-defined role). In doing this collaborative learning technique, a productive discussion among team members was stimulated.
After a short introduction (15-20 minutes), the next class time was mostly devoted to discussion of the solution to the practice problem by both the students and the instructor. About 50 minutes were spent in order for each of the 10 spokespersons (5 minutes each) to debrief and provide team's explanations for the case and calculations. After that, all students were encouraged to initiate a critical discussion on teams' conclusions for the remaining time (about 45 minutes). At this point, the instructor served as a moderator, provoking the debate among students and clarifying any doubt or correcting misleading viewpoints.
During the last 15 minutes of the session, students completed a quiz consisting of questions related to the subject, but emphasizing previously solved problems. After that, they were asked to complete a minute paper for classroom assessment.
Content
A brief description of content from this topic (handouts are distributed to students) is provided. As part of an introductory session, the extraction ratio (E) is presented as a measure of the organ efficiency to remove a fraction of the total amount of drug entering into the eliminating organ (usually hepatic, but also renal, etc), in a single-pass, from the blood perfusing the organ. Accordingly, values for this metric range from 0 to 1. High values of E mean high organ clearance and thus a reduced amount of drug gets through unchanged, whereas low values of E mean that only a small fraction will change. Hepatic E is mainly a measure of extent of metabolism. Based on the conceptual framework, extraction ratio is estimated by means of the incoming (Cin) and outgoing (Cout) drug concentrations to and from a clearing organ as
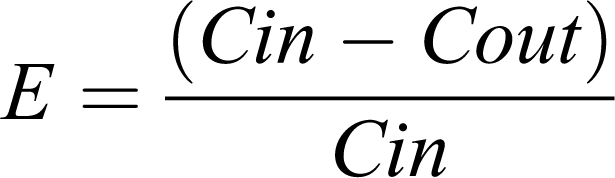 |
but for practical reasons, this metric is usually expressed by its relationship to 2 more relevant parameters: hepatic clearance (CLh) and liver blood flow or perfusion rate (LBF) as
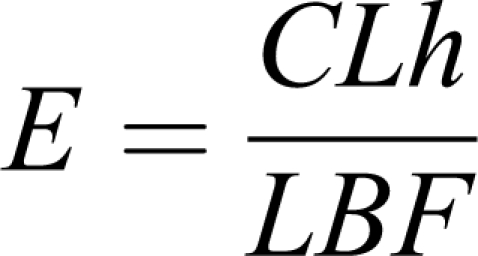 |
The underlying assumptions for this are that complete absorption takes place and the hepatic clearance is the major route of elimination. Solving at steady-state conditions, an equation for hepatic clearance is obtained,
 |
where CLint is the maximum organ capacity to clear the drug with no blood flow limitations and f u is the free (unbound) fraction of drug in blood. CLint is estimated from enzyme activity by summing up all parallel metabolic pathways (approximately under linear conditions).
 |
This so-called “well-stirred” or venous equilibrium model of organ clearance can consider the effect of each relevant parameter on overall hepatic clearance and consequently helps student understand inter-subjects variations in drug concentrations and effects after events such as drug-drug interactions due to protein-binding displacement, disease-state, concomitant medication, physiological alterations, habits (eg, smoking), among others.2-4, 11
Simplifications of the venous equilibrium model for extreme cases are usually well-accepted. In this sense, the clearance of those drugs showing the highest hepatic extraction ratio (E >0.70) is reduced to the equation
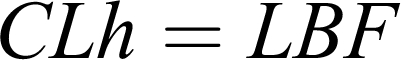 |
whereas the clearance of those with the lowest E values (E<0.3) is limited to
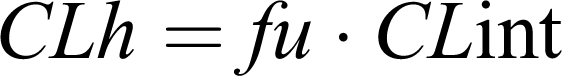 |
In the same way, F (systemic bioavailability) is reduced to
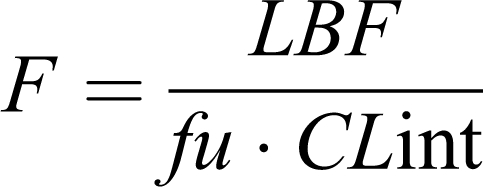 |
for drug with E >0.70 and F≈1 for drugs with E<0.3. Drugs belong to the former group are considered flow-limited drugs because any change in hepatic drug clearance is blood-flow dependent, but relatively independent of alterations in intrinsic clearance and the fraction of unbound drug.2-4, 11 Lidocaine, morphine, and propranolol are some examples with this pattern. Drugs in the second group are considered capacity-limited because overall hepatic clearance is mainly determined by the enzymatic capacity of the liver to metabolize free drug in plasma water.
