

Abstract
Background
Doxorubicin (DOX) is used to treat childhood and adult cancer. DOX treatment is associated with both acute and chronic cardiotoxicity. The cardiotoxic effects of DOX are cumulative, limiting its chemotherapeutic dose. Free radical generation and p53-dependent apoptosis are thought to contribute to DOX-induced cardiotoxicity.
Methods and Results
Adult transgenic (MHC-CB7) mice expressing cardiomyocyte-restricted dominant-interfering p53 and their non-transgenic (NON-TXG) littermates were treated with DOX (20 mg/kg cumulative dose). NON-TXG mice exhibited reduced left ventricular (LV) systolic function (pre-DOX Fractional Shortening, FS, = 61 ± 2%, post-DOX FS = 45 ± 2%, mean +/- SEM, p<0.008), reduced cardiac mass, and high levels of cardiomyocyte apoptosis 7 days after the initiation of DOX treatment. In contrast, DOX-treated MHC-CB7 mice exhibited normal LV systolic function (pre-DOX FS = 63 ± 2%, post-DOX FS = 60 ± 2%, p>0.008), normal cardiac mass, and low levels of cardiomyocyte apoptosis. Western blot analyses indicated mTOR signaling was inhibited in DOX-treated NON-TXG mice, but not in DOX-treated MHC-CB7 mice. Accordingly, transgenic mice with cardiomyocyte-restricted constitutively active mTOR expression (MHC-mTORca) were studied. LV systolic function (pre-DOX FS = 64 +/- 2%, post-DOX FS 60 +/- 3%, p>0.008) and cardiac mass were normal in DOX-treated MHC-mTORca mice, despite similar levels of cardiomyocyte apoptosis as seen in DOX-treated NON-TXG mice.
Conclusions
These data suggest that DOX treatment induces acute cardiac dysfunction and reduces cardiac mass via p53-dependent inhibition of mTOR signaling, and that loss of myocardial mass, and not cardiomyocyte apoptosis, is the major contributor to acute DOX cardiotoxicity.
Keywords: heart failure, apoptosis, myocytes
Introduction
Anthracyclines such as doxorubicin (DOX), daunomycin, epirubicin and idarubicin, are widely used and highly successful anti-cancer chemotherapeutics. Unfortunately, these drugs also induce acute cardiotoxicity which is characterized by hypotension, tachycardia, arrhythmia and transient depression of left ventricular function.1-4 In addition, high cumulative doses are associated with late-onset cardiomyopathy that is refractory to standard treatment. It is widely thought that free radical-induced mitochondrial damage contributes to DOX-induced cardiotoxicity.5 In addition, DOX can induce DNA damage, inhibit DNA and protein synthesis, promote myofiber degeneration, inhibit transcription of specific gene programs, and induce cardiomyocyte apoptosis via a caspase-3 dependent mechanism. Because DOX can interfere with many different intracellular processes, it has proven difficult to determine the molecular etiologies which give rise to acute and chronic cardiotoxicity.
Numerous studies have shown that DOX-induced cardiomyocyte apoptosis is associated with increased expression of the p53 tumor suppressor protein. Moreover, reduction of p53 activity via genetic deletion6 or chemical inhibition7 is cardioprotective during acute DOX-treatment. To further characterize the role of p53 in acute DOX-induced cardiotoxicity, MHC-CB7 mice (which express dominant-interfering p53 in cardiomyocytes)8 were studied 7 days after the initiation of treatment. Cardiac function was improved, with a concomitant reduction in cardiomyocyte apoptosis, in the MHC-CB7 mice as compared to their DOX-treated non-transgenic (NON-TXG) siblings. Surprisingly, expression of the MHC-CB7 transgene also markedly blunted the DOX-induced reduction of cardiac mass observed in NON-TXG mice. Western blot analyses indicated that DOX treatment reduced the level of activated mammalian Target of Rapamycin (mTOR) in NON-TXG mice. mTOR is a serine/threonine protein kinase that regulates protein translation and cell growth.9 Expression of the MHC-CB7 transgene blocked DOX-induced reduction of mTOR activity. To establish the role of mTOR signaling in DOX-induced cardiotoxicity, mice expressing constitutively active mTOR in the myocardium (MHC-mTORca mice)10 were subjected to DOX-treatment. Expression of the MHC-mTORca transgene was sufficient to block DOX-induced cardiac dysfunction and mass reduction, but not cardiomyocyte apoptosis. These data suggest that acute DOX-induced cardiotoxicity results from p53-dependent dysregulation of the mTOR pathway, and furthermore that cardiomyocyte apoptosis does not contribute significantly to cardiac dysfunction during the acute response to DOX treatment.
Methods
Mice
This study utilized MHC-CB7 mice8 (n=56), MHC-mTORca mice10 (n=50) and their non-transgenic littermates (n=136). Mice were maintained in a DBA/2J genetic background. Adult mice received DOX (2 intraperitoneal injections of 10mg/kg at 3-day intervals, 20 mg/kg cumulative dose) or vehicle (saline), and were sacrificed 7 days after the initial injection. All animal protocols were approved by the Indiana University School of Medicine Institutional Animal Care and Research Advisory Committee.
Echocardiography
Mice were lightly anesthetized with 1.5% isofluorane until the heart rate stabilized at 400 to 500 beats per minute. Two-dimensional short-axis images were obtained using a high resolution Micro-Ultrasound system (Vevo 770, VisualSonics Inc., Toronto, Canada) equipped with a 40-MHz mechanical scan probe. Fractional shortening (FS), ejection fraction (EF), left ventricular internal diameter during systole (LVIDs), left ventricular internal diameter during diastole (LVIDd), end systolic volume (ESV) and end diastolic volume (EDV) were calculated using the Vevo Analysis software (version 2.2.3) as described.10 LVIDs and LVIDd were measured from M-mode recording at the level of the mid-papillary muscle, while ESV and EDV were measured with B-Mode recording in a plane containing the aortic and mitral valves.
