

Abstract
The term “biological complexes” broadly encompasses particles as diverse as multisubunit enzymes, viral capsids, transport cages, molecular nets, ribosomes, nucleosomes, biological membrane components and amyloids. The complexes represent a broad range of stability and composition. Atomic force microscopy offers a wealth of structural and functional data about such assemblies. For this review, we choose to comment on the significance of AFM to study various aspects of biology of selected nonmembrane protein assemblies. Such particles are large enough to reveal many structural details under the AFM probe. Importantly, the specific advantages of the method allow for gathering dynamic information about their formation, stability or allosteric structural changes critical for their function. Some of them have already found their way to nanomedical or nanotechnological applications. Here we present examples of studies where the AFM provided pioneering information about the biology of complexes, and examples of studies where the simplicity of the method is used toward the development of potential diagnostic applications.
Keywords: atomic force microscopy, protein complexes, protein structure, allostery, proteasome, GroEL, amyloid, nanomedicine
1. Introduction
Biological complexes as diverse as multimeric enzymes, biological cages, viruses, amyloids or ribosomes are considered excellent subjects for a method like atomic force microscopy for several reasons. The complexes are large but not always stable in vitro. Such properties do not restrict AFM applications, however they are disadvantageous for NMR or X-ray crystallography. Importantly, AFM provides opportunity to study particles without fixing, in aqueous environment, and – thanks to the advance of oscillation-mode methods - in mechanically noninvasive manner. Therefore, the most interesting features of biological complexes: their assembly patterns, dynamic behavior or ability to interact with other molecules, all can be followed by AFM, a feature not easily achievable with other structural methods including EM or crystallography. Moreover, AFM can provide overall topographical and mechanical, to some extend time-resolved, information about particles. Such generality of structural characteristic distinguishes AFM from fluorescence or CD spectroscopies. Summarizing, AFM can offer a unique insight into both structure and mechanism of action of biological complexes. At the dawn of the method, collecting images representing the outer topography of the particles under liquid or in air was the desired outcome. Then, the processes involving interactions of multiple molecules came into focus, with the insight into their mechanism being in the center of interest. In this review, we will comment on the use of AFM in studies of nonmembrane protein assemblies. We will follow major groups of processes especially well fitted for the technique: assembly of multimeric complexes, aggregation of amyloidtype particles, and intrinsic structural dynamics. Finally, we will briefly review the actual and potential nanomedical and nanotechnological uses of AFM in the context of biological complexes.
2. AFM and biological complexes: a perfect match, thanks to the technique
The physical basis behind AFM determines its usefulness for studying biologically functional complexes. The settings favorable for intact biological activity imply low invasiveness of the imaging method: no need for chemical modification of the bioparticles, no mechanical damage, the opportunity of imaging in aqueous liquid at the physiologically relevant temperature and pH, and opportunity of repeated scanning of the same sample under distinct conditions. AFM imaging offers all of that. In short, the AFM probe, which consists of a small and very sharp tip mounted on a cantilever, scans the sample. The mechanical properties of the cantilever change depending on the topographical features of a scanned object. These changes: deflection of the cantilever or modifications of its oscillations, result in variations in the distance between the tip and the scanned object. The distance is constantly corrected thanks to the feedback loop between a sample/tip positioning system and a computer-controlled piezo element. The image with a three-dimensional topographical information is generated by plotting the z (vertical direction) correction signal from the feedback loop against the x and y plane [1]. Thus, the practical resolution of the method is limited only by properties of the probe, fidelity of the computer control over image creation (pixel size) and dynamic behavior of the imaged sample. All the factors combined translate into 1 nm or even less in an apparent lateral resolution and about 0.1 nm in apparent vertical resolution. The lateral dimensions in unprocessed images are enlarged by the “tip broadening effect”, originating in the geometry of the apical part of the AFM probe. The effect can be removed by correcting software or calculations, given that the tip radius have been estimated, for example by scanning an object of well-known dimensions [2].
The physical basics of AFM allow for exceptionally broad array of applications. The only required condition is that the object will be immobilized on a flat surface strongly enough to prevent its detachment by the scanning probe. Such surface, the AFM substrate, needs to be atomically flat in the range of micrometers. Since the scanning, especially in oscillation mode, involves only low force interactions, even the gentle electrostatic attachment will suffice. The muscovite mica is the most popular and convenient substrate and it allows for electrostatic attachment of bioparticles. The mineral is built from thin crystalline plates, which are easily peeled off to expose a clean and flat surface, which is negatively charged in aqueous liquids. The charge and the resulting electrostatic force is sufficient to keep most of protein complexes in place since in the pH close to neutral (physiological) most of proteins are positively charged. So far, no significant changes in biological activity or structure of protein assemblies were noted after such relatively gentle immobilization. If a particle happens to be of negative charge, the mica still could be used, but should be pretreated with positive ions, for example Ni2+ [3]. Glass coverslips, plain or silanized, graphite (HOPG) or gold are also used with AFM [4–6λ]. If necessary, the substrate may be derivatized to attach the particles by chemical cross-linking, affinity binding or embedding in a prepared lipid bilayer.
Both the object and the probe may be immersed in a liquid of choice under a physiologically relevant and controllable temperature. The liquid can be exchanged between scans, and ligands can be added to the scanned sample. This “wet mode” option is one of the major advantages of AFM over other structural methods, however “dry mode” is used as well, especially for more sturdy particles. In the dry mode, the opportunity of direct observation of biologically active molecules is lost. On the other hand, the AFM image of a dried mixture of, for example, DNA and DNA-binding protein shows a snapshot of the specific binding reaction and allows for fast and easy assessment of the yield and structural details of the binding [7]. Alternatively, the particles can be fixed with glutaraldehyde and imaged in liquid [8]. Both glutaraldehyde fixing and dry mode imaging may allow for better quality of images by elimination of internal dynamic of living particles. Cryo-AFM offers similar advantage by drastic lowering of the temperature of scanning. In cryo-AFM the molecules can show traces of biological activity, similarly to cryo-EM. However, one of the big advantages of AFM method, its simplicity, obviously suffers with the need of super-low temperature and high-vacuum.
All three major modes of operation of AFM: contact, oscillating and force, are used with biological complexes. The contact mode, where the tip and the atoms of the sample are in direct contact leading to the cantilever deflection, works well with dried or fixed samples, or when dissection or displacement of a particle is desired [8]. In the oscillating mode the tip touches the sample only very briefly and with a low force, offering nearly noninvasive conditions [9]. The probe vibrates vertically while scanning, with vibrations activated acoustically (“tapping mode”) or magnetically. The proximity of the atoms of scanned sample causes changes in the amplitude of oscillations of the probe due to van der Waals and electrostatic forces. Changes in the amplitude are translated into a topography image offering a three-dimensional map of outer surface of the object. Instead of amplitude, changes in the phase of the vibrating cantilever may be recorded, usually using faster rates and lower force than with amplitude change detection. Such “phase imaging” is useful for monitoring elasticity, viscosity or charge on the surface of samples [2]. Although the phase images are devoid of height information, they do have a 3D-like appearance with a high contrast between the background surface and the attached particles. The newest addition to the set of modes is contact oscillation mode, where the tip oscillates while maintaining constant contact with the sample [10λ λ].
The third AFM mode of operation, the force mode, also called force spectroscopy, does not provide Cartesian coordinates of the sample surface. Instead, the strength of the interaction between the tip and the sample is measured in the order of picoNewtons [11]. For biological applications, the tip is usually modified with a receptor - specific ligand. Not always the force data has to be devoid of the topography information, however. The force spectroscopy and AFM imaging can be combined as a single-molecule recognition imaging microscopy, useful for studying complex mixtures of biomolecules [12λλ]. In this technique the tip is modified with an antibody and scans a heterogenous sample. Specific interactions of the antibody with the antigens manifest in cantilever deflection change, as in contact mode, and yield an AFM image where non-antigenic particles remain in the background, while the topography of antigens is well resolved [12,13]. Another variation on the AFM technique, the cross between force spectroscopy and tapping mode imaging called jumping mode or pulsed force microscopy provides both the topographic and adhesion force images, useful for probing mechanical properties of macromolecules [14].
3. Basic characteristics of protein complexes: morphology, morphometry and assembly processes
Over the years, AFM proved to be a very useful tool for assessment of the outer topography and for studying the assembly processes of biological complexes. The changes of shape and size of objects during assembly are usually straightforward to detect in AFM images and the technique allows testing multiple conditions or stages of assembly with an ease. Imaging with ligands or labels helps with specific binding domains mapping. On many occasions, adsorption of particles to a variety of surfaces is assessed, a prelude to nanotechnological applications. Very often the structure of particles is already known from EM or even X-ray crystallography studies before the AFM attempts. It does not undermine the importance of AFM imaging, just to opposite, helps to extract maximum structural, functional and dynamic information from all the methods. For some well-known biological complexes, relatively limited AFM studies have been conducted, and ribosomes are a good example. Prokaryotic and eukaryotic ribosomal subunits and whole ribosomes were imaged by AFM in air and under liquid, with the shape-size data results generally consistent with EM and crystal structure models [15–20]. AFM images allowed to distinguish between ribosomes from different species [17], and provided useful information about binding sites of elongation factors to the 40S subunit from rat [21]. Imaging of native polysomes from yeast under native conditions showed both the complete ribosomes and a smaller component, likely the small ribosomal subunit, lined on the RNA strand [22]. Below we focus of several assemblies for which the most extensive and enlightening AFM studies were conducted.
3.1 Building up and tearing down the proteasomes
“Proteasome” is a common name for giant proteolytic assemblies sharing a catalytic core. The EM images of proteasomes from many organisms have been obtained and the crystal structures of core proteasome from archaebacteria, baker’s yeast and bovine were solved thus setting a stage for subsequent AFM studies [23–25]. The enzyme is a modular, multisubunit nanomachine essential for intracellular controlled protein degradation in all Eukaryotic cells. All assemblies named the “proteasome” contain the 20S, 700 kDa core particle (CP). The CP is a barrel-shaped structure, about 11 nm in diameter and 15 nm in length. CP is built from 28 subunits arranged into four heptameric rings, organized in an α–β–β–α manner. The catalytic chamber is formed inside β rings, and the channel to the chamber leads from the gate in the middle of the external α ring. The outer surface of α rings, the α face, provides an area for attachment of additional regulatory modules. The most physiologically important module is the 19S “cap” (regulatory particle; RP), resembling a dragon’s head attached to the core “trunk”.
Several groups imaged the stable core particles. In the most noninvasive imaging attempt, we used the tapping mode AFM in liquid with proteasomes electrostatically attached to mica. By adjusting protein concentration, generally in the nanomolar range, it was possible to obtain a layer of densely packed particles or a sparsely populated field [2]. Both top-view (“standing”) and side-view (“lying”) cylinders were imaged, easily distinguishable by their shape (Fig. 1, 2). The particles withstood more than an hour-long repeated scanning without detachment, proving the strength of electrostatic binding [26λ]. Other AFM in liquid studies explored the 20S complexes immobilized on lipid supports. For example, His-tagged archaebacterial proteasomes were specifically attached to a mica-supported chelator lipid membrane loaded with nickel ions. Position of His-tag forced the side-view orientation of particles [27]. Interactions of the core particles with different lipid bilayers were explored for bovine proteasome [28]. The CP specifically bound in a top-view position to lipid membranes containing phosphatidylinositol, but not to those with phosphatidylcholine, phosphatidic acid or dioleotrimethylammonium propane. It remains to be established if the phosphatidylinositol – specific interactions are physiologically relevant, for example during CP binding to endoplasmic reticulum membranes. A fluid lipid film was used as a support for two-dimensional crystals of “standing” His-tagged archaebacterial proteasomes [29]. The 2D crystals, which are easier to form and more stable than 3D crystals, are often used for electron microscopy or AFM of membrane proteins [30]. Analysis of the images revealed that the proteasomes optimized their packing in a crystal by interlocking both laterally and vertically, ring-to-waist. Contact mode AFM was used to successfully image the shear – resistant 2D crystals [29].
Fig. 1. “Building up” the proteasome assembly.

The particles electrostatically adsorbed to mica were imaged in tapping mode in liquid (Nanoscope IIIa; Veeco). (A) Representative top-view images of 20S core particle (CP) and core particle extended with a “stent” created from Rpn2 or Rpn1-Rpn2 dimer, presented below. The images were zoomed-in from 600 nm × 600 nm fields, flattened and plain-fitted. Color bars represent the height scale, different for CP and free stent components. (B) Height distribution of 20S CP in mixture with Rpn1 and Rpn2 is shown as relative number of molecules, which fall within a height range of 0.25 nm wide bins. Heights were analyzed with grain analysis function (SPIP v. 4.3.2.0, Image Metrology, Denmark). To fit the experimental frequency data, normal distribution of particle height was assumed for each type of particles, with the exception of the broad peak (presumably mostly CP-Rpn2), for which the height distribution curve was fit using an asymmetric double sigmoidal function within the OriginPro Peak Fitting module (modified from Rosenzweig, Osmulski, Gaczynska, Glickman, submitted).
Fig. 2. Two conformations of the 20S core proteasome.