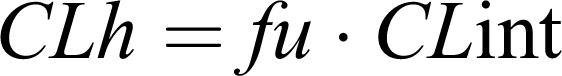 |
This latter group can also be subclassified as binding sensitive (≥90% bound) or binding insensitive (<90% bound), depending on the percentage of drug bound to plasma proteins.2-4, 11-12 Examples of these drugs are mentioned phenytoin and warfarin.
For restrictively cleared drugs (E ≤ f u), clearance process is described as limited by protein binding (altered by changes in f u). On the other hand, for nonrestrictive drug clearance (E>f u), students are advised that the protein binding does not protect the drug from elimination. In this context, protein binding is viewed as a delivery system for drug elimination.2-4, 11
For drugs inefficiently extracted (ie, showing a restrictive clearance):
for which E ≤ f u (restrictive, f available = f u)
Assume f u is both the measured plasma unbound drug fraction and the drug fraction available for metabolism (f available).
If the main elimination pathway for a hypothetical drug is the hepatic metabolism, it means that renal clearance is negligible and CL nonrenal is mainly the hepatic clearance (CLh). Accordingly, total body clearance for restrictively cleared drug is:
 |
Therefore, changes in f u will result in proportional changes in total body clearance for very low extraction drugs or restrictively cleared drug such as phenytoin in nephrotic syndrome. Using the pharmacokinetic relationships for average total drug concentration at steady state, the following equation is obtained for restrictively cleared drug:
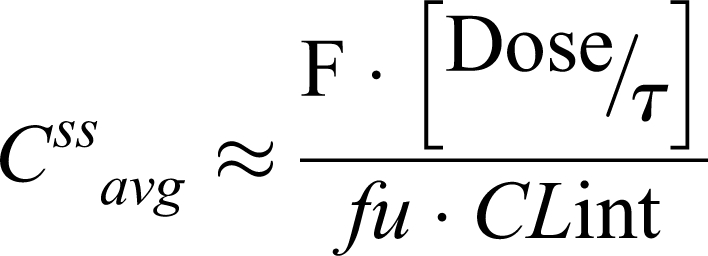 |
Where [Dose/τ] represents the dose rate (DR) or dosing regimen, and F is the oral bioavailability.
However, f u cancels out from the corresponding equation for determining the unbound drug concentration at steady state:
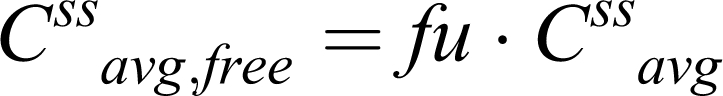 |
Hence,
 |
Consequently, students are taught that changes in f u will have no effect on unbound drug concentration, except if there is an effect on intrinsic clearance (CLint) via enzyme induction or inhibition and/or liver disease. Thus, sudden f u increase will cause only a transient rise in Css avg, free that is rapidly offset by free drug elimination.