Histology
Hearts were harvested, cryoprotected in 30% sucrose, embedded and sectioned at 10 microns using standard techniques. To quantitate minimal cardiomyocyte fiber diameter, images from Sirius red / Fast green stained sections were captured, digitized, and analyzed with NIH Image 1.36b software as described.11 At least 400 randomly selected cardiomyocytes from each animal were analyzed. To quantitate cardiomyocyte apoptosis, four transverse sections from each heart, sampled from the midpoint between the apex and base, were post-fixed in 4% paraformaldehyde and screened for anti-activated caspase-3 immune-reactivity (antibody #G7481, Promega, Madison, WI), followed by a horseradish peroxidase-conjugated secondary antibody; signal was visualized with a diaminobenzidine reaction as described previously.8
Western blot analyses
Proteins were extracted and quantitated using the Coomassie Blue method (Pierce, Rockford, IL) as described.8 Samples were solubilized in sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) loading buffer for 5 min at 95°C and resolved on 7 or 10% SDS-PAGE gels. Fractionated proteins were then electrotransferred from the gel to nitrocellulose (Amersham) filters in Towbin buffer at 200-mA constant current and analyzed by Western blotting. The filters were stained with 0.1% naphthol blue-black in 45% methanol, 10% acetic acid to assess the efficiency of transfer. Antibodies used recognized PARP (catalogue #9542, Cell Signaling Technology, Danvers, MA), Bcl-xL (catalogue #sc-7195, Santa Cruz Biotechnology, Santa Cruz, CA), p53 (catalogue #PC-35, EMD Chemicals, Gibbstown, NJ), AU-1 tag (catalogue #MMS-130R, Covance Inc., Emeryville, CA), P-mTOR[ser2481] (catalogue #2974, Cell Signaling Technology), P-mTOR[ser2448] (catalogue #2971, Cell Signaling Technology), total mTOR, (catalogue #2972,Cell Signaling Technology), P-eIF4G[ser1108] (catalogue #2441, Cell Signaling Technology), total eIF4G (catalogue #SC-11373, Santa Cruz Biotechnology), P-eIF4E[ser209] (catalogue #9741, Cell Signaling Technology), total eIF4E (catalogue #9742, Cell Signaling Technology), 4EBP1 (catalogue #9452, Cell Signaling Technology), P-P70S6 kinase[thr421/ser424] (catalogue #9204, Cell Signaling Technlogy), total P70S6 kinase (catalogue #9202, Cell Signaling Technlogy), and total TSC2 (catalogue #SC-893, Santa Cruz Biotechnology). Signal was visualized by the ECL method according to the manufacturer’s protocol (Amersham). Western signal was digitized and quantitated using ImageJet software.
Statistic analysis
All values are presented as mean ± SEM. Statistical significance (p<0.01) was determined by Student’s t-test (for groups of two) or by one way ANOVA with Bonferroni correction (p<0.008 was considered significant, 6 pair-wise comparisons).
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
Results
DOX induces acute cardiotoxicity via a p53-dependent pathway
MHC-CB7 mice (which express dominant-interfering p53) and their non-transgenic (NON-TXG) littermates were used to study the role of p53 in acute DOX cardiotoxicity. Prior to DOX administration, LV systolic function was similar in NON-TXG and MHC-CB7 mice (Figure 1A; see also Table 1, Supplemental Data). Seven days after the initiation of DOX treatment, Fractional Shortening (FS) was markedly reduced in the NON-TXG mice (Figure 1A). Increased left ventricular inner dimension during systole (LVIDs) and increased end systolic volume (ESV) were also noted (Table 1, Supplementary Data). In contrast, LV systolic function and cardiac dimensions in MHC-CB7 mice were not altered by DOX treatment (Figure 1A; see also Table 1, Supplemental data). Representative echocardiograms from the NON-TXG and MHC-CB7 mice are shown in Figure 1B.
Figure 1.
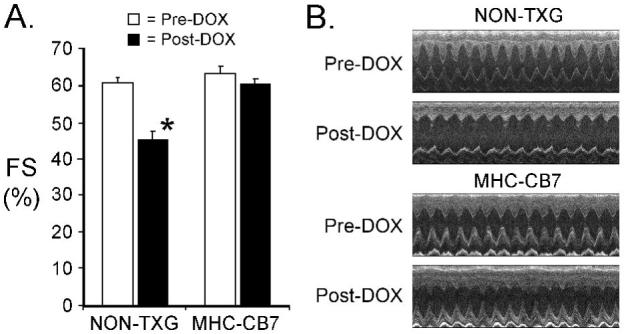
Cardiac function in NON-TXG and MHC-CB7 before and after DOX treatment. (A) Fractional Shortening (FS [%]) was measured prior to (Pre-DOX) and after (Post-DOX) treatment with DOX. ’*’ indicates p<0.008 vs. NON-TXG Pre-DOX. (B) Representative short axis echocardiograms from the NON-TXG and MHC-CB7 mice.
Activated caspase-3 was used as a marker to quantitate cardiomyocyte apoptosis. Since activated caspase-3 immune-reactivity is present in the cytoplasm, cardiomyocytes at early stages of apoptosis can be identified in histologic sections based on cell size and shape.8 DOX treatment resulted in a marked increase in the number of activated caspase-3 immune-reactive cardiomyocytes in NON-TXG animals (Figure 2A). Although DOX-treatment induced cardiomyocyte activated caspase-3 immune-reactivity in MHC-CB7 mice, the level of induction was approximately 4-fold reduced as compared to DOX-treated NON-TXG mice (Figure 2A). Cleavage of poly (ADP-ribose) polymerase (PARP, a substrate of caspase-3) was also monitored. DOX treatment in NON-TXG mice resulted in PARP cleavage (Figure 2B, upper panel), consistent with the observed increase in activated caspase-3 immune-reactivity. In contrast, PARP cleavage product was not detected in DOX-treated MHC-CB7 mice. Expression of the MHC-CB7 transgene was associated with a modest induction of Bcl-xL, a pro-survival member of the Bcl-2 gene family (Figure 2B). Expression of several other Bcl-2 family members (Bax, Bak and Bcl-2) were not altered (data not shown). Anti-p53 Western blots confirmed expression of the MHC-CB7 transgene (Figure 2B). The antibody used recognizes both CB7 and wild-type p53; in agreement with other studies,7 increased levels of endogenous p53 were observed in longer exposures of Western blots from DOX-treated NON-TXG hearts (Figure 2B, lower panel).