The fission yeast proteasome was electrostatically adsorbed to mica and imaged by tapping mode AFM in liquid [2]. Particles in two stable conformational states were detected: with a cavity in a ring (“open”) or with smooth a face (“closed”) and with a barrel-like or a more elongated (“cylinder”) shape. The representative images were zoomed-in from 1 μm × 1 μm fields and are presented as side-plots (tilted images).
With regard to the higher-order proteasomal assemblies, we imaged the human core with 19S RP by tapping mode in liquid (Fig. 3). The “dragon’s head” shape of 19S cap was strongly resembling familiar EM images [31]. The power of AFM imaging, however, proved invaluable for dissecting the components of the regulatory particle and the topography of their attachment to the core. The 19S cap is composed of two multisubunit parts: a base (lower “jaw” of the dragon) interacting directly with the α face, and the lid. The base is composed of six regulatory particle ATP-ases (Rpt1–6), presumed to form a ring. Co-purifying with the ATPases are two large proteins, Rpn1 and Rpn2 (regulatory particle non-ATPases), and the hinge subunit Rpn10.
Fig. 3. The imaged native 26S proteasome assembly binds a substrate and assumes distinct conformations.
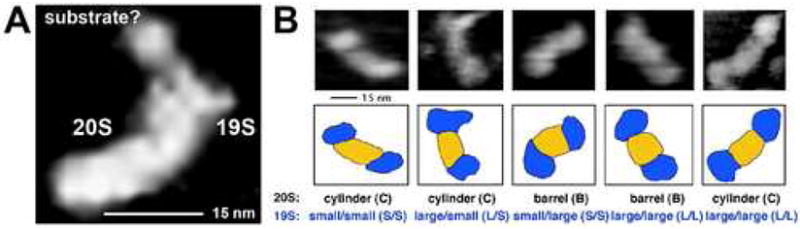
(A) The human assembly with a single 19S cap (“half-26S”) was imaged with a particle bound to the lid of the cap, presumably a substrate (polyubiquitinated lysozyme), which was added to the proteasome sample. (B) Images of bakers yeast 26S assemblies with diverse conformations of the core particle (see Fig. 2) and the 19S caps. The presumed dynamical behavior of the 26S particle is now under investigation. Tapping mode in liquid and electrostatic adsorption to mica were used. The images were zoomed-in from 1 μm × 1 μm fields (Osmulski and Gaczynska, unpublished observations).
The position of Rpn1 and Rpn2 was unknown until the newest AFM studies with yeast (S. cerevisiae) proteasomes (Rosenzweig, Osmulski, Gaczynska, Glickman, submitted). AFM imaging (Fig. 1A) confirmed the earlier theoretical predictions that the proteins may form toroids of about 5 nm in diameter and 3 nm in height [32]. When these apparently monomeric Rpn1 and Rpn2 were mixed in an equimolar ratio, a population of twice-the-height rings was found, strongly suggesting a formation of stacked heterodimers. When in turn the heterodimers were mixed with the core proteasomes, a new class of particles: cylinders extended by a stent or chimney protruding from the middle of the α ring, was detectable (Fig. 1A,B). Analysis of images of mixtures of either Rpn1 or Rpn2 with the 20S core allowed establishing that only the Rpn2 toroid interacts directly with the α face. The “building-up” of proteasome assemblies was continued with addition of Rad23, an adaptor protein imaged as a “bead” decorating the stent. The complementing biochemical and imaging data confirmed the model of proteasome core particle with a central channel extended by a molecular stent, capable to bind adaptor proteins (Rosenzweig, Osmulski, Gaczynska, Glickman, submitted).
We hypothesized that the Rpt ring surrounds the Rpn1/2 stent, which is a novel feature in the proteasomal structure. To confirm such predicted arrangement, 26S assemblies purified from yeast cells were gradually dissected by biochemical methods. Again, the AFM and biochemical data complemented each other in these “tearing-down” experiments. Images of samples where 20S subunits, plus Rpn1 and Rpn2 were detectable biochemically indeed showed core particles with the stent, undistinguishable from reconstructed species. Importantly, images of the fraction containing the base subunits: Rpt1–6 and Rpn1–2, showed the predicted new architecture: the stent surrounded by a large ring (Rosenzweig, Osmulski, Gaczynska, Glickman, submitted). The next call for interplay of biochemical and AFM studies would involve dissection of a role of the stent and surrounding subunits in the uptake and processing of substrates.
3.2 Dissecting the GroEL assembly
The architecture of core proteasome is in many aspects similar to bacterial chaperonin GroEL. The cylindrical GroEL, 14.5 nm high, with a 13.5 nm diameter, is built from two stacked rings, each with seven identical subunits, as determined with crystallography, EM and NMR studies [33]. There are spacious chambers inside each ring. A smaller, homoheptameric ring GroES sits atop GroEL, adding about 4 nm to its height. The cycles of energy-driven binding and releasing GroES underlie the function, which is assisting in polypeptide folding. The GroEL and GroES are one of most popular protein complexes studied by AFM, even if they are rather damage-prone. They were imaged by AFM with high resolution in liquid in contact mode [8,34] or oscillation mode [35,36], cross-linked with glutaraldehyde [8,34,37λ] or without fixation [35,36]. It was reported that, unless fixed with glutaraldehyde after the adsorption to mica, the GroEL/GroES complex was destroyed and GroEL was dissected into single rings by the contact mode probe [8,34]. Even in the gentle oscillation mode, it took optimization of binding and scanning conditions to obtain a stable layer of GroEL electrostatically adsorbed to mica [35,36]. Still, the perfectly symmetrical GroEL may find its use as a potential “living” calibration sample, especially when adsorbed in a dense, organized monolayer of two-dimensional crystals [36]. With such nanoscale metrology attempts, it was noted that the height measurements of loosely dispersed single particles of GroEL were suppressed by a constant value, in contrast to perfectly correct measurements for densely packed particles [38]. Such unexplained phenomenon was also observed under some conditions for the 20S proteasomes (Gaczynska and Osmulski, unpublished observations).
3.3 Molecular cages and nets of superb mechanical strength
Closed protein cages protecting organic or inorganic cargo or branched nets capable to catch bioparticles belong to this group of complexes. The AFM technique has been used not only to characterize their topography with nanometer-scale resolution, but most of all to follow their assembly and test their mechanical and physicochemical properties.
3.3.1 Lacy spheres with diverse filling
The cages used for intracellular protein trafficking are elaborate assemblies of multiple protein units. One type from this group, the clathrin, was studied by AFM. The unit of clathrin, a three-legged “triskelion”, is a trimer built from heavy chains reinforced by light chains [39]. A minimum of 36 triskelions can fit together to form a perfect lattice sphere, with a diameter in the range of 100 nm, coating a lipid vesicle with a protein to transport, as known from extensive structural studies [39]. In the earliest study by AFM, the clathrin-coated vesicles were immobilized by binding amino groups of the protein cage to a molecular “carpet” formed by dithiobis (succinimidylundecanoate) monolayer covalently chemisorbed to gold surface by thiolate bonds [40]. Such clathrin spheres withstood long, repeated scanning in contact mode in liquid. After two hours of incubation of the complexes with disassembly-promoting buffer, only single triskelia bound to the underlying monolayer were detectable [40]. In another study, the elasticity of clathrin spheres was measured by AFM [41]. The clathrin-coated vesicles were adsorbed to a mica surface pretreated with Ca2+ and imaged in liquid. Apparently, the pretreatment prevented collapse of adsorbed spheres imaged by low-force tapping mode in liquid. The contact mode, in turn, was used to apply compression force to the cages, in the range of 40 – 300 pN. The dimensions of compressed vesicles were computed and the model of elastic bending of the particles was developed. Apparently, the combination of outside protein shell with inside lining of lipid vesicle increases bending rigidity of the structure by 20-fold, comparing with a protein lattice or a vesicle alone, a feature critical for the cage intended to protect its cargo [41].
Another molecular cage, ferritin, does not deal with a protein cargo. The spherical apoferritin is formed by 24 subunits of light chain and/or heavy chain type, depending on cell type and species. The thick shell of an external diameter of about 13 nm is used to store iron in the form of ferric hydroxide-phosphate in its internal cavity of about 8 nm in diameter. The crystal structures of several types of ferritins have already been known when the assemblies, both with and without the iron load, were imaged by AFM in water or in air, in the form of single particles, monolayers, two-dimensional or three-dimensional crystals, adsorbed on mica, gold, HOPG, polypeptide-functionalized electrode surface, antibody-functionalized silicon surface or multicomponent lipid monolayers [6,42–47]. The specialized nature of the cages’ cargo, mandated the use of conductive probe atomic force microscopy [4,6]. Ferritin shells, both empty (apoferritin) and filled with iron deposits (holoferritin) are conductive. When the particles were adsorbed to conductive surface like gold [6] or HOPG [4], a current-voltage measurements on the single dried molecules could be executed with a metal-coated oscillation mode tips performing voltage scanning. Expectable, the conductance of apoferritin was found to be at least an order of magnitude lower than conductance of holoferritin [4,6λ]. Like the clathrin cages, the ferritin cages were subjected to mechanical studies. An empty shell of apoferritin was found to be prone to collapse under the AFM tip, in contrast to the much more rigid holoferritin [4].
3.3.2 Viral capsids: the crates for arduous journeys
There is one more class to de facto molecular cages, which are obvious subjects for AFM studies: viral capsids. They protect the load of a nucleic acid core, which together with the protein capsid forms the complete viral particle (virion). As molecular containers, viral capsids are more robust and versatile than clathrin or ferritin. They often have to withstand hostile environmental conditions; the packing of cargo has to be economical, and they have to assist in complicated entry-egress processes where precision of interactions with host cell membrane proteins is critical. Viral capsids, with diameters ranging from 10 to more than 100 nm, assemble into a single-layer cage from units - capsomeres. The capsomeres are built from just one or very few kinds of proteins. The shape of capsids can be spherical, spherocylindrical, rod-like or conical. There are more than seventy distinct capsid structures belonging to more than 20 families. Some viral families, for example herpesviruses or retroviruses, have their core and capsid additionally protected by a lipoprotein envelope, with lipid bilayer “borrowed” from the host’s cell membrane and envelope proteins responsible for the entry into a host cell. Numerous viruses from all major groups were imaged by AFM, mostly with recording their shape, size and special features. Even simple morphometric cataloguing makes perfect sense when considering AFM as a diagnostic technique. Selected studies from the vast body of literature are cited or briefly presented below as examples. For instance, details of arrangement of protomers in capsids were elucidated with purified viruses [48–50] or in vitro reassembled and glutaraldehyde-fixed shells [51]. AFM helped to reveal the unusually complex architecture of plant closteroviruses [52] and to find a new structural features in the coat of potyviruses [53]. Often, the viruses were imaged directly on the surface of glutaraldehyde-fixed host cells [54–57λ]. Labeling with specific antibodies decorated with gold nanoparticles helped to map the arrangement of entry proteins on envelopes of retroviruses HIV and MuLV (murine leukemia virus) [58]. The best resolution of details of capsid structure was achieved with 2D or 3D crystals, and many studies followed the growth of viral crystals or details of capsid symmetry [59–62].
A group of studies attempted to elucidate the mechanism of capsid formation. For example, tapping mode AFM in air and light scattering techniques were used to assess efficiency of immature coat formation by HIV-1. Apparently, pH higher than 6 and a high ionic strength promoted self-assembly, whereas the presence of cyclophilin A did not affect the process. Therefore, the putative role for the cyclophilin A is placed into the mature (spherical) capsid disassembly rather than the assembly of immature (conical) coat [63]. In another study, the function of murine leukemia virus protein required for capsid morphogenesis was tested. When the protein was mutated to resist glycosylation, aberrantly shaped capsids were detected on the surface of fixed host cells with a tapping mode in air [54]. To promote in vitro coat protein aggregation of the Moloney murine leukemia virus, the protein was His-tagged and deposited on a synthetic bilayer containing nickel-chelating lipid [64]. The resulting protein array was subjected to high-resolution tapping mode scans in liquid leading to creation of a two-dimensional model of the capsid architecture [64]. The processes reverse to self-assembly: a gradual degradation of virions or capsids was monitored in crude preparations of herpesviruses [65], in detergent – treated samples of HIV [56] and in preparations of Turnip Yellow Mosaic Virus [66].
The viral coats not only protect their cargo in transit, but also assist in injecting the genetic material into and releasing the new virions from the host cell in the entry and egress processes, respectively. In the case of enveloped viruses, the entry depends on fusion-promoting glycoproteins of the envelope. Trimeric hemagglutinin (HA) is one of such proteins. Tapping mode in liquid was used for in situ imaging of self-assembly of two distinct domains of influenza HA under variable pH conditions on mica-adsorbed synthetic lipid vesicles [67]. The results prompted speculations about specialized roles of distinct HA domains and their spatial organization in viral entry [67]. In a more recent study interactions of N-terminal fragments of gp41 with lipid bilayers were studied by real-time AFM in liquid [68]. Gp41 is a fusion protein derived from HIV-1. Two kinds of synthetic membranes were tested in order to model inner and outer surfaces of the plasma membrane. AFM images revealed membrane pore-forming activity of selected gp41-derived peptides and allowed for precise mapping of the fusion domain of gp41. The electronegative phospholipids of the inner membrane were proposed to control the fusion-promoting conformational change of the gp41 polypeptides [68].