Finally, because the drug's effect is determined by the unbound drug concentrations, students learned that no significant changes in patient outcomes are expected as a result of drug displacement from protein binding, except that the displacing compound also inhibits its liver metabolism.1-3, 5, 11
Problem: The following problem (hypothetical example) is solved in class to demonstrate how to apply this knowledge, which was derived from organ clearance concepts and principles that were discussed in the first portion of this topic, in order to manage clinical situations:
WT is a 25-year-old, 93 kg male patient (height, 188 cm) who returns to the hospital for a follow-up visit after beginning treatment one month ago with oral extended phenytoin sodium (Dilantin Kapseals; F=1; Salt Form Correction Factor, S=0.92) 100mg tid, for primary generalized epilepsy. He reports that the phenytoin makes him feel drunk and unsteady and his vision is blurred. Past medical history shows that WT was diagnosed with ankylosing spondylitis one year ago; he was initially treated with ibuprofen, then switched 6 months ago to phenylbutazone (Butazolidin), which appears to relieve the pain more effectively. After measuring, total blood phenytoin level was set at 16 mcg/mL.
Questions:
(1) How do you explain the apparent signs and symptoms of phenytoin toxicity at a total phenytoin level within the usual therapeutic range (10 – 20 mcg/mL)?
(2) Considering that 300 mg a day is the usual starting dose rate for an otherwise typical patient, what should have been done in this case and why?
(3) What changes, if any, do you expect to happen in the following relevant parameters (LBF; CLint; f u; Css,avg; Css,avg,free; F (bioavailability); CL; Vd; Effect; Dose Rate (DR = Dose/τ)? Hint: According to Benet et al., phenytoin shows a low hepatic extraction ratio of E=0.03.5 It is also highly bound, (≥90%).5
(4) Is this drug showing a restrictive clearance? Explain.
Also, what would happen if the above patient contracts viral myocarditis that causes reduced cardiac output (CO) and therefore slower LBF was discussed. Accordingly, the analysis of all the above parameters was repeated considering the additional information (disease state).
The graphic technique showed in the textbook Applied Clinical Pharmacokinetics was used to illustrate the differences that may occur in the relevant parameters, the steady-state drug concentration, and the pharmacological effects among patients with altered plasma protein binding of phenytoin.13 In the example, it is assumed that all physiologic, pharmacokinetic, and drug effect parameters (y axis) are initially stable. On the x axis, an arrow indicates the plasma protein binding of phenytoin decreases and that the unbound fraction increases in the patient; an assumption made is that any changes in the parameters are instantaneous. An increase in a parameter is denoted as an uptick in the line, and a decrease in the parameter is shown as a downtick in the line.13
Briefly, in the hypothetical example discussed, phenylbutazone is a displacing agent and, at the same time, an inhibitor of phenytoin metabolism.14-16 Consequently, the increase of the unbound phenytoin pool cannot be efficiently corrected by the elimination process while this mechanism is blocked. Phenytoin accumulates and higher systemic exposure leads to increased risk of adverse (toxic) events as described in the example. Notice that total phenytoin concentrations at steady state will remain the same and within the therapeutic range, but the displacing agent will definitely increase the unbound pool. Furthermore, the nonlinear nature of phenytoin disposition and its narrow therapeutic margin should be taken into account.13
Phenytoin is eliminated primarily by means of hepatic metabolism (>95%).13, 17-18 Thus, total body clearance is mainly determined by phenytoin hepatic clearance, which increases when the unbound fraction rises
 |
However, the unbound steady-state concentration remains unaltered because the free fraction of drug in the blood is higher than it was before the increase in unbound fraction
Accordingly, students were advised that when only total steady-state phenytoin concentrations are measured, at this point in time, clinicians will be under the impression that an increase in phenytoin dosage may be in order. But, if unbound (free) steady-state phenytoin concentrations are simultaneously measured, it will be found that these concentrations at steady state have not changed and consequently no changes in the pharmacological/toxic effect will occur. This outcome can be unexpected for the decrease in plasma protein binding, especially because the total steady-state concentration of phenytoin decreases. Accordingly, students need to be aware of situations such as this because the total drug concentration (bound + free) can be misleading and cause an unwarranted increase in drug dosage. Unbound (free) concentrations of phenytoin should be used to persuade clinicians that an increase in phenytoin dosage is not needed despite a decrease in total concentrations caused by this interaction.
In this context, students were taught that any drug interactions that displace plasma protein binding of phenytoin will only be clinically relevant if the displacing agent is also altering the intrinsic phenytoin clearance.