Figure 2.
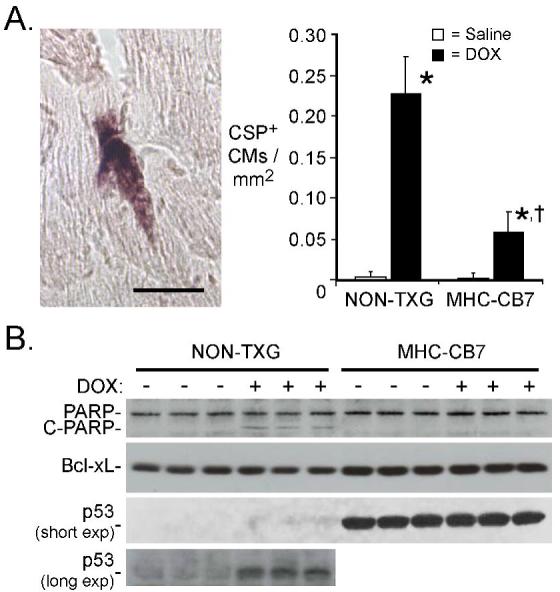
Cardiomyocyte apoptosis in NON-TXG and MHC-CB7 mice following saline or DOX treatment. (A) The left panel shows a representative caspase-3 immune-reactive cardiomyocyte from a DOX-treated NON-TXG mouse (bar=40 μm). The right panel indicates the number of activated caspase-3 immune-reactive cardiomyocytes (CSP+) per mm2 following saline or DOX treatment. “*” indicates p<0.008 vs. saline-treated NON-TXG mice; “†” indicates p<0.008 vs. DOX-treated NON-TXG mice; n = 5 mice per group. (B) Western blot analysis of apoptosis-related proteins in NON-TXG and MHC-CB7 mice following saline or DOX treatment (see Supplemental Data for densitometric signal quantitation; short and long exposures of the anti-p53 blot are shown).
DOX treatment had a dramatic impact on heart size in NON-TXG mice. Total heart weight (HW) was reduced by ca. 30% (Figure 3A; a reduction in body weight was also noted, see Table 2, Supplemental Data). Previous studies have shown that cardiomyocyte minimal fiber diameter (MFD) measurements can be used to quantitate myocyte hypertrophy and atrophy.12 A reduction in cardiomyocyte MFD was apparent following DOX-treatment of NON-TXG mice, suggesting that the reduction in heart weight largely reflected DOX-induced reduction of cardiomyocyte size (Figure 3B; see also Table 2 Supplemental Data). In contrast, heart weight and cardiomyocyte MFD in MHC-CB7 mice were largely unaffected by DOX treatment (Figure 3A, B), although a marked reduction in body weight was observed (Table 2, Supplemental Data). Control dose-response experiments with NON-TXG mice revealed that the onset of decreased cardiac mass and cardiomyocyte apoptosis (as indicated by PARP cleavage) were first detected at the same dose of DOX (Figure 6, Supplemental Data). Collectively, these data indicate that acute DOX-induced cardiac dysfunction, cardiomyocyte apoptosis, and cardiac mass reduction occur via a p53-dependent pathway, and that these characteristics of acute DOX-induced cardiotoxicity can effectively be blocked by expression of dominant-interfering p53.
Figure 3.
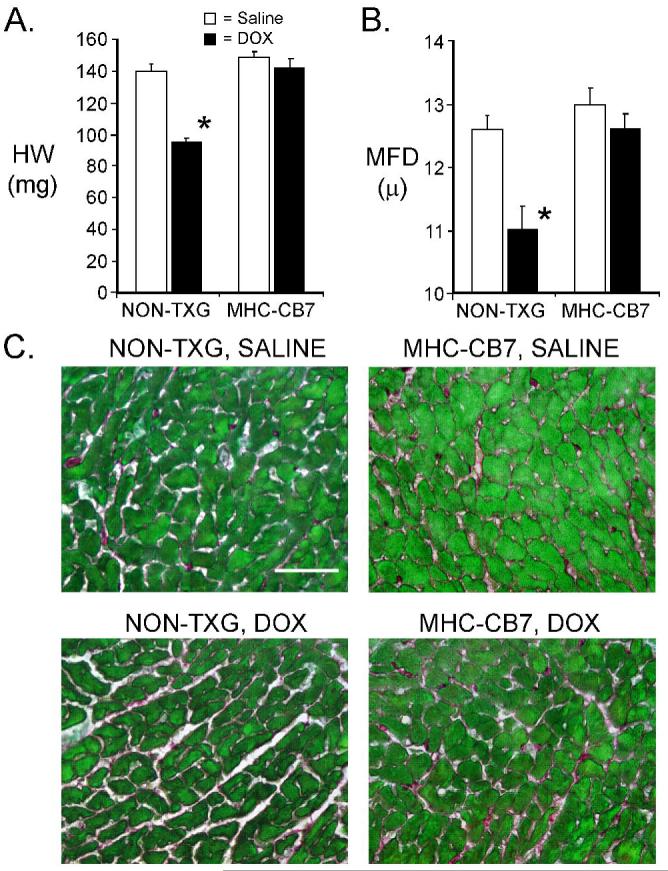
Characterization of NON-TXG and MHC-CB7 hearts following saline or DOX treatment. (A) Heart weight in milligrams (HW [mg]) in NON-TXG and MHC-CB7 mice treated with saline or DOX. ’*’ indicates p<0.008 vs. saline-treated NON-TXG mice. (B) Cardiomyocyte minimal fiber diameter (MFD [μ]) measurements in NON-TXG and MHC-CB7 mice treated with saline or DOX. ’*’ indicates p<0.008 vs. saline-treated NON-TXG mice. (C) Sections from saline- or DOX-treated NON-TXG and MHC-CB7 hearts stained with Sirius red / fast green (bar = 50 microns).