Revealing the origins of very good mechanical resistance of viral capsids inspired numerous AFM studies. One rather obvious result was that virions are stiffer and less prone to collapse than capsids, similarly to empty and filled ferritin [66,69–72]. Indentation studies with AFM tip showed that the mature shell of murine leukemia virus (MLV) was more brittle than the immature shell, a feature possibly related to the infectivity [73]. Recent studies using jumping mode (pulsed force) AFM in liquid showed that the core and the capsid closely collaborate in making the virion a masterpiece of nanoengineering. For example, the parvovirus minute viruses of mice (MVM) were adsorbed to silanized glass and the indentation study was carried out to compare stiffness of the relatively simple icosahedral shell of MVM [69λ]. The reinforcement of the capsid by the DNA cargo was found to be highly anisotropic [69λ]. An anisotropic elastic response was also documented for the empty shells of bacteriophage φ29 [74]. In a more recent study by the same group, the deformation of the WT λ phage capsid packed with DNA of various lengths and adsorbed on hydrophobic glass was monitored [70λ]. The results of indentation experiments showed that internal osmotic pressure of the wild type DNA with hydrating water molecules is in perfect balance with mechanical properties of the capsid. A higher pressure would break the capsid whereas lower pressure leaves the shells less mechanically resistant and incapable to effectively inject DNA to the host bacteria [70]. In yet another study the Young modulus was calculated for tobacco mosaic viruses (TMV) adsorbed to a mica or polycarbonate ion-etched membranes in ethanol [75]. Not surprisingly, the wealth of AFM studies contributed significantly to attempts of mechanical modeling of viral capsids [76].
3.3.3 Protein nets from the blood clotting cascade
Specialized proteins may assemble not only into cages, but also into nets. In the process of hemostasis and thrombosis, a cascade of proteins takes part in formation of thrombus. Self-assembly of fibrinogen into a fibrin clot and von Willebrand factor (VWF) into a platelet-catching mesh are examples of formation of biological nets. Proteins participating in the blood clotting cascade were extensively explored by AFM, first by contact mode in liquid or air then by oscillation mode [77,78]. Many studies were limited to visualizing the components of the system [78,79]. The most interesting, however, are experiments exploring self-assembly and conformational changes of net-forming complexes in a dynamic manner. Fibrinogen (factor I) is the most extensively studied protein from the hemostatic cascade, by AFM and other methods. This large (340 kDa) glycoprotein circulating in blood is a hexamer built from a pair of three polypeptide chains, twisted and disulfide-bound together to form a rod-like, trinodular, octaglobular structure of about 46 nm in length and 9 nm in diameter [80]. X-ray crystallography and NMR studies explored the fibrinogen architecture in great detail, and high-resolution AFM imaging added to the pool of knowledge [78,81–84]. Proteolytic processing of fibrinogen by thrombin exposes polymerization sites and promotes a formation of the insoluble fibrin net [80]. The fibrin mesh and aggregated platelets are the main constituents of a blood clot, however fibrin provides also a cellular scaffold during wound healing and angiogenesis. To directly visualize fibrin assembly by tapping mode AFM in liquid, the fibrinogen was adsorbed on the substrate, followed by sequential addition of thrombin and protofibrils [85]. Hydrophobic HOPG and hydrophilic mica substrates were used. The process of formation and propagation of fibrin strands in the course of several rounds of addition of fibrin monomers was monitored in a series of time-lapse images spanning several hours. The fibrin clot on HOPG propagated from a few short, linear strands to a dense branched net covering a vast area [85λ]. To the contrary, mica did not provide a good thrombogenic surface. Conformational changes in an adsorbed protein monolayer during thrombin-catalyzed conversion of fibrinogen to fibrin were monitored by AFM in another study. The gold-coated glass was used as a substrate and micropatterned with fibrinogen by stamping. Upon conversion to fibrin, the protein film contracted and adhered stronger to the hydrophobic surface, a phenomenon observed with other methods as well [86]. Numerous other AFM studies tested a broad range of various conditions like a type of surface, pH, charge, ionic strength on the conformation and adsorption properties of fibrinogen [87–92λ]. Generally, hydrophobic surfaces promoted spreading of the fibrinogen molecules [87,91]. Phenomenon reverse to fibrin polymerization, a fibrinolysis of the clot by plasmin, was monitored by AFM as well, and images showed thinning and cutting the fibrin strands [79,93,94]. Interestingly, the fibrinolysis processes more efficiently on thick strands than on thin ones [79,94].
AFM imaging of a giant glycoprotein complex, von Willebrand factor, helped to understand the mechanism of its assembly. Large, multidomain protomers (309 kDa) of VWF are 120 nm in length. More than 40 protomers can polymerize into the largest known soluble globular protein, of molecular weight up to 20 × 106, although smaller molecules are also found. VWF multimers bind to collagen and possibly other components at the vessel wall at injury site and promote platelet adhesion and then clot formation. The unique feature of VWF is its functional dependence on the shear stress. The VWF binding and subsequent platelet tethering is facilitated by high shear force, which occurs at the wound site. It has been proposed that globular VWF multimers circulating in plasma may change the conformation to fibrous upon shear stress, assisting this way the self-assembly [95]. This hypothesis was tested with AFM. In the most recent study, a solution of VWF multimers was applied to a silicanized mica surface under controlled, physiologically relevant shear stress conditions [93]. The adsorbed particles were then dried and subjected to contact mode AFM imaging in air. Apparently, the VWF multimers assembled into long fibers (up to 300 μm), oriented in direction of shear flow. Numerous filamentous multimers formed the massive fibers, with occasional globular multimers attached to the large strings. The molecular net was fully functional and able to bind platelets, as was visualized by AFM. The AFM and immunofluorescence microscopy imaging helped to establish that three factors are necessary for the fiber formation: available high concentration of soluble VWF, high shear stress (at least 21 dyn/cm2) and suitable binding surface of low hydrophobicity. Problems with protein concentration may have tampered some earlier attempts to characterize the putative shear stress [96]. In other AFM studies, the high shear force indeed was found to induce uncoiling of the globular VWF multimers with the impressive, up to five-fold, extension in length. The uncoiling was also observed after applying force to the globular particles adsorbed to mica and tackled by the contact mode probe [97], and after spin-stretching of denatured multimers [98]. Even if high shear stress is necessary for molecular net formation, adsorption of VWF multimers to a proper thrombogenic surface can be sufficient for inducing the platelet-binding capacity. Changes in VWF conformation were explored by nanogold immunolabeling of the glycoprotein Ib binding domain (GPIb), a region critical for platelet tethering to VWF multimers [99]. The exposure of GPIb was detected upon adhesion of VWF to both collagen-coated and synthetic hydrophobic surfaces [99]. In earlier studies, adsorption of VWF to hydrophilic mica and to hydrophobic octadecyltrichlorosilane - modified glass were compared, with a conclusion that a highly hydrophobic substrate did not support the extended conformation [100]. The result is consistent with the latest observation that a surface of low hydrophobicity was preferred over that of high hydrophobicity for the VWF net formation [93].
4. Aggregation: another face of assembling
Aggregation of proteins under proper conditions leads to formation of two distinct types of fibrils: elastomeric or amyloid. Elastin type aggregates form a basis of extensible tissues like skin, blood vessels and ligaments, whereas amyloids commonly are associated with tissue degenerative diseases, although functional amyloids have been also recently identified. Amyloids share an extended cross β sheet structure with a 4.7Å molecular spacing resulting from alterations within the protein native fold, rod like linear extracellular filaments, and propensity to bind specific hydrophobic dyes.
It is suggested that an outcome of the aggregation depends on a relative content of Pro and Gly residues in the protein sequence with their high content promoting elastomeric fibrils [101]. Predictably, the actual formation of fibrils is much more complicated and it is controlled by a complex interplay of physical and chemical factors. Despite enormous efforts, the mechanism of aggregation, the order of steps in fibrillation, changes in structure and properties of intermediates and products or even exact influence of particular residues in fibril formation are still poorly understood. The faster progress in this area is restricted by the unusual solid state-like properties of aggregates challenging to study with the relatively limited array of analytical tools. Attempts to subject the aggregates to rigorous analysis are plagued by their low solubility, tendency to form amorphous bodies, and instability. Additionally, poorly defined in vivo conditions supporting aggregation often are impossible to recreate in vitro. Although several techniques have been successfully applied to study selected aspects of the process, for example electron microscopy, NMR or X-ray diffraction, comprehensive characterization of morphology and dynamics of the fibrils is achieved only with AFM. The relative simplicity of the sample preparation and topography measurements makes the method a perfect choice, in particular to examine molecular consequences of the introduced mutations within fibril forming proteins, influence of co-solvents, ions, other proteins and participation of a supporting surface as a catalyst. So far, AFM technique has been used to study aggregates of numerous amyloidogenic proteins, like α-synuclein, Ab-peptides, amylin, IgG light chain, insulin, lysozyme, tauprotein, transthyretin and at least 40 more other proteins. About a half of them form amyloid type aggregates that are connected to distinct pathologies and these were subjected to numerous AFM studies. Many of these amyloidogeneses are broadly associated with neurodegenerative diseases like Alzheimer, Parkinson, and Huntington disease. However, aggregation of the most of nonpathogenic proteins still awaits closer AFM inspection, probably with an exception of intensely examined lysozyme. It seems that most of proteins that are capable to form the cross β structure and self assemble can produce amyloid fibrils, even as unlikely as lysozyme, lactalbumin or myoglobin. However, such fibrils are not always detectable under physiologically relevant conditions [102]. Even if these amyloidogenic proteins are not formed in vivo, they still provide plethora of information on the mechanism of fibril formation and their properties.
AFM has been applied to study three basic aspects of protein aggregation: morphology of fibrils, their growth/assembly and mechanism of action. The majority of earlier studies focused on description of fibrils topography. Such use of AFM imaging is fully justified since it delivers a rich set of morphometric data: length, diameter (height), characterization of branching, and fibril periodicity. Taking into account a high morphological and physical diversity of fibrils, it is surprising that the vast analytical potential of AFM to measure elasticity, viscosity or hydrophobicity practically has not been utilized in such studies. A tapping mode AFM with protein samples dried on mica is preferred since it “freezes” further changes in fibril morphology. This snapshot approach is useful as long as the aggregation is a relatively slow and a direct observation of the process is unnecessary. Here we will briefly discuss only few selected aggregating proteins to show how AFM technology was applied. Strikingly, most of the aggregates show high morphological similarity in organization of fibrils despite different primary sequence and completely distinct propensity to form βstructures, like SH3 domains and lysozyme [103]. Amylin is major component of pancreatic amyloid deposits in type II diabetes mellitus. It forms AFM detectable polymorphic structures that slowly converge to more uniform fibrils when they age [104]. b2 microglobulin (β2m) belongs to Ig superfamily and is a part of MHC class I. It is built from a β-sandwich stabilized by a single disulfide bond. β2m amyloids are often formed in patients receiving prolonged hemodialysis. Similarly to amylin, a high morphological diversity of β2m fibrils was detected. Their origin in part is traced to the existence of competing polymerization pathways starting either from nucleated or not nucleated material [105] and distinct templates [106]. Interestingly, collagen is suggested as associating with b2m fibrils and as a catalyst of their formation under physiological conditions. These ex vivo observations may explain an exclusive formation of b2m amyloids in skeletal tissues [107]. In general, the branching of fibrils is evident only at later stages of fibrils growth. However, Htt protein of Huntington’s disease forms extensively branched fibrils even during early stages of aggregation [108].
α-Synuclein (α-syn) fibrils are commonly associated with the Parkinson disease. In vitro grown fibrils of α-syn typically vary in length from about 500 nm to 3 mm when deposited on mica. Taking an advantage of AFM capability to measure a height of objects with unparalleled precision, it was possible to distinguish three different classes of typical fibrillar species: protofilaments, protofibrils, and fibrils [109]. Their overall dimensions are similar to those measured with EM. Here in comparison with the EM, the application of AFM is purely analytical but bringing an advantage of investigation of the morphological dynamics leading to the end products. Moreover, it is possible with AFM to distinguish fibrillation from formation of other types of aggregates, for example spherical assemblies in an A30P mutant of α-syn [110λλ]. Formation of intermediary oligomers shaped as spheres, chains and rings resembles aggregates detected in Alzheimer disease [110λλ].
The AFM technique is particularly well suited to follow the growth of polymers, including nucleation, elongation, branching, and lateral association of protofibrils. Visualization of fibril growth is most often followed with tapping AFM but in this case samples are kept in liquid rather than dry. For example, it was demonstrated that early stages of fibril formation are decisive for the further amylin aggregation showing two distinct phases: initial lateral growth followed by polymer elongation [111]. Interestingly, the same authors could detect with AFM the smallest oligomers consisting of only 16 molecules. Time lapse AFM was also successfully applied to compare the fibril formation of mutant amylins, where three Pro residues presumed to control aggregation were targeted [112]. Additionally, it was possible to determine that protofibrils elongate bidirectionally at a rate of 1.1nm/min [113].