For drugs such as phenytoin that have a low hepatic extraction ratio (E≤30%), changes in the LBF parameter (due to viral myocarditis that causes reduced CO) will have limited effect on the hepatic drug clearance.13
Because it is so highly metabolized in the liver by CYP2C9 (and also by CYP2C19), phenytoin is prone to drug interactions that inhibit hepatic microsomal enzymes.19 Cimetidine, valproic acid, amiodarone, chloramphenicol, isoniazid, disulfiram, omeprazole, and phenylbutazone have been reported to inhibit phenytoin metabolism and increase the blood concentration of phenytoin.13 Phenytoin also is a broad-based hepatic enzyme inducer that affects most cytochrome P450 systems. Drugs with narrow therapeutic ranges that can have their metabolism increased by concurrent phenytoin administration include carbamazepine, phenobarbital, cyclosporine, tacrolimus, and warfarin.13 When phenytoin therapy is added to the medication regimen for a patient, a comprehensive review for drug interactions should be conducted. Valproic acid, aspirin (>2 g/day), some highly protein-bound nonsteroidal anti-inflammatory drugs, and warfarin can displace phenytoin from plasma protein binding sites. This necessitates monitoring of unbound phenytoin concentrations.
Bringing Theory to the Clinical Setting: Application to a Real Case
The valproic acid-phenytoin interaction requires special attention considering its complexity and because these 2 anticonvulsants are regularly used together for the management of seizures.20-22 The valproate-phenytoin interaction involves displacement of phenytoin binding to albumin and inhibition of phenytoin intrinsic clearance by valproate. What makes this interaction so difficult to notice and understand is the fact that these 2 events do not occur at the same time. Consequently, the timing of the observation will determine further interpretation and decision-making regarding this drug interaction.
For example, a 61-year-old female patient, weight 56 kg , height, 162 cm, suffering from tonic-clonic seizures received 100 mg tid of extended phenytoin sodium (Dilantin Kapseals capsules) by mouth (F=1, S=0.92) for 1 month to start therapy. She had normal liver and renal functions after routine laboratory tests (total bilirubin, 0.8 mg/dl; albumin, 5.1 g/dl; serum creatinine, 0.4 mg/dl). Steady-state total concentration of phenytoin was 8.7 mcg/mL at the time of that visit (steady state is attained in 7 to 14 days). The dosage was then increased to 400 mg daily for an additional month to adjust to individual patient response, and the resulting drug concentration at steady-state was then 12.3 mcg/mL. Nonetheless, the patient complained about this regimen because optimal seizure control was lacking so the clinician added valproic acid (Depakene, 1000 mg twice daily by mouth) to the regimen.
As valproate accumulates, the first interaction is a displacement of phenytoin plasma protein binding to albumin because both drugs compete for the same binding site on albumin. Upon this first drug interaction, there is a transient increase in unbound phenytoin fraction (f u) that leads to an increase in phenytoin clearance (remember that phenytoin is a low extraction ratio drug).5,13 As a result, a decrease in total plasma phenytoin concentration is observed, but the free phenytoin concentration remains unaltered. Once plasma valproate concentrations reach the steady-state conditions, the higher amounts of valproate strike the microsomal hepatic CYP 450 enzymes and inhibit the metabolism of phenytoin. This second stage of drug interaction decreases intrinsic clearance and thus the hepatic clearance of phenytoin is significantly reduced, so both unbound and total phenytoin concentrations increase. When phenytoin concentrations finally equilibrate and reach steady state under the new plasma protein binding and intrinsic clearance conditions imposed by concomitant valproate levels, the total phenytoin concentration often is at about the same level as before the drug interaction occurred, but unbound phenytoin concentrations are much higher. If total phenytoin concentrations were measured at this point, the clinician would observe any relevant change and drug interaction could be overlooked. However, if unbound phenytoin concentration were measured, the clinician would find these concentrations rose dramatically and that the free fraction of phenytoin in plasma is twice or more the baseline amount (≥ 20%).13,23 In such a situation, the above mentioned patient would be exposed to toxic levels of phenytoin and a decrease in phenytoin dosage would be needed. The dosage adjustment would depend on how much has changed the phenytoin clearance after inhibition of its metabolism by valproate. Students were also encouraged to keep in mind the nonlinear nature of phenytoin metabolism. Additionally, dose correction using the salt form correction factor (S) was suggested as sodium phenytoin is being administered instead of phenytoin acid (phenytoin dose = 0.92 × sodium phenytoin dose). In this case, a 1.2-fold reduction in the phenytoin dosage was requested. This is an example of how real-life health care professionals use this knowledge to manage critical patient situations.