DOX alters mTOR pathway activity via a p53-dependent mechanism
The marked heart weight reduction in DOX-treated NON-TXG mice was somewhat surprising. Previous studies have shown that the mTOR pathway plays a critical role in the regulation of protein translation and cell growth.9 We therefore compared the activity and total level of mTOR in hearts from saline- and DOX-treated NON-TXG mice. mTOR undergoes an auto-phosphorylation event at serine residue 2481, and the presence of P-mTOR[ser2481] is often used as an indicator of mTOR activity.13 DOX treatment resulted in a ca. 4-fold reduction of P-mTOR[ser2481] levels in the hearts of NON-TXG, mice but did not impact total mTOR levels (Figure 4; see Supplemental Data for densitometric quantitation). In contrast, no changes in P-mTOR[ser2481] levels were observed in DOX-treated MHC-CB7 hearts (Figure 4).
Figure 4.
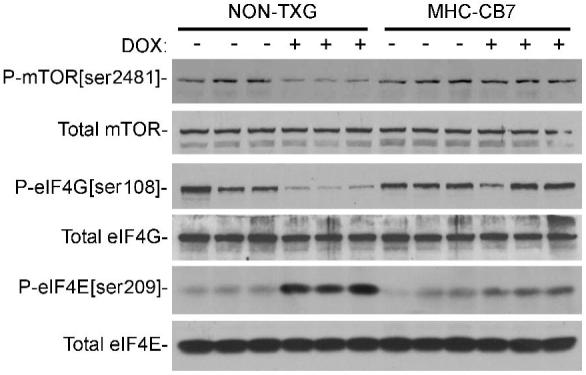
Western blot analysis of the mTOR signaling pathway in hearts from NON-TXG and MHC-CB7 mice treated with saline or DOX (see Supplemental Data for densitometric signal quantitation).
To determine the potential consequences of decreased levels of active mTOR, the canonical downstream effectors were analyzed. No changes in the total level or phosphorylation status of 4EBP1 and p70S6 kinase were apparent in DOX-treated NON-TXG hearts (see Figure 7, Supplemental Data). mTOR activation also promotes the phosphorylation of protein translation initiation factor eIF4G at serine residue 1108 (P-eIF4G[ser1108]), as well as the dephosphorylation of eIF4E at serine residue 209 (P-eIF4E[ser209]). These post-translational modifications have been shown to enhance protein translation initiation under some experimental circumstances (see Discussion). DOX-treatment decreased P-eIF4G[ser1108] levels and increased P-eIF4E[ser209] levels in NON-TXG mice, without impacting the total level of the initiation factors (Figure 4). In contrast, no changes were apparent in P-eIF4G[ser1108] or P-eIF4E[ser209] levels in DOX-treated MHC-CB7 hearts (Figure 4).
Constitutively active mTOR blocks acute DOX cardiotoxicity
The data presented above suggest that acute DOX cardiotoxicity occurs via p53-dependent modulation of mTOR signaling. To directly test this hypothesis, we used transgenic mice that express a mutant mTOR in which the auto-inhibition domain located between amino acid residues #2430 and #2450 was deleted.14 These animals (designated MHC-mTORca) exhibit constitutively elevated mTOR activity in the heart.10 An AU1 epitope tag was engineered in the N-terminus to facilitate identification of the transgene-encoded protein via Western blot analysis. Adult MHC-mTORca mice and their non-transgenic littermates were injected with DOX and subjected to functional, histologic and molecular analyses as described above. DOX treatment resulted in a marked reduction in FS in NON-TXG mice, but had no impact on cardiac function in the MHC-mTORca animals (Figure 5). Increased LVIDs was apparent in the DOX-treated NON-TXG mice, but not in the DOX-treated MHC-mTORca mice (Table 3, Supplementary Data).
Figure 5.
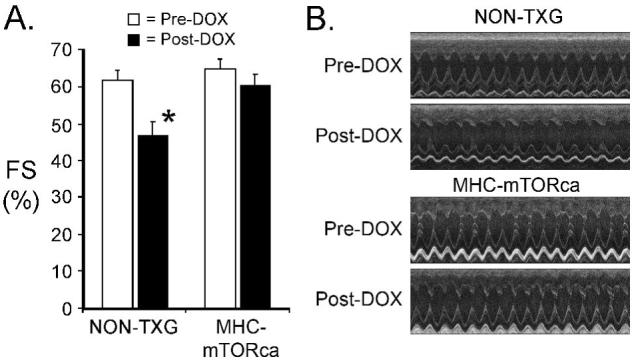
Cardiac function in NON-TXG and MHC-mTORca before and after DOX treatment. (A) Fractional Shortening (FS [%]) was measured prior to (Pre-DOX) and after (Post-DOX) treatment with doxorubicin. ’*’ indicates p<0.008 vs. Pre-DOX NON-TXG mice. (B) Representative short axis echocardiograms from the NON-TXG and MHC-mTORca mice.