On the basis of morphometric studies and time lapse AFM imaging of topographic changes in several protein aggregates, a hierarchical model was developed to describe amyloid formation [114,115λ]. The model called Hierarchical Assembly Model (HAM) proposes that partially folded precursors polymerize to form protofilaments that further grow by adding monomers at the ends. Intertwining of two or three protofilaments leads to formation of a protofibril. Interlace of a pair of protofibrils in turn creates a fibril. The validity of the model has been further confirmed with a non contact AFM using α-syn, insulin and B1 domain of G protein [109]. Unexpectedly, the assumptions set forward for the model still hold even for the aggregation process initiated under conditions not physiologically relevant, like low pH, high ionic strength or presence of hydrophobic co-solvents.
AFM technology was also instrumental in exploring the mechanisms of physiologically normal and toxic actions of aggregates. For example, it was found that amylin disrupts a lipid bilayer. Unexpectedly, no pores are formed but small membrane defects are propagated as detected with time lapse AFM [116]. It is suspected that such defects may constitute a basis for cytotoxicity of amylin. It is hypothesized that beyond the normal function as phospholipase D2 inhibitor, α-syn fibrils have neuroprotective functions since a prevention of the fibril formation, for example by a ring closing, is toxic or even pathogenic [110]. Indeed, it was found that α-syn interacts with acid phospholipids [117λ], disrupts POPC/POPS lipid bilayer by poking holes in it or by forming aggregates with lipids. An α-syn A53T mutant exhibits a diminished lipid binding as it forms a β-sheet as opposed to the helical wild type molecules.
Comparison of β amyloid fibril formation on mica where pseudomicellar aggregates are created and graphite where linear assemblies prevail suggests that Alzheimer aggregates form on the interface of aqueous and hydrophobic phases [118]. Furthermore, the AFM detected winding of protofilaments was instrumental in understanding the spatial organization of fibrils and possible mechanism of the transition from proto- to filaments [119]. These observations should aid the design of compounds preferable targeting the plaque formation at a presymptomatic stage of the disease. Such an approach is of particular importance in the light of an early observation that a nucleation step is slow followed by a fast elongation with rapid conversion of protofibrils to fibrils [120].
Insulin can assemble under proper conditions into either toxic fibrils or relatively benign amorphous aggregates. Understanding of the process is vital since the aggregates play a role in developing type II diabetes and they interfere with insulin production in pharmaceutical industry. Aggregation of insulin was followed with several techniques, notably also with AFM [121]. Generally, the process follows the heterogeneous coagulation mechanism leading to formation of fibril bundles [122], where the prefibrillar steps with initial globular oligomers [123] are controlled via the coagulation-evaporation mechanism [124]. The overall aggregation is controlled by solvation that perturbs not only monomers folding but the fibrils morphology as well [125]. Interestingly, insulin fibrils show a mechanical strength similar to spider silk likely controlled by a single domain [126]. The same authors also showed that the internal breakage of the polymers is an indispensable part of the normal aggregation. Unexpectedly, under a high hydrostatic pressure, the amyloidogenic pathway of insulin leads to formation of unusual circular amyloids [127λ]. This finding opened a door for in depth exploration of structural consequences of such perturbation with the possibility to influence the distribution of oligomers with specific drugs.
Testing forces responsible for filaments interaction in a fibril, their unwinding and related studies classified as single molecule AFM force measurements are rare. AFM force measurements of pulling apart interacting monomers indicate that the formation of protofilaments is a highly cooperative process but surprisingly, it is also reversible [128]. It seems that the cooperativity constitutes a universal mechanism of fibril assembly since it was detected in the course of aggregation of several structurally unrelated proteins. In most cases, the process of aggregation is initiated with a crucial remodeling of secondary structure with a notable exception of acylphosphatase. It is suggested that in this case the fold adjustment is executed later during the amyloid formation [129].
Finally, as a novel application, AFM was utilized to isolate single chain antibodies against α-syn using a phage display [130,131]. Here a mixture of antibodies and oligomeric α-syn was deposited on a mica surface and collected images allowed to select antibodies directed toward particular type of oligomer morphology.
5. Intrinsic dynamics of protein complexes
By virtue of thermodynamic laws, biological complexes are dynamic structures. The motions start with vibrations of bonded atoms in sub-angstrom scale, and extend to rotations of protein side chains and to hinge bending. The spatial extent and amplitude of the motions approach the nanometer range detectable by AFM. However, their pico-or nanosecond time-scale prevents direct AFM observation and causes the blurring of AFM images of living molecules. The local and global conformational fluctuations may manifest in conformational diversity defined as a ligand-independent existence of more than one conformation of a single protein [132]. The conformational diversity, in turn, is postulated to determine catalytic functions and allosteric behavior understood broadly as a coupling of conformational changes between two widely separated sites [133,134]. It takes allosteric transitions to reach the nanometer spatial range and millisecond -second temporal range suitable for AFM analysis. AFM is especially useful for studies of allostery of large heterooligomeric assemblies, when NMR and molecular modeling are confronted with overly complicated systems.
5.1 The dynamic gate to the proteasome
The catalytic core of the proteasome provides an example of the use of AFM in concert with other structural methods to detect and characterize molecular dynamics. The allosterically driven dynamics of a proteasome gate were detected and characterized by AFM [26,135λ]. The crystal structure of eukaryotic core particles provided a detailed picture of the architecture of the enzyme, with its tightly interwoven subunits and internal cavities [23,25]. The most puzzling feature was, however, the lack of suitable entrance for the polypeptide substrate and exit for peptide products. Such results were in stark contrast to the earlier-solved structure of the simpler, but homologous archaebacterial core particle, where α rings were open in the center, providing unobstructed access to the central channel and catalytic chamber [24].
To probe the gate area of native, unfixed core particles, we imaged the assemblies with the oscillation mode AFM in liquid [2]. Imaging of top-view proteasomes from T. acidophilus indeed showed a crater-like cavity in a center of the α face, consistent with the presence of a channel entrance (Gaczynska & Osmulski, unpublished observations; archaebacterial proteasomes a courtesy of A. L. Goldberg). The crater instead of deep hole was an expected result, since a standard AFM tip cannot penetrate deep channels in protein assemblies, imaging holes as craters. Imaging of eukaryotic core particles provided a distinct picture. The topography of the upper part (the α face) of most of the top-view particles presented a smooth surface, consistent with the crystal structure. A significant fraction of top-view particles, however, showed a well-resolved middle cavity. Intriguingly, when the same set of particles was scanned multiple times, each proteasome molecule was able to assume both the “closed” and “open” conformations [135λ]. Analysis of lateral dimensions of the side-view particles revealed that there are two populations as well, one resembling barrel (short and wide) and another more cylindrical (long and narrow). We hypothesized that a closed barrel and an open cylinder represent allosteric forms analogous to R (relaxed) and T (tense) forms in a classical two-state model of allosteric transitions [26λ]. We concluded that the Eukaryotic core proteasome displays a conformational diversity detectable by AFM, with X-ray crystallography being able to portray the more stable closed barrel structure. Meantime, both the X-ray crystallography and AFM showed that recombinant core particles missing a major part of the gate were permanently open [2,136]. Moreover, a recent NMR studies of the archaebacterial proteasome revealed high dynamics of residues in the central channel entrance area, in tune with the AFM supported hypothesis of a dynamic gate in Eukaryotic proteasome [137].
Interestingly, an addition of protein or peptide substrates to the enzymatically active particles already settled on the mica resulted in a dramatic increase in the occurrence of the open cylinder conformation [135λ]. The observation led to a hypothesis that latent core proteasome occasionally strikes the open-gate conformation, thus allowing a substrate molecule to enter. A productive interaction of a substrate with the enzyme leads to the allosteric gate opening allowing in turn more substrate molecules to enter the catalytic chamber and setting a positive-feedback loop activating the catalytic core. Subsequent studies with competitive inhibitors engaging different parts of the active centers and with recombinant proteasomes with active centers modified by mutations allowed to pinpoint the most likely source of allosteric signal as tetrahedral intermediate stage of the catalytic act (Osmulski et al., submitted). The hypothesis proposing a direct coupling of the catalytic act with a partition of the conformers was additionally confirmed by a comparison of the catalytic parameters for one of the active centers calculated using data gathered with the conventional biochemical assay and with a single-molecule AFM-based test. Apparently, the abundance (%) of open form was an accurate measure of a catalytic turnover at the involved active center. Since the core particle is built from two halves, an additional conclusion from the experiment was that blocking one entrance to the 20S cylinder by direct interactions with the mica did not affect catalytic performance of the active centers. A similar conclusion that a single functional orifice is sufficient for both substrate entry and product exit, was reached by a study with archaebacterial proteasomes [29].
Influence of ligands other than substrates were also tested with the proteasomes. We monitored with AFM a conformation of top-view human and yeast core proteasomes treated with proline and arginine - rich peptides, a group of anti-inflammatory agents derived from a natural product [140,141]. Strikingly, AFM images of the top-view 20S proteasomes treated with PR peptides revealed locking of the α ring conformation in an unusual “shaky-open” position. The 26S assembly treated with PR peptides change the outer shape as well, in a manner that is as dramatic, as it is unexplained [141]. High-speed, time-resolved AFM analysis of the peptide-treated assemblies should shed light on the nature of induced dynamic structural changes. Noncompetitive inhibition is the general mechanism of action of a natural protein inhibitor of the proteasome, PI31. The protein was postulated to bind in the gate area on the α face and to interfere with the entry of substrates to the central channel [142]. In the AFM images of yeast 20S complex mixed with human PI31, the gate is apparently in open position, suggesting either a “foot in the door” mechanism or allosteric – type opening “from the distance” (Gaczynska, Osmulski, DeMartino, unpublished observations). The ligand protein was not well resolved, however the proteasomal cylinder was extended by about 2 nm upon addition of PI31, suggesting the expected ligand binding on the α face. Another uncharted area is dynamics of the 26S assembly. We noticed that the core particle still could bear the barrel or cylinder shape, and that the caps could be of distinct dimensions (Fig. 2, 3). The classification of the conformants is currently in progress.
5.2 GroEL: hats on and off
Studies on conformational diversity and ligand-induced dynamics of the proteasome emerged only in recent years. There are many more assemblies, though, for which the dynamic motions are already well established. Bacterial chaperonin GroEL/GroES is a perfect example of a protein complex with the acknowledged large-scale dynamic changes of its structure. The robust movements underlie the function of the assembly: assisting in protein folding by binding the substrate and then stretching and releasing it in order to induce desired structure folds. The ATP-driven catalytic cycle of the chaperonin is based on an asymmetric behavior of the two rings. The ring with ATP bound to seven positively cooperative sites is in a position to bind the polypeptide substrate, when the other ring, with ADP bound, is capped with GroES. An exchange of GroES between rings promotes a nanometer-scale elevation and twist of the ring with the substrate bound in its cavity. Subsequent hydrolysis of ATP is followed by the release of GroES and the folded polypeptide [33]. A GroEL ring cycles between GroES-capped R state and the free-ring T state. The 2 nm - upward movement of the GroEL ring in R state and the changes of the size of central opening detected by other techniques were confirmed with AFM of glutaraldehyde-fixed complexes [8]. The formation and dissociation of the GroEL-GroES was observed in real time with a small cantilever in an oscillation mode AFM in liquid [143]. The GroEL was electrostatically adsorbed to mica in an end-up position, and then GroES, ATP and Mg2+ were added to the sample. A specially prepared small cantilever assured a low force of interactions, not interfering with the GroES binding and releasing. An original one-dimensional scanning, with a slow scan axis disabled half way, enabled real time monitoring of the association status of several particles, which were repeatedly crossed by the imaging probe with high speed, faster than 20 μm/s. The temporal resolution of the method was 100 ms, whereas with the standard two-dimensional scanning the probe would return to the same molecule only within tens of seconds. On this basis the average lifetime of the GroEL-GroES complexes was estimated at 5 seconds [143λλ].
In another dynamics-oriented studies, the up-right position of glutaraldehyde-fixed GroEL on mica was confirmed with tapping mode imaging in liquid, followed by force spectroscopy measurements of interactions of substrate polypeptides with the chaperonin under different conditions [37λ]. Interestingly, the interaction forces were weaker in the presence of ATP than in its absence. Additionally, they were lower between a folded polypeptide and GroEL than between the fully denatured substrate and the enzyme [37λ].
6. AFM of protein complexes in nanomedicine and nanobio technology
The use of AFM as a fast, simple and versatile structural method in nanomedicine and nanotechnology seems more than obvious. After all, the method can provide high-resolution data with very small amounts of material, in little time and with relatively low-cost equipment. From a simple testing of quality of biomaterials to screening for pathogens or building nanobiomachines, AFM of protein complexes is in a good position to be one of the most useful methods in nanomedicine and nanotechnology.