In order to reinforce these statements, students were asked to think about any difference in phenytoin dosage requirements between pregnant woman and the elderly. In contrast to elderly individuals (older than 65 years, who have a decreased capacity to metabolize phenytoin because of age-related losses in liver functionality),24-25 pregnant women taking phenytoin have increased dosage requirements, particularly during the third trimester.26-29 The main reason for this change is that, although both have decreased protein binding due to low albumin concentrations (hypoalbuminemia) and consequently increased unbound fraction in plasma, pregnant women usually show increased metabolism of phenytoin (apart from a decreased bioavailability due to malabsorption). Because of these changes, clinicians tend to prescribe lower initial doses of phenytoin to older patients but higher initial doses to pregnant women. Students were encouraged to follow aggressive monitoring of phenytoin serum concentration in these patients, including measurement of unbound concentrations. This example offered an opportunity to capitalize on the students' desire to energize the class with education relevant to their future needs.
Assessment Methods
Classroom assessment techniques such as application cards and minute papers were used to provide specific feedback on students' understanding and progress toward the intended learning objectives.30-31 Once students had received instruction about organ clearance concepts and principles, they were provided with a half-sheet of paper and given 5-7 minutes to write down at least one possible application for this new knowledge.
In addition, students individually prepared a minute paper at the end of the topic, after completing the scheduled quiz but before leaving the classroom, by briefly answering the following questions: What's the most important thing you learned in this topic? What important question remains unanswered? Did you find this topic useful (yes/no)? Why? Students were encouraged to answer each question in 1 sentence.
Additionally, the student's comprehension and understanding of the material presented as part of this topic was assessed utilizing a multiple-choice examination (although, students were required to briefly explain their choices).
ASSESSMENT
The application card technique prompted students to think about possible real-world applications for what they had just learned about organ clearance concepts and, as a consequence, to connect this newly learned concept with prior knowledge from nonlinearity and clinical pharmacokinetic principles such as dosing rate, free drug concentration at steady-state, etc, that were previously taught in the course. At the beginning of the next session, an opportunity was provided for students to comment about these possible applications.
Concerning the minute paper, overall student responses were overwhelmingly positive. Student responses to the questions included: “Yes, because this is the best experience I've ever been engaged in terms of translating basic concepts into a clinical scenario” “Absolutely, I think it was excellent and this learning-enhanced activity should be copied while teaching other courses/topics” “Yes. It was great because this is the way we're supposed to fill in the gap between in-class instruction and real-world performance”
Last year, students scored an average of 88% on the examination given at the end of the course and 87% of the quiz pertaining specifically to this material.
DISCUSSION
The first 10 minutes of the next session were used to deal with unanswered questions (notably, most unanswered questions were about the clinical significance –if any- of drug-drug interactions due to protein binding displacement). Sometimes, a face-to-face (student-instructor) contact at extra-class time was also scheduled to attend any individual question.
Despite its simplicity, the assessment technique measured more than simply students' ability to recall information. That is, students had to first evaluate what they recall in order to select the most important thing they have just learned. Secondly, they had to self-assess and ask themselves how well they understood what they just heard.