Cardiomyocyte apoptosis was quantitated by activated caspase-3 immune-reactivity. As was observed above, DOX treatment resulted in a marked increase in the level of cardiomyocyte activated caspase-3 immune-reactivity in NON-TXG mice (Figure 6A). Surprisingly, comparable levels were observed in DOX-treated NON-TXG and DOX-treated MHC-mTORca animals. Western blot analyses revealed similar levels of cleaved PARP in the DOX-treated NON-TXG and MHC-mTORca mice (Figure 6B, upper panel), consistent with the activated caspase-3 results. Moreover, no induction of Bcl-xL was observed in the transgenic hearts. Transgene expression was confirmed via Western analysis with an anti-AU1 antibody. A 26% reduction in HW and a 12% reduction in cardiomyocyte MFD were observed in DOX-treated NON-TXG mice (Figure 7A and B; see also Table 4, Supplemental Data). In contrast, DOX-treatment had no impact on these parameters in MHC-mTORca mice. Tritiated thymidine incorporation analyses11 indicated that DOX-treatment did not induce cell cycle activity in the MHC-mTOR mice (data not shown); thus cardiomyocyte replacement cannot account for the preservation of cardiac mass. Interestingly, normal cardiac function and mass were retained for as long as three weeks post DOX-treatment in the MHC-mTOR mice (See Table 5, Supplemental Data). Collectively, these data indicate that activation of the mTOR pathway effectively blocks acute DOX-induced cardiac dysfunction and mass reduction, but not acute DOX-induced cardiomyocyte apoptosis.
Figure 6.
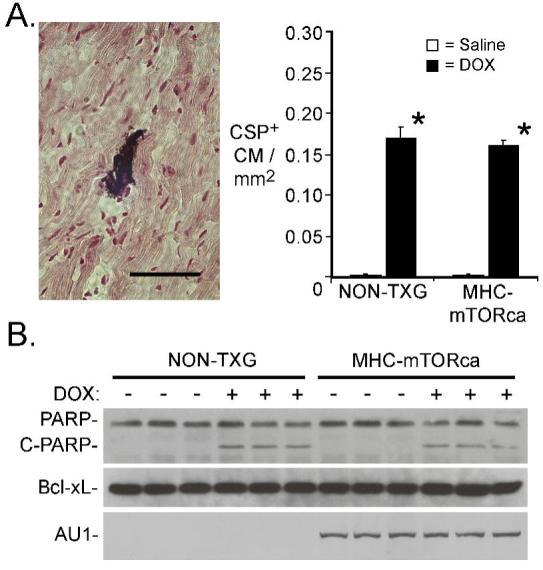
Cardiomyocyte apoptosis in NON-TXG and mTORca mice following saline or DOX treatment. (A) The left panel shows a representative caspase-3 immune-reactive cardiomyocyte from a DOX-treated NON-TXG mouse (bar=40 μm). The right panel indicates the number of activated caspase-3 immune-reactive cardiomyocytes (CSP+) per mm2 following saline or DOX treatment. “*” indicates p<0.008 vs. saline-treated NON-TXG mice; n = 5 mice per group. (B) Western blot analysis of apoptosis-related proteins in NON-TXG and mTORca mice following saline or DOX treatment (see Supplemental Data for densitometric signal quantitation).
Figure 7.
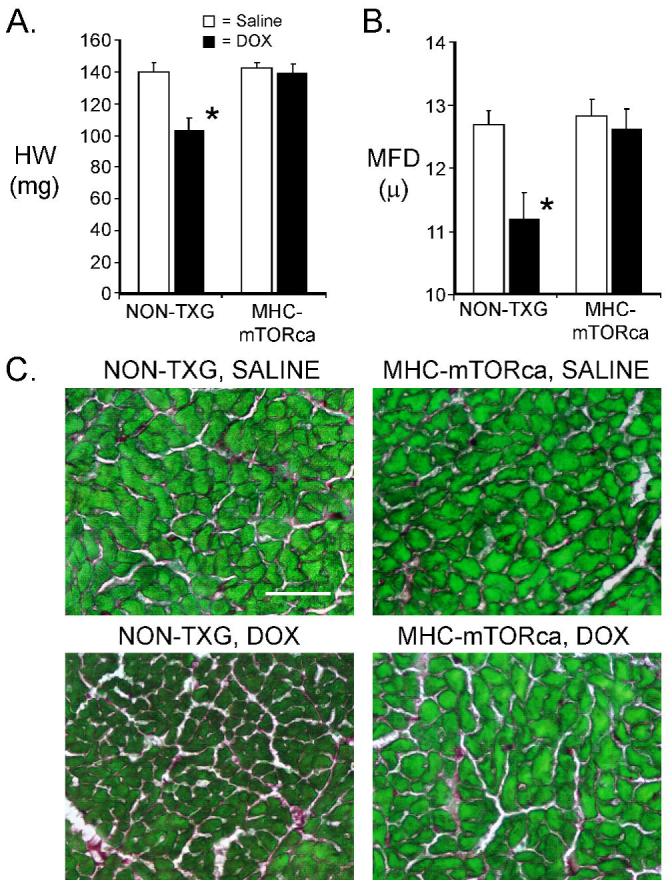
Characterization of NON-TXG and MHC-mTORca hearts following saline or DOX treatment. (A) Heart weight in milligrams (HW [mg]) in NON-TXG and MHC-mTORca mice treated with saline or DOX. ’*’ indicates p<0.008 vs. saline-treated NON-TXG mice. (B) Cardiomyocyte minimal fiber diameter (MFD [μ]) measurements in NON-TXG and MHC-mTORca mice treated with saline or DOX. ’*’ indicates p<0.008 vs. saline-treated NON-TXG mice. (C) Sections from saline- or DOX-treated NON-TXG and MHC-mTORca hearts stained with Sirius red / fast green (bar = 50 microns).