6.1 Nanomedical screening
The use of atomic force microscopy for screening is well established in electronic industry. When the technique matured and a high-throughput set-up became available, morphometric testing of data storage media or processor elements emerged as a fast and convenient method of quality control. In the context of protein complexes, nanomedical screening with AFM should easily find a use in diagnostics of pathological viruses, when a simple present/absent answer is required. The sturdy virions can be imaged dried or rapidly fixed in air, with minimal time and workload effort. There are two approaches to perform an efficient screening: to identify particles solely by their morphometric features or to capture them on a prepared surface with specific virus-binding molecules and then to visualize. Both options are seriously considered, however the former has the advantage of simplicity. Since viral capsids have well-defined shapes, it is relatively easy to detect and classify them by AFM in very small amounts of crude samples, including the surface of fixed cells [58,144,145]. The VIPER database of icosahedral capsids (http://viperdb.scripps.edu) provides the X-ray crystallography and cryoEM data on several hundreds of structures, which should be useful to guide interpretation of AFM data [146]. Moreover, high-quality AFM topography images of more than a dozen of pathological viruses were published to date [51,144,145,147]. Automated image analysis is very helpful in extracting the structural information, as was shown for rotaviruses [148]. The drawback of this approach may be the necessity of development of good surface adsorption methods working well for distinct pathogens [149]. Fortunately, the adsorption properties of many viruses on several surfaces were studied extensively [150–152]. The latter bypasses the adsorption peculiarities of viral species. It requires a chip with a low-roughness surface coated with virus-binding molecules: antibodies or cell surface receptors, ideally in the nanoarray format. One such device is already on the market under the name ViriChip™ [153,154]. This solid – phase affinity substrate is a silicon wafer functionalized with an ultramicroarray of antibodies with an ink-jet technology. It is capable to capture intact viruses from body fluids or environmental samples. AFM not only detects viruses bound to specific antibodies on the array but also provides information on the size and shape of the particles [155–157λ]. Since the viruses on the chip are intact, they can be used for subsequent PCR studies [158]. In other, not yet commercialized studies, chips with antibodies against feline calicivirus were developed for screening with both AFM and surface-enhanced Raman scattering (after coupling the captured viruses with extrinsic Raman label), and both methods gave satisfactory results with cell culture media [159]. In turn, an original method of dip-pen nanolithography on metal-ion template was used to create a functional antibody array used to capture the influenza virus [160]. In a non-antibody based affinity assay, a silicon wafer was functionalized with heparan sulfate, a cell surface polysaccharide known as a receptor for many infectious viruses. The chip was used to characterize binding properties of several serotypes of adeno-associated virus [161]. Microreaction wells were fabricated on the silicon chip by coating it with the polydimethylsiloxane (PDMS) and then boring holes with steel tubing to create PDMS -free wells. PDMS resist protein adsorption and can be peeled off the chip before AFM analysis [161]. An interesting variations of the specific-capture chip: a virus stamping and molecular imprinting, were described by [162λ]. First, tobacco mosaic viruses (TMV) adsorbed to glass were used as a stamp. Such stamp was pressed into prepolymerized surface of soft polyurethane. After polyurethane hardening and washing-out the template viruses, a chip with tailor-made pits and trenches for TMV was ready for treatment with crude plant sap samples. Alternatively, an acrylate polymer was set to self-organize around template TMV particles, again forming cavities precisely mimicking the shape of TMV. Viruses from the sap of infected plants filled the cavities in the chip, as detected by AFM in a contact or tapping mode. Such bioimprinted sensor did not require expensive anti-virus antibodies, was reusable, sturdy and worked well with crude plant extracts [162λ].
The potential for AFM - based medical screening is obviously not restricted to viruses. Advances in micropatterning and producing ultra-small arrays enable multiple high-throughput AFM applications. Every large enough particle of diagnostic relevance like protein markers of disease or amyloid aggregates is suitable for this type of tests. An extension of such analysis is a search for protein-protein interactions on a chip printed with a receptor or ligand. The protein-protein interactions lie in the heart of recognition imaging, where the presence of a macromolecule of interest can be assessed even in a heterogenous sample. Development of a true structural screen would immediately impact drug discovery technology, where a simple identification of conformational changes of protein complexes, for example proteasome, treated with noncompetitive regulators [142] is urgently needed.
6.2 Biocompatibility of materials
Artificial materials introduced into human body: implants, stents, catheters, sensors, have to be tissue compatible and hemocompatible but they must not induce inflammation or blood clotting. The ideal surface should be non-fouling that is resistant to nonspecific adhesion of proteins or microbes. Since forming of the fibrin net is prerequisite to thrombosis, adhesion of fibrinogen is an excellent subject for AFM studies of high nanomedical relevance. Synthetic polymers, stainless steel, titanium or tantalum, gold or silver with different surface preparation: polishing, etching, coating with synthetic polymers, were tested. The nonspecific binding of fibrinogen and often also other proteins or platelets was directly visualized by AFM [163–165λ]. Alternatively, strength of fibrinogen interaction with the surface was tested with force spectroscopy [166–169]. Another variation of a biocompatibility test was described by Siedlecki et al. [170λ]. Here, mica or PDMS substrates were stamped with a plasma protein of one kind (bovine serum albumin) and then incubated with another plasma protein (fibrinogen) to fill in the patterned areas. AFM-based detection of fibrinogen was carried out by monitoring adhesion forces or by visualizing gold-labeled anti-fibrinogen antibodies. The experiment demonstrated that surface adhesion of plasma proteins and inter-protein interactions could be monitored by AFM in multicomponent protein films created by rounds of stamping and filling [170λ]. It is worth to mention that, in addition to protein-adhesion studies, AFM was often used to characterize physicochemical properties of the medically relevant surfaces [163,171–175].
6.3 Micropatterning with protein complexes
AFM is a staple method in creating and quality testing of arrayed nanostructures. Protein complexes emerged as convenient tools and materials for micropatterning. Recently, proteasome was used as one of components in AFM-based native protein nanolithography [10λλ]. His-tagged archaebacterial 20S proteasome particles were first arranged into a uniformly oriented array on a chelator-coated gold glass coverslips. AFM tip working in a novel contact oscillation mode was used to “erase” parts of the array by displacing the proteasomes. Addition of fresh particles, either proteasomes or other His-tagged proteins, filled the free spaces in a “rewriting” process. If the replacing protein was not a proteasome, the “re-written” passages were “read” by the AFM tip by monitoring the differences in height within the array. Importantly, all the procedures of “writing, erasing, rewriting and reading” were carried out in liquid, maintaining the proteins in native condition, suitable for interaction screens of diagnostic value [10λλ]. Another kind of nanolithography involving the proteasome was carried out under native conditions as well. Here, a monolayer of small protein lysozyme was adsorbed on mica. The lysozyme was previously modified by polyubiquitination, which rendered the protein a suitable substrate for the human 26S proteasome. Tapping mode AFM in liquid was used to scan the uniform layer of polyubiquitinated lysozyme. When the purified 26S proteasomes were injected into the AFM chamber, after several minutes numerous randomly positioned proteasome particles, easily identified by their shape, settled on the layer of substrate covering most of its surface. During an hour of continuous scanning most of proteasomes gradually disappeared leaving large patches of bare mica. Subsequent addition of fresh proteasomes did not result in significant adsorption of the enzyme to the mica patches. We interpret these real time observations as indications of: (i) specific binding of 26S proteasome to a substrate protein adsorbed on mica, and (ii) nanolithography-type protein degradation process monitored by AFM scanning and imaging (Gaczynska and Osmulski, unpublished results).
A different type of bio-nanoarrays can be created with molecular cages used as containers. Ferritin is especially well suited for nanotechnology applications due to its ability to accommodate various inorganic cargo. For example, ferritin particles were self-assembled into a 2D array on air-water interface, transferred to silica surface and heat-treated to remove the protein shell, leaving iron oxide particles regularly patterned on the substrate, a prototype array for potential use in nanoelectronics [176λ]. In turn, ferritin cages reassembled with distinct cores and characterized by AFM were organized in multilayered arrays as bio-nanobattery [172]. Ferritin molecules reconstituted with iron, cobalt, manganese, platinum or nickel were used to create regularly patterned quantum dots suitable for direct manipulation with AFM tip [177]. The ferritin - templated quantum dots exceeded in uniformity those created with physicochemical methods [177]. Such quantum dots are required for construction of quantum logic gates, a fundamental building block for quantum computing. Other cage-like protein complexes and also assemblies with inner compartment of restricted - access type, like the proteasome, may find a way into nanotechnology as vehicles and containers [142]. Definitely, the future will bring many more exciting applications for protein assemblies and atomic force microscopy in nanomedicine and nanotechnology.
Abbreviations
- AFM
atomic force microscopy
- CD
circular dichroism
- EM
electron microscopy
- HOPG
highly oriented pyrolytic graphite
- NMR
nuclear magnetic resonance
- PI31
proteasome inhibitor of molecular weight 31,000 kDa
- Rpn
regulatory particle non-ATPase subunit
- Rpt
regulatory particle ATPase subunit
- STM
scanning tunneling microscopy
- vWF
von Willebrand factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Fotiadis D, Scheuring S, Mu?ller SA, Engel A, Muller DJ. Imaging and manipulation of biological structures with the AFM. Micron. 2002;33:385. doi: 10.1016/s0968-4328(01)00026-9. [DOI] [PubMed] [Google Scholar]
- 2.Osmulski PA, Gaczynska M. Atomic force microscopy of the proteasome. Methods in Enzymology. 2005;398:414. doi: 10.1016/S0076-6879(05)98034-8. [DOI] [PubMed] [Google Scholar]
- 3.Rivetti C, Guthold M, Bustamante C. Scanning force microscopy of DNA deposited onto mica: equilibration versus kinetic trapping studied by statistical polymer chain analysis. Journal of Molecular Biology. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 4.Axford DN, Davis JJ. Electron flux through apo-and holoferritin. Nanotechnology. 2007:18. [Google Scholar]
- 5.Caruso F, Furlong DN, Kingshott P. Characterization of ferritin adsorption onto gold. Journal of Colloid and Interface Science. 1997;186:129. doi: 10.1006/jcis.1996.4625. [DOI] [PubMed] [Google Scholar]
- 6λ.Xu D, Watt GD, Harb JN, Davis RC. Electrical conductivity of ferritin proteins by conductive AFM. Nano Letters. 2005;5:571. doi: 10.1021/nl048218x. A less-standard use of AFM not only for topography scanning but for conductance measurements. [DOI] [PubMed] [Google Scholar]
- 7.Gaczynska M, Osmulski PA, Jiang Y, Lee JK, Bermudez V, Hurwitz J. Atomic force microscopic analysis of the binding of the Schizosaccharomyces pombe origin recognition complex and the spOrc4 protein with origin DNA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17952. doi: 10.1073/pnas.0408369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mou J, Sheng S, Ho R, Shao Z. Chaperonins GroEL and GroES: views from atomic force microscopy. Biophysical Journal. 1996;71:2213–2221. doi: 10.1016/S0006-3495(96)79422-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansma PK, Cleveland JP, Radmacher M, Walters DA, Hillner PE, Bezanilla M, Fritz M, Vie D, Hansma HG, Prater CB, et al. Tapping Mode Atomic Force Microscopy In Liquids. Applied Physics Letters. 1994;64:1738–1740. [Google Scholar]