The process of measuring outcome expectation includes student self-assessment of performance in the stated professional abilities. One of the most interesting and rewarding long-term outcomes of the lectures and related activities on this topic was students' decision to include what they learned in this session as part of their portfolios at the end of the academic year. Actually, by self-assessment, at least 3 students referred to activities in this topic (particularly, the practice problems) as evidence of their performance at a higher level in professional abilities such as pharmaceutical care; problem solving and decision making; critical thinking; and self-learning and professional development. These abilities are among a core of 10 general/professional abilities that have been developed as a set of expectations at 3 levels of progress through the curriculum. The abilities are contextualized in the disciplines and practice that comprise the pharmacy profession.
Student performance on summative evaluations of student achievement showed the degree to which our goals for this topic were attained at the conclusion of the course. Table 2 summarizes the achievement of learning outcomes. Although there is not time enough to apply methods for the formative use of outcome data within this topic, based on evaluation process, the use of CATs as well as reading assignments with thought-provoking questions help this instructor identify some student deficiencies and concerns and use this feedback to modify later course activities. For instance, the controversy about clinical significance of protein-binding displacement that raised while discussing the thought-provoking questions (ie, why some drugs are affected but most are not influenced by changes in plasma protein binding), was then used to prepare related examples and case discussions such as the interaction between valproate and phenytoin. The pharmacokinetic unit of the FARM 7325 course is consistently ranked as a favorite section by second-year students. This is reflected in student evaluation of the course, with good assessments and comments as can be corroborated in Table 3.
Table 2.
Percentage (%) of PharmD Students Who Achieved the Expected Learning Outcomes After Taking Both the Scheduled Examination and In-Class Quiz

these learning outcomes were evaluated in the in-class quiz
Table 3.
Pharmacy Students' Responses on an Evaluation of a Course in Which “Organ Clearance Concepts: Modeling and Clinical Applications” Was Taught (N = 23)a

Only 23 students were available at the time of completing this survey for course/professor evaluation
SUMMARY
The learning tools used in teaching the topic Organ Clearance Concepts: Modeling and Clinical Applications to PharmD students included assigned reading and thought-provoking questions, enhanced lectures, a handout containing practice problems as the basis of class discussion, 2 classroom assessment techniques, assignments to stimulate collaborative learning and small group discussions, an in-class quiz, and a multiple-choice examination. This unit in the pharmacy curriculum was designed to provide core knowledge and assist in the building of a strong foundation for future learning in other courses such as practicum. Student performance on the assessments suggests achievement of course objectives as they begin to understand how organ clearance concepts and models may be applied to the clinical practice of pharmacy. Students find the opportunity to apply their knowledge academically rewarding.
ACKNOWLEDGEMENT
I would like to thank Dr. Luz M Gutierrez-Collazo, Dr. Elga E Vega-Maldonado and Ms Rafaela Mena for their revision of this article and their useful comments. The collaborative support of Dr. Jorge R Miranda-Massari is highly appreciated. The relevant information about the case herein discussed was kindly provided by a faculty member of the UPR-SP, who is also a practitioner at the Caribbean Healthcare System in San Juan, Puerto Rico.
Appendix 1. The example below is one out of 10 different practice problems that were provided in class to be solved by small group discussions
For the following scenario determine the change in LBF (QH); CLint; f u; Css,avg; Css,avg,free; F (bioavailability); CL; Vd; Effect; Dose Rate (DR = Dose/τ)
AF is a 52-year-old, 64-kg, woman who presents to ER with complaints of hypotension, impaired coordination, dizziness, hangover-like symptoms of being drowsy, having a headache, being sluggish and irritable after waking up and episodes of anterograde amnesia during the last week. Her past Hx indicates a 2-year history of absence seizures (petit mal) and anxiety disorders treated with clonazepam, and a 1-year history of ankylosing spondylitis treated with ibuprofen and ASA (hint: displaces clonazepam from plasma protein binding). Because she was feeling depressed over last month, she was prescribed with nefazodone hydrochloride (a blocker of post-synaptic serotonin type-2A receptors). Medications on admission are Clonazepam (Klonopin®, Roche) 1-mg bid, nefazodone (Serzone, Bristol-Myers Squibb) 300 mg twice daily, ibuprofen (Advil) 1,200 mg daily dose, and aspirin 325mg every day. Notice that clonazepam hepatic extraction ratio is E=0.002. The serum clonazepam levels rose at this visit with respect to 15 month ago from 7.2 ng/mL to 29.4 ng/mL.