Western blot analyses were performed to determine the impact of MHC-mTORca transgene expression on post-translational modification of the downstream effectors of the mTOR signaling pathway. As was observed above, DOX treatment in NON-TXG mice resulted in decreased levels of P-mTOR[ser2481] and P-eIF4G[ser1108] and increased levels of P-eIF4E[ser209] (Figure 8, see Supplemental Data for densitometric quantitation). Expression of the MHC-mTORca transgene resulted in an increase in total mTOR levels, and also a ca. two-fold increase in the level of P-mTOR[ser2481]. This constitutive mTOR activity resulted in increased levels of P-eIF4G[ser1108] and decreased levels of P-eIF4E[ser209] in saline treated animals. DOX treatment had no impact on the levels of these phosphorylation events in MHC-mTORca mice (Figure 8).
Figure 8.
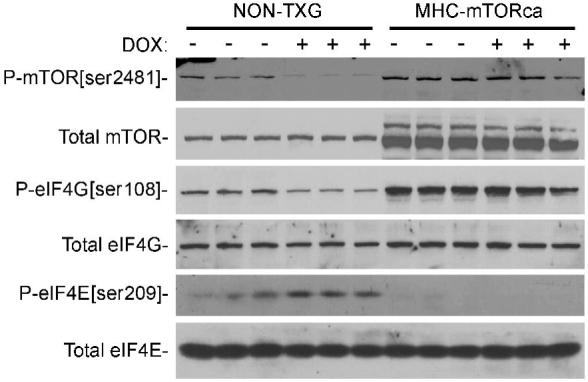
Western blot analysis of the mTOR signaling pathway in hearts from NON-TXG and MHC-mTORca mice treated with saline or DOX (see Supplemental Data for densitometric signal quantitation).
Discussion
The data presented here support a model wherein acute DOX-induced cardiac systolic dysfunction and cardiac mass loss occur via p53-dependent modulation of the mTOR pathway. The data also demonstrate that acute DOX-induced cardiomyocyte apoptosis occurs via a p53 dependent pathway, but is independent of mTOR signaling. The presence of similar levels of cardiomyocyte activated caspase-3 immune-reactivity in DOX-treated NON-TXG and MHC-mTORca hearts suggests that cardiomyocyte apoptosis per se does not contribute significantly to decreased cardiac function during acute DOX cardiotoxicity. Rather, inhibition of the mTOR signaling pathway appears to be the predominant contributor to acute DOX cardiotoxicity.
Previous studies have implicated the p53 pathway in DOX-induced cardiotoxicity. For example, reduced levels of cardiomyocyte apoptosis and concomitant improvements in cardiac function were observed in DOX-treated p53 null mice as compared to their wild-type littermates.6 Similarly, pretreatment with pifithrin-α (a chemical which inhibits nuclear translocation of p53) reduced DOX-induced cardiomyocyte apoptosis and cardiac dysfunction in vivo.7 Thus, the reduced level of cardiomyocyte apoptosis observed in DOX-treated MHC-CB7 mice was not unexpected. This phenotype may result in part from the concomitant albeit modest increase in steady state levels of Bcl-xL (Figure 2), as other studies have demonstrated that p53 can regulate cell apoptosis in part by interacting with Bcl-xL.15
In contrast, the impact of p53 inactivation on DOX-induced cardiac mass reduction was not expected, nor was it noted in previous studies. The observation that DOX treatment decreased mTOR activity (as evidenced by decreased P-mTOR[ser2481] levels) in NON-TXG mice but not in MHC-CB7 mice, coupled with the marked cardioprotection observed in DOX-treated MHC-mTORca mice, strongly implicates a causal relationship between p53 and mTOR signaling during acute DOX-induced cardiotoxicity. Indeed, recent studies in non-cardiomyocytes have shown that p53 can negatively regulate the mTOR pathway via the Tuberous Sclerosis complex (TSC) 2 - RHEB G-protein axis.16-19 Although DOX treatment failed to alter steady-state levels of TSC2 in hearts from DOX-treated mice (Figure 7, Supplemental Data), a recent study suggests that post-translational modifications might be more relevant for regulation of TSC2 activity in muscle.20
The mTOR pathway has been implicated previously in the regulation of cardiomyocyte growth. For example, alcohol decreases the level of mTOR phosphorylation and activity in the myocardium, with a concomitant reduction of cardiac mass. Interestingly, phosphorylation was reduced predominately at P-mTOR[ser2481] following acute alcohol administration,21 and predominantly at P-mTOR[ser2448] following chronic alcohol administration.22 mTOR signaling is also important for the induction of cardiac hypertrophy. Physiologic cardiac hypertrophy induced by treadmill exercise is associated with activation of the mTOR pathway.23 Pathophysiologic cardiac hypertrophy induced by aortic banding is also associated with rapamycin-sensitive activation of mTOR at early, but not late, timepoints.23, 24 Thyroid hormone-induced hypertrophy also activates mTOR signaling and is rapamycin sensitive.25
Although these studies indicate that mTOR is important for the regulation of myocardial growth, they also illustrate that its signaling through downstream effector molecules is very complex and at times contradictory. This fact is underscored by the absence of overt changes in the canonical mTOR downstream effectors p70S6 kinase and 4EBP-1 in hearts from DOX-treated NON-TXG mice (Figure 7, Supplemental Data). In this regard, it is of interest to note that previous studies using non-phosphorylatable point mutants have shown that dephosphorylation at mTOR serine residue #2448 (but not #2481) mediates rapamycin-induced inhibition of p70S6 kinase and 4EBP-1 activity.13,26 Indeed, marked dephosphorylation of mTOR serine residue #2448 and concomitant reduction of p70S6 kinase and 4EBP-1 phosphorylation was observed in hearts from rapamycin-treated mice, but not from DOX-treated mice (Figure 7, Supplemental Data). Thus, our data supports the previous studies indicating that post-translational modulation of mTOR residue #2448 is responsible for regulating p70S6 kinase and 4EBP-1 activity. Conversely, the observation that co-administration of DOX and rapamycin did not result in reduced cardiac mass in MHC-CB7 mice, and did not further exacerbate DOX-induced cardiac mass in NON-TXG mice (Table 6, Supplemental Data), supports the importance of mTOR residue #2481 in acute DOX-induced cardiotoxicity. The complexity of feed-back regulation of the mTOR pathway is further underscored by the absence of overt cardiac hypertrophy in the MHC-mTORca mice (Figure 7), and the absence of a reduction in baseline cardiac mass in rapamycin-treated rats.25
The observed changes in P-eIF4G[ser1108] and P-eIF4E[ser209] levels in DOX-treated NON-TXG mice, coupled with the absence of changes in DOX-treated MHC-CB7 and MHC-mTORca mice, raises the possibility that these molecules may be downstream mTOR effectors during acute DOX cardiotoxicity. eIF4G is a scaffold protein that links eIF3 and the 43S initiation complex.27, 28 Serum-stimulation promotes rapamycin-sensitive increases in P-eIF4G[ser1108], and concomitant protein synthesis, in cultured HEK293 cells.29 Moreover, rats exhibit rapamycin-sensitive increases in P-eIF4G[ser1108] in the myocardium following controlled food intake.30 Thus, mTOR-mediated phosphorylation (either directly or indirectly) of eIF4G at serine residue 1108 is associated with anabolic growth in the heart. Of interest, activation of a temperature sensitive p53 transgene in murine erythroleukemia cells decreased P-mTOR[ser2481] and P-eIF4G[ser1108] levels and promoted cellular atrophy, 31 a sequale which is remarkably similar to that observed in DOX-treated myocardium.