- 10λλ.Tinazli A, Piehler J, Beuttler M, Guckenberger R, Tampe R. Native protein nanolithography that can write, read and erase. Nature Nanotechnology. 2007;2:220. doi: 10.1038/nnano.2007.63. Another development in protein nanolitography methods, this time performed with immobilized proteasome arrays with the use of an original "contact oscillation mode" with diverse manipulation capabilities. [DOI] [PubMed] [Google Scholar]
- 11.Radmacher M, Cleveland JP, Fritz M, Hansma HG, Hansma PK. Mapping interaction forces with the atomic force microscope. Biophysical Journal. 1994;66:2159. doi: 10.1016/S0006-3495(94)81011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12λλ.Stroh C, Wang H, Bash R, Ashcroft B, Nelson J, Gruber H, Lohr D, Lindsay SM, Hinterdorfer P. Single-molecule recognition imaging microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12503. doi: 10.1073/pnas.0403538101. A method combining force spectroscopy with topography imaging can be used in biomedical nanoarrays for specific biomolecule recognition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Bash R, Lohr D. Two-component atomic force microscopy recognition imaging of complex samples. Analytical Biochemistry. 2007;361:273. doi: 10.1016/j.ab.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreno-Herrero F, Colchero J, Go?mez-Herrero J, Baro AM. Atomic force microscopy contact, tapping, and jumping modes for imaging biological samples in liquids. Physical Review E - Statistical, Nonlinear, and Soft Matter Physics. 2004:69. doi: 10.1103/PhysRevE.69.031915. [DOI] [PubMed] [Google Scholar]
- 15.Fritzsche W, Henderson E. Ribosome substructure investigated by scanning force microscopy and image processing. Journal of Microscopy. 1998;189:50. [Google Scholar]
- 16.Fritzsche W, Trepte HH, Jovin TM. Comparative visualization of giant polysomes from salivary gland cells of Chironomus pallidivittatus by scanning force microscopy and electron microscopy. Probe Microscopy. 1997;1:81. [Google Scholar]
- 17.Malyuchenko NV, Tonevitsky EA, Agapov II, Pevzner IB, Bykov VA, Kirpichnikov MP, Tonevitsky AG. A study of E. coli and T. maritima ribosomes by atomic force microscopy. Biophysics. 2006;51:385. [PubMed] [Google Scholar]
- 18.Matsuura T, Tanaka H, Matsumoto T, Kawai T. Atomic force microscopic observation of Escherichia coli ribosomes in solution. Bioscience, Biotechnology and Biochemistry. 2006;70:300. doi: 10.1271/bbb.70.300. [DOI] [PubMed] [Google Scholar]
- 19.Wu XH, Liu WY, Xu L, Li MQ. Topography of ribosomes and initiation complexes from rat liver as revealed by atomic force microscopy. Biological Chemistry. 1997;378:363. doi: 10.1515/bchm.1997.378.5.363. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida T, Wakiyama M, Yazaki K, Miura KI. Transmission electron and atomic force microscopic observation of polysomes on carbon-coated grids prepared by surface spreading. Journal of Electron Microscopy. 1997;46:503. doi: 10.1093/oxfordjournals.jmicro.a023550. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Liu W, Xu L, Li M. Secondary structure of rat ribosomal RNAs studied by atomic force microscope. Progress in Biochemistry and Biophysics. 1997;24:434. [Google Scholar]
- 22.Mikamo E, Tanaka C, Kanno T, Akiyama H, Jung G, Tanaka H, Kawai T. Native polysomes of Saccharomyces cerevisiae in liquid solution observed by atomic force microscopy. Journal of Structural Biology. 2005;151:106. doi: 10.1016/j.jsb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 24.Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A? resolution. Science. 1995;268:533. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 25.Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, Tsukihara T. The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure. 2002;10:609. doi: 10.1016/s0969-2126(02)00748-7. [DOI] [PubMed] [Google Scholar]
- 26λ.Osmulski PA, Gaczynska M. Nanoenzymology of the 20S proteasome: Proteasomal actions are controlled by the allosteric transition. Biochemistry. 2002;41:7047. doi: 10.1021/bi0159130. Tapping mode AFM in liquid was used to reveal dynamic motions of the 20S proteasome complex, not detectable with other available methods. [DOI] [PubMed] [Google Scholar]
- 27.Dorn IT, Eschrich R, Seemu?ller E, Guckenberger R, Tampe R. High-resolution AFM-imaging and mechanistic analysis of the 20 S proteasome. Journal of Molecular Biology. 1999;288:1027. doi: 10.1006/jmbi.1999.2714. [DOI] [PubMed] [Google Scholar]
- 28.Furuike S, Hirokawa J, Yamada S, Yamazaki M. Atomic force microscopy studies of interaction of the 20S proteasome with supported lipid bilayers. Biochimica et Biophysica Acta - Biomembranes. 2003;1615:1. doi: 10.1016/s0005-2736(03)00227-x. [DOI] [PubMed] [Google Scholar]
- 29.Thess A, Hutschenreiter S, Hofmann M, Tampe R, Baumeister W, Guckenberger R. Specific orientation and two-dimensional crystallization of the proteasome at metal-chelating lipid interfaces. Journal of Biological Chemistry. 2002;277:36321. doi: 10.1074/jbc.M202145200. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Wang H, Erie DA. Quantitative characterization of biomolecular assemblies and interactions using atomic force microscopy. Methods. 2003;29:175. doi: 10.1016/s1046-2023(02)00308-0. [DOI] [PubMed] [Google Scholar]
- 31.Voges D, Zwickl P, Baumeister W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annual Review of Biochemistry. 1999;68:1015. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 32.Kajava AV. What curves? -solenoids? Evidence for an α-helical toroid structure of Rpn1 and Rpn2 proteins of the 26 S proteasome. Journal of Biological Chemistry. 2002;277:49791. doi: 10.1074/jbc.M204982200. [DOI] [PubMed] [Google Scholar]
- 33.Horwich AL, Farr GW, Fenton WA. GroEL-GroES-mediated protein folding. Chemical Reviews. 2006;106:1917. doi: 10.1021/cr040435v. [DOI] [PubMed] [Google Scholar]
- 34.Mou J, Czajkowsky DM, Sheng S, Ho R, Shao Z. High resolution surface structure of E. coli GroES oligomer by atomic force microscopy. FEBS Letters. 1996;381:161. doi: 10.1016/0014-5793(96)00112-3. [DOI] [PubMed] [Google Scholar]
- 35.Valle F, DeRose JA, Dietler G, Kawe M, Pluckthun A, Semenza G. Imaging the native structure of the chaperone protein GroEL without fixation using atomic force microscopy. Journal of Microscopy. 2001;203:195. doi: 10.1046/j.1365-2818.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- 36.Valle F, DeRose JA, Dietler G, Kawe M, Pluckthun A, Semenza G. AFM structural study of the molecular chaperone GroEL and its two-dimensional crystals: An ideal "living" calibration sample. Ultramicroscopy. 2002;93:83. doi: 10.1016/s0304-3991(02)00149-3. [DOI] [PubMed] [Google Scholar]
- 37λ.Vinckier A, Gervasoni P, Zaugg F, Ziegler U, Lindner P, Groscurth P, Pluckthun A, Semenza G. Atomic force microscopy detects changes in the interaction forces between GroEL and substrate proteins. Biophysical Journal. 1998;74:3256. doi: 10.1016/S0006-3495(98)78032-4. A dynamic aspect of the chaperone at work revealed by AFM, perfectly complementing other structural methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelhady HG, Allen S, Ebbens SJ, Madden C, Patel N, Roberts CJ, Zhang J. Towards nanoscale metrology for biomolecular imaging by atomic force microscopy. Nanotechnology. 2005;16:966. [Google Scholar]
- 39.Young A. Structural insights into the clathrin coat. Seminars in Cell and Developmental Biology. 2007;18:448. doi: 10.1016/j.semcdb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Wagner P. Covalent anchoring of proteins onto gold-directed NHS-terminated self-assembled monolayers in aqueous buffers: SFM images of clathrin cages and triskelia. FEBS Letters. 1994;356:267. doi: 10.1016/0014-5793(94)01296-2. [DOI] [PubMed] [Google Scholar]
- 41.Jin AJ, Prasad K, Smith PD, Lafer EM, Nossal R. Measuring the elasticity of clathrin-coated vesicles via atomic force microscopy. Biophysical Journal. 2006;90:3333. doi: 10.1529/biophysj.105.068742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tominaga M, Soejima K, Matsumoto M, Taniguchi I. Electrostatic modification of ferritin onto polypeptide-functionalized indium oxide electrode surfaces: Electrochemical and AFM studies. Journal of Electroanalytical Chemistry. 2005;579:51. [Google Scholar]
- 43.Du X, Hlady V, Britt D. Langmuir monolayer approaches to protein recognition through molecular imprinting. Biosensors and Bioelectronics. 2005;20:2053. doi: 10.1016/j.bios.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 44.Mollica V, Borassi A, Relini A, Cavalleri O, Bolognesi M, Rolandi R, Gliozzi A. An atomic force microscopy investigation of protein crystal surface topography. European Biophysics Journal. 2001;30:313. doi: 10.1007/s002490100165. [DOI] [PubMed] [Google Scholar]
- 45.Ohnishi S, Hara MH, Furuno T, Okada T, Sasabe H. Direct visualization of polypeptide shell of ferritin molecule by atomic force microscopy. Biophysical Journal. 1993;65:573. doi: 10.1016/S0006-3495(93)81125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perrin A, Elai?ssari A, Theretz A, Chapot A. Atomic force microscopy as a quantitative technique: Correlation between network model approach and experimental study. Colloids and Surfaces B: Biointerfaces. 1998;11:103. [Google Scholar]
- 47.Ohnishi S, Hara M, Furuno T, Sasabe H. Imaging the ordered arrays of water-soluble protein ferritin with the atomic force microscope. Biophysical Journal. 1992;63:1425. doi: 10.1016/S0006-3495(92)81719-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuznetsov YG, Gurnon JR, Van Etten JL, McPherson A. Atomic force microscopy investigation of a chlorella virus, PBCV-1. Journal of Structural Biology. 2005;149:256. doi: 10.1016/j.jsb.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 49.Kuznetsov YG, Low A, Fan H, McPherson A. Atomic force microscopy investigation of isolated virions of murine leukemia virus. Journal of Virology. 2005;79:1970. doi: 10.1128/JVI.79.3.1970-1974.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda H. An atomic force microscopy study for the assembly structures of tobacco mosaic virus and their size evaluation. Langmuir. 1997;13:4150. [Google Scholar]
- 51.Kuznetsov YG, Ulbrich P, Haubova S, Ruml T, McPherson A. Atomic force microscopy investigation of Mason-Pfizer monkey virus and human immunodeficiency virus type 1 reassembled particles. Virology. 2007;360:434. doi: 10.1016/j.virol.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 52.Peremyslov VV, Andreev IA, Prokhnevsky AI, Duncan GH, Taliansky ME, Dolja VV. Complex molecular architecture of beet yellows virus particles. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5030. doi: 10.1073/pnas.0400303101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torrance L, Andreev IA, Gabrenaite-Verhovskaya R, Cowan G, Makinen K, Taliansky ME. An unusual structure at one end of potato potyvirus particles. Journal of Molecular Biology. 2006;357:1. doi: 10.1016/j.jmb.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 54.Kuznetsov YG, Datta S, Kothari NH, Greenwood A, Fan H, McPherson A. Atomic force microscopy investigation of fibroblasts infected with wild-type and mutant murine leukemia virus (MuLV) Biophysical Journal. 2002;83:3665. doi: 10.1016/S0006-3495(02)75366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuznetsov YG, Low A, Fan H, McPherson A. Atomic force microscopy investigation of wild-type Moloney murine leukemia virus particles and virus particles lacking the envelope protein. Virology. 2004;323:189. doi: 10.1016/j.virol.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 56.Kuznetsov YG, Victoria JG, Robinson WE, Jr, McPherson A. Atomic Force Microscopy Investigation of Human Immunodeficiency Virus (HIV) and HIV-Infected Lymphocytes. Journal of Virology. 2003;77:11896. doi: 10.1128/JVI.77.22.11896-11909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57λ.Low A, Datta S, Kuznetsov Y, Jahid S, Kothari N, McPherson A, Fan F. Mutation in the glycosylated gag protein of murine leukemia virus results in reduced in vivo infectivity and a novel defect in viral budding or release. Journal of Virology. 2007;81:3685. doi: 10.1128/JVI.01538-06. A good example of the use of AFM to reveal mechanisms of viral infectivity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuznetsov Yu G, Victoria JG, Low A, Robinson WE, Jr, Fan H, McPherson A. Atomic force microscopy imaging of retroviruses: Human immunodeficiency virus and murine leukemia virus. Scanning. 2004;26:209. doi: 10.1002/sca.4950260409. [DOI] [PubMed] [Google Scholar]
- 59.Makino DL, Larson SB, McPherson A. Preliminary analysis of crystals of panicum mosaic virus (PMV) by X-ray diffraction and atomic force microscopy. Acta Crystallographica Section D: Biological Crystallography. 2005;61:173. doi: 10.1107/S090744490403077X. [DOI] [PubMed] [Google Scholar]
- 60.Malkin AJ, Kuznetsov YG, Land TA, DeYoreo JJ, McPherson A. Mechanisms of growth for protein and virus crystals. Nature Structural Biology. 1995;2:956. doi: 10.1038/nsb1195-956. [DOI] [PubMed] [Google Scholar]
- 61.McPherson A, Kuznetsov YG, Malkin AJ, Plomp M. Macromolecular crystal growth investigations using atomic force microscopy. Journal of Synchrotron Radiation. 2004;11:21. doi: 10.1107/s0909049503023914. [DOI] [PubMed] [Google Scholar]