Additional questions
1. How do you explain the raise in the drug levels?
2. The physician wants to decrease the dosage to get the desired level (between 6.5–13.5 ng/mL). What would the consequence be if this were done? How should the dose of clonazepam be adjusted (provide calculations)?
3. In your opinion, should the doctor consider any alternative for clonazepam as well? Is this drug showing a restrictive clearance (YES/NO)? Explain.
REFERENCES
- 1.Rowland M. Protein binding and drug clearance. Clin Pharmacokinet. 1984;9:10–7. doi: 10.2165/00003088-198400091-00002. [DOI] [PubMed] [Google Scholar]
- 2.Tozer TN, Rowland M. Introduction to Pharmacokinetics and Pharmacodynamic: The Quantitative Basis of Drug Therapy. 1st ed. Baltimore, MD: Lippincott Williams & Wilkins; 2006. pp. 92–7. [Google Scholar]
- 3.Kashuba ADM, Park JJ, Persky AM, Brouwer LR. Drug Metabolism, Transport and the influence of hepatic disease. Chapter 7. In: Burton ME, Shaw LM, Schentag JJ, Evans WE, editors. Applied Pharmacokinetics and Pharmacodynamics, Principles of Therapeutic Drug Monitoring. 4th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2006. p. 133. [Google Scholar]
- 4.Shargel L, Wu-Pong S, Yu ABC. Applied Biopharmaceutics and Pharmacokinetics. 5th ed. New York, NY: McGraw-Hill; 2005. pp. 342–343. [Google Scholar]
- 5.Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002;71:115–21. doi: 10.1067/mcp.2002.121829. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson GR, Shand DG. Commentary: a physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975;18:377–90. doi: 10.1002/cpt1975184377. [DOI] [PubMed] [Google Scholar]
- 7.Zimmerman CL. Physiological approach to the hepatic clearance of drugs. Am J Pharm Educ. 1989;53:157–60. [Google Scholar]
- 8.Sahin S, Rowland M. Evaluation of route of input on the hepatic disposition of diazepam. J Pharmacol Exp Ther. 2000;295:836–43. [PubMed] [Google Scholar]
- 9.Express-Scripts Drug-Digest® website, Check Interactions. Available at: http://www.drugdigest.org/DD/Interaction/ChooseDrugs/1,4109,00.html Accessed December 3, 2007
- 10.David WA Bourne's Pharmacokinetic and Pharmacodynamic Resources page. Available at: http://www.boomer.org/c/p4/ja/Fig1702/Fig1702.html Accessed December 3, 2007.
- 11.Bauer LA. Drug dosing in special populations: Renal and hepatic disease, dialysis, heart failure, obesity, and drug interactions. In: Bauer LA, editor. Applied Clinical Pharmacokinetics. New York, NY: McGraw Hill; 2001. pp. 57–90. [Google Scholar]
- 12.Gabrielsson J, Weiner D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications. 2nd ed. Stockholm, Sweden: Swedish Pharmaceutical Press; 1997. pp. 100–6. [Google Scholar]
- 13.Bauer LA. Anticonvulsants: Phenytoin. In: Bauer LA, editor. Applied Clinical Pharmacokinetics. New York, NY: McGraw Hill; 2001. pp. 447–56. [Google Scholar]
- 14.Miners JO. Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45:525–38. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pond SM, Birkett DJ, Wade DN. Mechanism of inhibition of tolbutamide metabolism: phenylbutazone, oxyphenbutazone, sulphaphenazole. Clin Pharmacol Ther. 1977;22:573–9. doi: 10.1002/cpt1977225part1573. [DOI] [PubMed] [Google Scholar]