The situation with eIF4E is less straightforward. eIF4E binds to the 5’ end cap structure of the mRNA and interacts with eIF4G.27, 28 Phosphorylation of eIF4E at serine residue 209 is rapamycin-sensitive,32 and has been reported to inhibit,33 enhance,34 or have no impact35 on protein translation, depending upon the cell type and experimental system employed. P-eIF4E[ser209] has reduced affinity for capped mRNA, which is thought to promote the release of initiation factors from existing translational complexes.36 Such activity could contribute in part to the reduction in cardiac mass observed in DOX-treated hearts. Ultimately, direct proof of the role of these (or any other) putative mTOR effector molecules in DOX-induced cardiotoxicity will require the generation of additional transgenic models with the relevant phosphomimetic and/or non-phosphorylatable mutations. However, as with all transgenic overexpression studies, the possibility that elevated mTOR activity is cardioprotective irrespective of the molecular etiology of DOX-induced ventricular induction in NON-TXG animals constitutes a limitation of the present study.
The p53/mTOR axis may regulate other pathways that contribute to acute DOX cardiotoxicity. For example, activation of the ubiquitin-proteasome system has been reported in DOX-treated cardiomyocytes.37 Interestingly, proteosome-mediated degradation of the p300 transcription co-activator has previously been implicated in negative regulation of myocardial transcription programs.38-40 Activation of the ubiquitinproteasome system has also been implicated in heart size reduction following chronic unloading,41, 42 albeit with a paradoxical increase in mTOR activity. It is also tempting to speculate that cardiomyocyte autophagy43, 44 may contribute to acute DOX cardiotoxicity. Although preliminary analysis failed to establish direct correlations between the presence of molecular markers for proteasome and/or autophagy activity and the presence of cardiotoxicity in DOX-treated mice under the conditions employed here (W. Zhu, unpublished observations), the potential role of these pathways in acute DOX cardiotoxicity cannot as yet be discounted.
In summary, the data reported here suggests that acute DOX-induced cardiotoxicity results from p53-dependent modulation of mTOR activity. The data also suggests that cardiomyocyte apoptosis does not contribute to decreased cardiac function at the time point studied. It has recently been reported that treatment with erythropoietin,45 thrombopoietin46 or granulocyte colony-stimulating factor47 can blunt DOX-induced cardiac dysfunction and mass reduction. It would be informative to determine if these molecules also work via alteration of mTOR signaling. Additionally, it would be of considerable interest to determine if upstream effectors which activate the mTOR pathway would be cardioprotective in the setting of acute DOX cardiotoxicity.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by grants from the NHLBI.
Footnotes
Disclosures
The authors have no disclosures to report.