- 62.Muller DJ, Engel A, Carrascosa JL, Velez M. The bacteriophage φ29 head-tail connector imaged at high resolution with the atomic force microscope in buffer solution. EMBO Journal. 1997;16:2547. doi: 10.1093/emboj/16.10.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.BonHomme M, Wong S, Carter C, Scarlata S. The pH dependence of HIV-1 capsid assembly and its interaction with cyclophilin A. Biophysical Chemistry. 2003;105:67. doi: 10.1016/s0301-4622(03)00063-2. [DOI] [PubMed] [Google Scholar]
- 64.Zuber G, Barklis E. Atomic force microscopy and electron microscopy analysis of retrovirus Gag proteins assembled in vitro on lipid bilayers. Biophysical Journal. 2000;78:373. doi: 10.1016/S0006-3495(00)76600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Plomp M, Rice MK, Wagner EK, McPherson A, Malkin AJ. Rapid visualization at high resolution of pathogens by atomic force microscopy: Structural studies of herpes simplex virus-1. American Journal of Pathology. 2002;160:1959. doi: 10.1016/S0002-9440(10)61145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuznetsov YG, McPherson A. Atomic force microscopy investigation of Turnip Yellow Mosaic Virus capsid disruption and RNA extrusion. Virology. 2006;352:329. doi: 10.1016/j.virol.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 67.Epand RF, Yip CM, Chernomordik LV, LeDuc DL, Shin YK, Epand RM. Self-assembly of influenza hemagglutinin: Studies of ectodomain aggregation by in situ atomic force microscopy. Biochimica et Biophysica Acta - Biomembranes. 2001;1513:167. doi: 10.1016/s0005-2736(01)00350-9. [DOI] [PubMed] [Google Scholar]
- 68.Korazim O, Sackett K, Shai Y. Functional and Structural Characterization of HIV-1 gp41 Ectodomain Regions in Phospholipid Membranes Suggests that the Fusion-active Conformation Is Extended. Journal of Molecular Biology. 2006;364:1103. doi: 10.1016/j.jmb.2006.08.091. [DOI] [PubMed] [Google Scholar]
- 69λ.Carrasco C, Carreira A, Schaap IAT, Serena PA, Gomez-Herrero J, Mateu MG, De Pablo PJ. DNA-mediated anisotropic mechanical reinforcement of a virus. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13706. doi: 10.1073/pnas.0601881103. AFM studies help to reveal the basis of unusual mechanical properties of viral capsids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70λ.Ivanovska I, Wuite G, Jonsson B, Evilevitch A. Internal DNA pressure modifies stability of WT phage. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9603. doi: 10.1073/pnas.0703166104. AFM indentation studies reveal the perfect equilibrium between DNA packing and properties of viral capsid. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Michel JP, Ivanovska IL, Gibbons MM, Klug WS, Knobler CM, Wuite GJL, Schmidt CF. Nanoindentation studies of full and empty viral capsids and the effects of capsid protein mutations on elasticity and strength. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6184. doi: 10.1073/pnas.0601744103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moloney M, McDonnell L, O’Shea H. Immobilisation of Semliki forest virus for atomic force microscopy. Ultramicroscopy. 2002;91:275. doi: 10.1016/s0304-3991(02)00109-2. [DOI] [PubMed] [Google Scholar]
- 73.Kol N, Gladnikoff M, Barlam D, Shneck RZ, Rein A, Rousso I. Mechanical properties of murine leukemia virus particles: Effect of maturation. Biophysical Journal. 2006;91:767. doi: 10.1529/biophysj.105.079657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ivanovska IL, De Pablo PJ, Ibarra B, Sgalari G, MacKintosh FC, Carrascosa JL, Schmidt CF, Wuite GJL. Bacteriophage capsids: Tough nanoshells with complex elastic properties. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7600. doi: 10.1073/pnas.0308198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmatulla A, Maghelli N, Marti O. Micromechanical properties of tobacco mosaic viruses. Journal of Microscopy. 2007;225:264. doi: 10.1111/j.1365-2818.2007.01741.x. [DOI] [PubMed] [Google Scholar]
- 76.Gibbons MM, Klug WS. Mechanical modeling of viral capsids. Journal of Materials Science. 2007;42:8995. [Google Scholar]
- 77.Drake B, Prater CB, Weisenhorn AL, Gould SA, Albrecht TR, Quate CF, Cannell DS, Hansma HG, Hansma PK. Imaging crystals, polymers, and processes in water with the atomic force microscope. Science. 1989;243:1586. doi: 10.1126/science.2928794. [DOI] [PubMed] [Google Scholar]
- 78.Marchant RE, Kang I, Sit PS, Zhou Y, Todd BA, Eppell SJ, Lee I. Molecular views and measurements of hemostatic processes using atomic force microscopy. Current Protein and Peptide Science. 2002;3:249. doi: 10.2174/1389203023380611. [DOI] [PubMed] [Google Scholar]
- 79.Blinc A, Fric J, Mus?evlc I. Atomic force microscopy of fibrin networks and plasma clots during fibrinolysis. Fibrinolysis and Proteolysis. 1998;12:26. [Google Scholar]
- 80.Lord ST. Fibrinogen and fibrin: Scaffold proteins in hemostasis. Current Opinion in Hematology. 2007;14:236. doi: 10.1097/MOH.0b013e3280dce58c. [DOI] [PubMed] [Google Scholar]
- 81.Marchant RE, Barb MD, Shainoff JR, Eppell SJ, Wilson DL, Siedlecki CA. Three dimensional structure of human fibrinogen under aqueous conditions visualized by atomic force microscopy. Thrombosis and Haemostasis. 1997;77:1048. [PubMed] [Google Scholar]
- 82.Sit SP, Marchant RE. Surface-dependent conformations of human fibrinogen observed by atomic force microscopy under aqueous conditions. Thrombosis and Haemostasis. 1999;82:1053. [PubMed] [Google Scholar]
- 83.Taatjes DJ, Quinn AS, Jenny RJ, Hale P, Bovill EG, McDonagh J. Tertiary structure of the hepatic cell protein fibrinogen in fluid revealed by atomic force microscopy. Cell Biology International. 1997;21:715. doi: 10.1006/cbir.1997.0216. [DOI] [PubMed] [Google Scholar]
- 84.Wigren R, Elwing H, Erlandsson R, Welin S, Lundstrom I. Structure of adsorbed fibrinogen obtained by scanning force microscopy. FEBS Letters. 1991;280:225. doi: 10.1016/0014-5793(91)80298-h. [DOI] [PubMed] [Google Scholar]
- 85λ.Sit PS, Marchant RE. Surface-dependent differences in fibrin assembly visualized by atomic force microscopy. Surface Science. 2001;491:421. Dynamic observation of the fibrinogen net formationin real time. [Google Scholar]
- 86.Evans-Nguyen KM, Fuierer RR, Fitchett BD, Tolles LR, Conboy JC, Schoenfisch MH. Changes in adsorbed fibrinogen upon conversion to fibrin. Langmuir. 2006;22:5115. doi: 10.1021/la053070y. [DOI] [PubMed] [Google Scholar]
- 87.Agnihotri A, Siedlecki CA. Time-Dependent Conformational Changes in Fibrinogen Measured by Atomic Force Microscopy. Langmuir. 2004;20:8846. doi: 10.1021/la049239+. [DOI] [PubMed] [Google Scholar]
- 88.Bai Z, Gilbert JL, Gettens R. Transactions - 7th World Biomaterials Congress. 2004. Self assembled fibrinogen structures film on selected surfaces in molecule level; p. 1740. [Google Scholar]
- 89.Gergely C, Voegel JC, Schaaf P, Senger B, Maaloum M, Ho?rber JKH, Hemmerle J. Unbinding process of adsorbed proteins under external stress studied by atomic force microscopy spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10802. doi: 10.1073/pnas.180293097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90λ.Hemmerle J, Altmann SM, Maaloum M, Horber JKH, Heinrich L, Voegel JC, Schaaf P. Direct observation of the anchoring process during the adsorption of fibrinogen on a solid surface by force-spectroscopy mode atomic force microscopy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6705. doi: 10.1073/pnas.96.12.6705. Real-time monitoring of the fibrinogen net formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marchin KL, Berrie CL. Conformational changes in the plasma protein fibrinogen upon adsorption to graphite and mica investigated by atomic force microscopy. Langmuir. 2003;19:9883. [Google Scholar]
- 92.Tsapikouni TS, Missirlis YF. pH and ionic strength effect on single fibrinogen molecule adsorption on mica studied with AFM. Colloids and Surfaces B: Biointerfaces. 2007;57:89. doi: 10.1016/j.colsurfb.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 93.Barg A, Ossig R, Goerge T, Schneider MF, Schillers H, Oberleithner H, Schneider SW. Soluble plasma-derived von Willebrand factor assembles to a haemostatically active filamentous network. Thrombosis and Haemostasis. 2007;97:514. [PubMed] [Google Scholar]
- 94.Blinc A, Magdic J, Fric J. Atomic force microscopy of fibrin networks and plasma clots during fibrinolysis. Fibrinolysis and Proteolysis. 2000;14:288. [Google Scholar]
- 95.Savage B, Sixma JJ, Ruggeri ZM. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:425. doi: 10.1073/pnas.012459599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Novak L, Deckmyn H, Damjanovich S, Harsfalvi J. Shear-dependent morphology of von Willebrand factor bound to immobilized collagen. Blood. 2002;99:2070. doi: 10.1182/blood.v99.6.2070. [DOI] [PubMed] [Google Scholar]
- 97.Siedlecki CA, Lestini BJ, Kottke-Marchant K, Eppell SJ, Wilson DL, Marchant RE. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 1996;88:2939. [PubMed] [Google Scholar]
- 98.Yokota H, Sunwoo J, Sarikaya N, Van Den Engh G, Aebersold R. Spin-stretching of DNA and protein molecules for detection by fluorescence and atomic force microscopy. Analytical Chemistry. 1999;71:4418. doi: 10.1021/ac9902695. [DOI] [PubMed] [Google Scholar]
- 99.Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thrombosis Research. 2007;119:731. doi: 10.1016/j.thromres.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 100.Raghavachari M, Tsai HM, Kottke-Marchant K, Marchant RE. Surface dependent structures of von Willebrand factor observed by AFM under aqueous conditions. Colloids and Surfaces B: Biointerfaces. 2000;19:315. doi: 10.1016/s0927-7765(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 101.Rauscher S, Baud S, Miao M, Keeley F, Pome?s R. Proline and Glycine Control Protein Self-Organization into Elastomeric or Amyloid Fibrils. Structure. 2006;14:1667. doi: 10.1016/j.str.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 102.Aso Y, Shiraki K, Takagi M. Systematic analysis of aggregates from 38 kinds of non disease-related proteins: Identifying the intrinsic propensity of polypeptides to form amyloid fibrils. Bioscience, Biotechnology and Biochemistry. 2007;71:1313. doi: 10.1271/bbb.60718. [DOI] [PubMed] [Google Scholar]
- 103.Chamberlain AK, MacPhee CE, Zurdo J, Morozova-Roche LA, Hill HAO, Dobson CM, Davis JJ. Ultrastructural organization of amyloid fibrils by atomic force microscopy. Biophysical Journal. 2000;79:3282. doi: 10.1016/S0006-3495(00)76560-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sedman VL, Allen S, Chan WC, Davies MC, Roberts CJ, Tendler SJB, Williams PM. Atomic force microscopy study of human amylin (20–29) fibrils. Protein and Peptide Letters. 2005;12:79. doi: 10.2174/0929866053406129. [DOI] [PubMed] [Google Scholar]
- 105.Gosal WS, Morten IJ, Hewitt EW, Smith DA, Thomson NH, Radford SE. Competing pathways determine fibril morphology in the self-assembly of? 2-microglobulin into amyloid. Journal of Molecular Biology. 2005;351:850. doi: 10.1016/j.jmb.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 106.Yamaguchi KI, Takahashi S, Kawai T, Naiki H, Goto Y. Seeding-dependent propagation and maturation of amyloid fibril conformation. Journal of Molecular Biology. 2005;352:952. doi: 10.1016/j.jmb.2005.07.061. [DOI] [PubMed] [Google Scholar]
- 107.Relini A, Canale C, De Stefano S, Rolandi R, Giorgetti S, Stoppini M, Rossi A, Fogolari F, Corazza A, Esposito G, et al. Collagen plays an active role in the aggregation of? 2- microglobulin under physiopathological conditions of dialysis-related amyloidosis. Journal of Biological Chemistry. 2006;281:16521. doi: 10.1074/jbc.M513827200. [DOI] [PubMed] [Google Scholar]
- 108.Dahlgren PR, Karymov MA, Bankston J, Holden T, Thumfort P, Ingram VM, Lyubchenko YL. Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomedicine: Nanotechnology, Biology, and Medicine. 2005;1:52. doi: 10.1016/j.nano.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 109.Khurana R, Ionescu-Zanetti C, Pope M, Li J, Nielson L, Ramirez-Alvarado M, Regan L, Fink AL, Carter SA. A general model for amyloid fibril assembly based on morphological studies using atomic force microscopy. Biophysical Journal. 2003;85:1135. doi: 10.1016/S0006-3495(03)74550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110λλ.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both?-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:571. doi: 10.1073/pnas.97.2.571. The first experimental proof that a-synuclein nonfibrillar oligomers could be a culprit in Parkinson’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Green JD, Goldsbury C, Kistler J, Cooper GJS, Aebi U. Human Amylin Oligomer Growth and Fibril Elongation Define Two Distinct Phases in Amyloid Formation. Journal of Biological Chemistry. 2004;279:12206. doi: 10.1074/jbc.M312452200. [DOI] [PubMed] [Google Scholar]
- 112.Green J, Goldsbury C, Mini T, Sunderji S, Frey P, Kistler J, Cooper G, Aebi U. Full-length rat amylin forms fibrils following substitution of single residues from human amylin. Journal of Molecular Biology. 2003;326:1147. doi: 10.1016/s0022-2836(02)01377-3. [DOI] [PubMed] [Google Scholar]