- 16.Lewis RJ, Trager WF, Chan KE, et al. Warfarin- stereochemical aspects of its metabolism and the interaction with phenylbutazone. J Clin Invest. 1974;53:1607–17. doi: 10.1172/JCI107711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Allen JP, Ludden TM, Burrow SR, Clementi WA, Stavchansky SA. Phenytoin cumulation kinetics. Clin Pharmacol Ther. 1979;26:445–8. doi: 10.1002/cpt1979264445. [DOI] [PubMed] [Google Scholar]
- 18.Grasela TH, Sheiner LB, Rambeck B, et al. Steady-state pharmacokinetics of phenytoin from routinely collected patient data. Clin Pharmacokinet. 1983;8:355–64. doi: 10.2165/00003088-198308040-00006. [DOI] [PubMed] [Google Scholar]
- 19.Hansten PD, Horn JR. Drug Interactions Analysis and Management. Vancouver, WA: Applied Therapeutics; 1999. p. 480. [Google Scholar]
- 20.Perucca E, Hebdige S, Frigo GM, Gatti G, Lecchini S, Crema A. Interaction between phenytoin and valproic acid: plasma protein binding and metabolic effects. Clin Pharmacol Ther. 1980;28:779–89. doi: 10.1038/clpt.1980.235. [DOI] [PubMed] [Google Scholar]
- 21.Frigo GM, Lecchini S, Gatti G, Perucca E, Crema A. Modification of phenytoin clearance by valproic acid in normal subjects. Br J Clin Pharmacol. 1979;8:553–6. doi: 10.1111/j.1365-2125.1979.tb01043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riva R, Albani F, Contin M, et al. Time-dependent interaction between phenytoin and valproic acid. Neurology. 1985;35:510–5. doi: 10.1212/wnl.35.4.510. [DOI] [PubMed] [Google Scholar]
- 23.Bauer LA. Clinical pharmacokinetics and pharmacodynamics. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM, editors. Pharmacotherapy: A Pathophysiologic Approach. 4th ed. Stamford, CT: Appleton & Lange; 1999. pp. 21–43. [Google Scholar]
- 24.Bauer LA, Blouin RA. Age and phenytoin kinetics in adult epileptics. Clin Pharmacol Ther. 1982;31:301–4. doi: 10.1038/clpt.1982.37. [DOI] [PubMed] [Google Scholar]
- 25.Bach B, Molholm Hasen J, Kampmann JP, Rasmussen SN, Skovsted L. Disposition of antipyrine and phenytoin correlated with age and liver volume in man. Clin Pharmacokinet. 1981;6:389–96. doi: 10.2165/00003088-198106050-00005. [DOI] [PubMed] [Google Scholar]
- 26.Chen SS, Perucca E, Lee JN, Richens A. Serum protein binding and free concentration of phenytoin and phenobarbitone in pregnancy. Br J Clin Pharmacol. 1982;13:547–52. doi: 10.1111/j.1365-2125.1982.tb01420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knott C, Williams CP, Reynolds F. Phenytoin kinetics during pregnancy and the puerperium. Br J Obstet Gynaecol. 1986;93:1030–7. doi: 10.1111/j.1471-0528.1986.tb07827.x. [DOI] [PubMed] [Google Scholar]
- 28.Kochenour NK, Emery MG, Sawchuk RJ. Phenytoin metabolism in pregnancy. Obstet Gynaecol. 1980;56:577–82. [PubMed] [Google Scholar]
- 29.Lander CM, Smith MT, Chalk JB, et al. Bioavailability and pharmacokinetics of phenytoin during pregnancy. Eur J Clin Pharmacol. 1984;27:105–10. [PubMed] [Google Scholar]
- 30.Angelo T. Classroom Assessment: Assessing to Improve Higher Learning in the Life Sciences. In: Modell HI, Michael JA, Richardson D, Carroll RG, editors. Promoting Active Learning in the Life Science Classroom. NY: Annals of the New York Academy of Sciences; 1993. p. 701. (12). [Google Scholar]
- 31.Angelo T, Cross PK. Classroom Assessment Techniques: A Handbook For College Teachers. 2nd ed. San Francisco: Jossey-Bass; 1993. [Google Scholar]