References
- 1.Bristow MR, Billingham ME, Mason JW, Daniels JR. Clinical spectrum of anthracycline antibiotic cardiotoxicity. Cancer Treat Rep. 1978;62(6):873–879. [PubMed] [Google Scholar]
- 2.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32(2):302–314. doi: 10.1002/1097-0142(197308)32:2<302::aid-cncr2820320205>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Lipshultz SE, Alvarez JA, Scully RE. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart. 2008;94(4):525–533. doi: 10.1136/hrt.2007.136093. [DOI] [PubMed] [Google Scholar]
- 4.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339(13):900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 5.Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001;61(2):771–777. [PubMed] [Google Scholar]
- 6.Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273(12):25–32. doi: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Chua CC, Gao J, Chen Z, Landy CL, Hamdy R, Chua BH. Pifithrin-alpha protects against doxorubicin-induced apoptosis and acute cardiotoxicity in mice. Am J Physiol Heart Circ Physiol. 2004;286(3):H933–939. doi: 10.1152/ajpheart.00759.2003. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94(12):1606–1614. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 9.Lee CH, Inoki K, Guan KL. mTOR pathway as a target in tissue hypertrophy. Annu Rev Pharmacol Toxicol. 2007;47:443–467. doi: 10.1146/annurev.pharmtox.47.120505.105359. [DOI] [PubMed] [Google Scholar]
- 10.Shen WH, Chen Z, Shi S, Chen H, Zhu W, Penner A, Bu G, Li W, Boyle DW, Field LJ, Abraham R, Liechty EA, Shou W. Cardiac restricted overexpression of kinase dead mTOR mutant impairs the mTOR-mediated signaling and cardiac function. J Biol Chem. 2008 doi: 10.1074/jbc.M801510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am J Physiol. 1994;266(4 Pt 2):H1439–1445. doi: 10.1152/ajpheart.1994.266.4.H1439. [DOI] [PubMed] [Google Scholar]
- 12.Dubowitz V, Sewry CA, Fitzsimons RB. Muscle biopsy: a practical approach. 2nd Baillière Tindall; London; Philadelphia: 1985. [Google Scholar]
- 13.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275(10):7416–7423. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 14.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60(13):3504–3513. [PubMed] [Google Scholar]
- 15.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 16.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7):3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 17.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 19.Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev. 2006;20(3):267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 20.McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol. 2008;586(6):1731–1741. doi: 10.1113/jphysiol.2007.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vary TC, Deiter G, Goodman SA. Acute alcohol intoxication enhances myocardial eIF4G phosphorylation despite reducing mTOR signaling. Am J Physiol Heart Circ Physiol. 2005;288(1):H121–128. doi: 10.1152/ajpheart.00440.2004. [DOI] [PubMed] [Google Scholar]
- 22.Vary TC, Deiter G, Lantry R. Chronic alcohol feeding impairs mTOR(Ser 2448) phosphorylation in rat hearts. Alcohol Clin Exp Res. 2008;32(1):43–51. doi: 10.1111/j.1530-0277.2007.00544.x. [DOI] [PubMed] [Google Scholar]
- 23.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214(2):316–321. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- 24.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107(12):1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 25.Kuzman JA, O’Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148(7):3477–3484. doi: 10.1210/en.2007-0099. [DOI] [PubMed] [Google Scholar]
- 26.Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280(27):25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 27.Clemens MJ, Bommer UA. Translational control: the cancer connection. Int J Biochem Cell Biol. 1999;31(1):1–23. doi: 10.1016/s1357-2725(98)00127-7. [DOI] [PubMed] [Google Scholar]
- 28.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 29.Raught B, Gingras AC, Gygi SP, Imataka H, Morino S, Gradi A, Aebersold R, Sonenberg N. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. Embo J. 2000;19(3):434–444. doi: 10.1093/emboj/19.3.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vary TC, Deiter G, Lynch CJ. Rapamycin limits formation of active eukaryotic initiation factor 4F complex following meal feeding in rat hearts. J Nutr. 2007;137(8):1857–1862. doi: 10.1093/jn/137.8.1857. [DOI] [PubMed] [Google Scholar]
- 31.Constantinou C, Clemens MJ. Regulation of the phosphorylation and integrity of protein synthesis initiation factor eIF4GI and the translational repressor 4E-BP1 by p53. Oncogene. 2005;24(30):4839–4850. doi: 10.1038/sj.onc.1208648. [DOI] [PubMed] [Google Scholar]
- 32.de Obanos MP Perez, Zabalza MJ Lopez, Prieto J, Herraiz MT, Iraburu MJ. Leucine stimulates procollagen alpha1(I) translation on hepatic stellate cells through ERK and PI3K/Akt/mTOR activation. J Cell Physiol. 2006;209(2):580–586. doi: 10.1002/jcp.20790. [DOI] [PubMed] [Google Scholar]
- 33.Knauf U, Tschopp C, Gram H. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol Cell Biol. 2001;21(16):5500–5511. doi: 10.1128/MCB.21.16.5500-5511.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19(3):1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morley SJ, Naegele S. Phosphorylation of eukaryotic initiation factor (eIF) 4E is not required for de novo protein synthesis following recovery from hypertonic stress in human kidney cells. J Biol Chem. 2002;277(36):32855–32859. doi: 10.1074/jbc.C200376200. [DOI] [PubMed] [Google Scholar]
- 36.Scheper GC, van Kollenburg B, Hu J, Luo Y, Goss DJ, Proud CG. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J Biol Chem. 2002;277(5):3303–3309. doi: 10.1074/jbc.M103607200. [DOI] [PubMed] [Google Scholar]
- 37.Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. Faseb J. 2005;19(14):2051–2053. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 38.Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol Cell Biol. 2000;20(23):8643–8654. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem. 1997;272(36):22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 40.Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependent degradation of p300 by doxorubicin-activated p38 mitogen-activated protein kinase in cardiac cells. Mol Cell Biol. 2005;25(7):2673–2687. doi: 10.1128/MCB.25.7.2673-2687.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB, Taegtmeyer H. Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation. 2003;108(20):2536–2541. doi: 10.1161/01.CIR.0000096481.45105.13. [DOI] [PubMed] [Google Scholar]
- 42.Sharma S, Ying J, Razeghi P, Stepkowski S, Taegtmeyer H. Atrophic remodeling of the transplanted rat heart. Cardiology. 2006;105(2):128–136. doi: 10.1159/000090550. [DOI] [PubMed] [Google Scholar]
- 43.Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4(3):322–329. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 45.Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Khai NC, Maruyama R, Ogino A, Minatoguchi S, Fujiwara T, Fujiwara H. Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation. 2006;113(4):535–543. doi: 10.1161/CIRCULATIONAHA.105.568402. [DOI] [PubMed] [Google Scholar]
- 46.Li K, Sung RY, Huang WZ, Yang M, Pong NH, Lee SM, Chan WY, Zhao H, To MY, Fok TF, Li CK, Wong YO, Ng PC. Thrombopoietin protects against in vitro and in vivo cardiotoxicity induced by doxorubicin. Circulation. 2006;113(18):2211–2220. doi: 10.1161/CIRCULATIONAHA.105.560250. [DOI] [PubMed] [Google Scholar]
- 47.Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Ogino A, Maruyama R, Nakagawa M, Minatoguchi S, Fujiwara T, Fujiwara H. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest. 2007;87(5):440–455. doi: 10.1038/labinvest.3700530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.