- 113.Goldsbury C, Kistler J, Aebi U, Arvinte T, Cooper GJS. Watching amyloid fibrils grow by time-lapse atomic force microscopy. Journal of Molecular Biology. 1999;285:33. doi: 10.1006/jmbi.1998.2299. [DOI] [PubMed] [Google Scholar]
- 114.Kad NM, Myers SL, Smith DP, Smith DA, Radford SE, Thomson NH. Hierarchical assembly of beta2-microglobulin amyloid in vitro revealed by atomic force microscopy. Journal of molecular biology. 2003;330:785. doi: 10.1016/s0022-2836(03)00583-7. [DOI] [PubMed] [Google Scholar]
- 115λ.Ionescu-Zanetti C. Monitoring the assembly of Ig light-chain amyloid fibrils by atomic force microscopy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13175. doi: 10.1073/pnas.96.23.13175. A model describing stepwise fibrillization proposed on a basis of AFM studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Green JD, Kreplak L, Goldsbury C, Li Blatter X, Stolz M, Cooper GS, Seelig A, Kistler J, Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. Journal of Molecular Biology. 2004;342:877. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 117λλ.Jo E, Darabie AA, Han K, Tandon A, Fraser PE, McLaurin J. alpha-Synuclein-synaptosomal membrane interactions: implications for fibrillogenesis. European Journal of Biochemistry. 2004;271:3180–3189. doi: 10.1111/j.1432-1033.2004.04250.x. AFM study of Ab demonstrating in situ process of aggregation. It is the first observation showing that the outcome of aggregation into fibrils depends on protein interaction at the interface resembling conditions at cell membranes. [DOI] [PubMed] [Google Scholar]
- 118.Kowalewski T, Holtzman DM. In situ atomic force microscopy study of Alzheimer’s? -amyloid peptide on different substrates: New insights into mechanism of?-sheet formation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3688. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harper JD, Lieber CM, Lansbury PT., Jr Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-? protein. Chemistry and Biology. 1997;4:951. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 120.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Assembly of A? amyloid protofibrils: An in vitro model for a possible early event in Alzheimer’s disease. Biochemistry. 1999;38:8972. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- 121.Grudzielanek S, Velkova A, Shukla A, Smirnovas V, Tatarek-Nossol M, Rehage H, Kapurniotu A, Winter R. Cytotoxicity of Insulin within its Self-assembly and Amyloidogenic Pathways. Journal of Molecular Biology. 2007;370:372. doi: 10.1016/j.jmb.2007.04.053. [DOI] [PubMed] [Google Scholar]
- 122.Mauro M, Craparo EF, Podesta A, Bulone D, Carrotta R, Martorana V, Tiana G, San Biagio PL. Kinetics of Different Processes in Human Insulin Amyloid Formation. Journal of Molecular Biology. 2007;366:258. doi: 10.1016/j.jmb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 123.Jansen R, Dzwolak W, Winter R. Amyloidogenic self-assembly of insulin aggregates probed by high resolution atomic force microscopy. Biophysical Journal. 2005;88:1344. doi: 10.1529/biophysj.104.048843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Podesta A, Tiana G, Milani P, Manno M. Early events in insulin fibrillization studied by time-lapse atomic force microscopy. Biophysical Journal. 2006;90:589. doi: 10.1529/biophysj.105.068833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grudzielanek S, Jansen R, Winter R. Solvational tuning of the unfolding, aggregation and amyloidogenesis of insulin. Journal of Molecular Biology. 2005;351:879. doi: 10.1016/j.jmb.2005.06.046. [DOI] [PubMed] [Google Scholar]
- 126.Smith JF, Knowles TPJ, Dobson CM, MacPhee CE, Welland ME. Characterization of the nanoscale properties of individual amyloid fibrils. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15806. doi: 10.1073/pnas.0604035103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127λ.Jansen R, Grudzielanek S, Dzwolak W, Winter R. High pressure promotes circularly shaped insulin amyloid. Journal of Molecular Biology. 2004;338:203. doi: 10.1016/j.jmb.2004.02.056. A short communication revealing formation of circular insulin amyloids under high hydrostatic pressure. A possibility to design compounds specifically forcing or interfering with such structures. [DOI] [PubMed] [Google Scholar]
- 128.McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, Lyubchenko YL. Protein interactions and misfolding analyzed by AFM force spectroscopy. Journal of Molecular Biology. 2005;354:1028. doi: 10.1016/j.jmb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 129.Soldi G, Bemporad F, Torrassa S, Relini A, Ramazzotti M, Taddei N, Chiti F. Amyloid formation of a protein in the absence of initial unfolding and destabilization of the native state. Biophysical Journal. 2005;89:4234. doi: 10.1529/biophysj.105.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barkhordarian H, Emadi S, Schulz P, Sierks MR. Isolating recombinant antibodies against specific protein morphologies using atomic force microscopy and phage display technologies. Protein Engineering, Design and Selection %R 10.1093/protein/gzl036. 2006;19:497–502. doi: 10.1093/protein/gzl036. [DOI] [PubMed] [Google Scholar]
- 131.Emadi S, Barkhordarian H, Wang MS, Schulz P, Sierks MR. Isolation of a Human Single Chain Antibody Fragment Against Oligomeric α-Synuclein that Inhibits Aggregation and Prevents? -Synuclein-induced Toxicity. Journal of Molecular Biology. 2007;368:1132. doi: 10.1016/j.jmb.2007.02.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.James LC, Tawfik DS. Conformational diversity and protein evolution--a 60-year-old hypothesis revisited. Trends in Biochemical Sciences. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 133.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 134.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 135λ.Osmulski PA, Gaczynska M. Atomic force microscopy reveals two conformations of the 20 S proteasome from fission yeast. Journal of Biological Chemistry. 2000;275:13171. doi: 10.1074/jbc.c901035199. The presence of a dynamic gate to the proteasome revealed by AFM. [DOI] [PubMed] [Google Scholar]
- 136.Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D. A gated channel into the proteasome core particle. Nature Structural Biology. 2000;7:1062–1067. doi: 10.1038/80992. [DOI] [PubMed] [Google Scholar]
- 137.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 138.Gaczynska M, Osmulski PA. Small-molecule inhibitors of proteasome activity. Methods in molecular biology (Clifton, NJ) 2005;301:3. doi: 10.1385/1-59259-895-1:003. [DOI] [PubMed] [Google Scholar]
- 139.Lin H, Clegg DO, Lal R. Imaging real-time proteolysis of single collagen I molecules with an atomic force microscope. Biochemistry. 1999;38:9956. doi: 10.1021/bi990800q. [DOI] [PubMed] [Google Scholar]
- 140.Tan X, Osmulski PA, Gaczynska M. Allosteric regulators of the proteasome: Potential drugs and a novel approach for drug design. Current Medicinal Chemistry. 2006;13:155. doi: 10.2174/092986706775197926. [DOI] [PubMed] [Google Scholar]
- 141.Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Proline- and arginine-rich peptides constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. 2003;42:8663. doi: 10.1021/bi034784f. [DOI] [PubMed] [Google Scholar]
- 142.Gaczynska M, Rodriguez K, Madabhushi S, Osmulski PA. Highbrow proteasome in high-throughput technology. Expert Review of Proteomics. 2006;3:115. doi: 10.1586/14789450.3.1.115. [DOI] [PubMed] [Google Scholar]
- 143λλ.Viani MB, Pietrasanta LI, Thompson JB, Chand A, Gebeshuber IC, Kindt JH, Richter M, Hansma HG, Hansma PK. Probing protein-protein interactions in real time. Nature Structural Biology. 2000;7:644. doi: 10.1038/77936. An original one-dimensional scanning method enabled analysis of fast movements of the GroEL/GroES assembly. [DOI] [PubMed] [Google Scholar]
- 144.Dubrovin EV, Drygin Y-F, Novikov VK, Yaminsky IV. Atomic force microscopy as a tool of inspection of viral infection. Nanomedicine: Nanotechnology, Biology and Medicine. 2007;3:128. doi: 10.1016/j.nano.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 145.Trindade GS, Vilela JMC, Ferreira JMS, Aguiar PHN, Leite JA, Guedes MIMC, Lobato ZIP, Madureira MC, da Silva MIN, da Fonseca FG, et al. Use of atomic force microscopy as a diagnostic tool to identify orthopoxvirus. Journal of Virological Methods. 2007;141:198. doi: 10.1016/j.jviromet.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 146.Shepherd CM, Borelli IA, Lander G, Natarajan P, Siddavanahalli V, Bajaj C, Johnson JE, Brooks CL, 3rd, Reddy VS. VIPERdb: a relational database for structural virology. Nucleic acids research. 2006:34. doi: 10.1093/nar/gkj032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kuznetsov YG, Malin AJ, Lucas RW, Plomp M, McPherson A. Imaging of viruses by atomic force microscopy. Journal of General Virology. 2001;82:2025. doi: 10.1099/0022-1317-82-9-2025. [DOI] [PubMed] [Google Scholar]
- 148.Venkataraman S, Allison DP, Qi H, Morrell-Falvey JL, Kallewaard NL, Crowe JE, Jr, Doktycz MJ. Automated image analysis of atomic force microscopy images of rotavirus particles. Ultramicroscopy. 2006;106:829. doi: 10.1016/j.ultramic.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 149.Dubrovin EV, Kirikova MN, Novikov VK, Drygin YF, Yaminsky IV. Study of the peculiarities of adhesion of tobacco mosaic virus by atomic force microscopy. Colloid Journal. 2004;66:673. [Google Scholar]
- 150.Kim HS, Jung SH, Kim SH, Suh IB, Kim WJ, Jung JW, Yuk JS, Kim YM, Ha KS. High-throughput analysis of mumps virus and the virus-specific monoclonal antibody on the arrays of a cationic polyelectrolyte with a spectral SPR biosensor. Proteomics. 2006;6:6426. doi: 10.1002/pmic.200600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suci PA, Klem MT, Douglas T, Young M. Influence of electrostatic interactions on the surface adsorption of a viral protein cage. Langmuir. 2005;21:8686. doi: 10.1021/la050217c. [DOI] [PubMed] [Google Scholar]
- 152.Knez M, Sumser MP, Bittner AM, Wege C, Jeske H, Hoffmann DMP, Kuhnke K, Kern K. Binding the Tobacco Mosaic Virus to Inorganic Surfaces. Langmuir. 2004;20:441. doi: 10.1021/la035425o. [DOI] [PubMed] [Google Scholar]
- 153.Henderson E. BioForce Nanoscience, Inc. Nanomedicine. 2007;2:391. doi: 10.2217/17435889.2.3.391. [DOI] [PubMed] [Google Scholar]
- 154.Huff JL, Lynch MP, Nettikadan S, Johnson JC, Vengasandra S, Henderson E. Label-free protein and pathogen detection using the atomic force microscope. Journal of Biomolecular Screening. 2004;9:491. doi: 10.1177/1087057104268803. [DOI] [PubMed] [Google Scholar]
- 155.Johnson JC, Nettikadan SR, Vengasandra SG, Henderson E. Analysis of solid-phase immobilized antibodies by atomic force microscopy. Journal of Biochemical and Biophysical Methods. 2004;59:167. doi: 10.1016/j.jbbm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 156.Nettikadan SR, Johnson JC, Mosher C, Henderson E. Virus particle detection by solid phase immunocapture and atomic force microscopy. Biochemical and Biophysical Research Communications. 2003;311:540. doi: 10.1016/j.bbrc.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 157λ.Nettikadan SR, Johnson JC, Vengasandra SG, Muys J, Henderson E. ViriChip: A solid phase assay for detection and identification of viruses by atomic force microscopy. Nanotechnology. 2004;15:383. The first commercially available chip for specific detection of viruses described. [Google Scholar]
- 158.Radke KM, Nettikadan SR, Johnson JC, Vengasandra SG, Henderson E. ViriChip enhances reverse transcriptase polymerase chain reaction in biological fluids and environmental samples. Analytical Biochemistry. 2004;330:350. doi: 10.1016/j.ab.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 159.Driskell JD, Kwarta KM, Lipert RJ, Porter MD, Neill JD, Ridpath JF. Low-level detection of viral pathogens by a surface-enhanced Raman scattering based immunoassay. Analytical Chemistry. 2005;77:6147. doi: 10.1021/ac0504159. [DOI] [PubMed] [Google Scholar]
- 160.Vega RA, Maspoch D, Shen CKF, Kakkassery JJ, Chen BJ, Lamb RA, Mirkin CA. Functional antibody arrays through metal ion-affinity templates. ChemBioChem. 2006;7:1653. doi: 10.1002/cbic.200600271. [DOI] [PubMed] [Google Scholar]
- 161.Negishi A, Chen J, McCarty DM, Samulski RJ, Liu J, Superfine R. Analysis of the interaction between adeno-associated virus and heparan sulfate using atomic force microscopy. Glycobiology. 2004;14:969. doi: 10.1093/glycob/cwh118. [DOI] [PubMed] [Google Scholar]
- 162λ.Dickert FL, Hayden O, Bindeus R, Mann KJ, Blaas D, Waigmann E. Bioimprinted QCM sensors for virus detection-screening of plant sap. Analytical and Bioanalytical Chemistry. 2004;378:1929. doi: 10.1007/s00216-004-2521-5. A chip for specific detection of viruses in crude extracts described. [DOI] [PubMed] [Google Scholar]
- 163.Cacciafesta P, Hallam KR, Watkinson AC, Allen GC, Miles MJ, Jandt KD. Visualisation of human plasma fibrinogen adsorbed on titanium implant surfaces with different roughness. Surface Science. 2001;491:405. [Google Scholar]
- 164.Kim YH, Yoo YM, Song DM, Kim SH, Jung Y, Ko YG, Jang Y, Chung KH. Transactions - 7th World Biomaterials Congress. 2004. Animal implantation study of surface modified stents grafted with sulfonated PEG: Blood compatibility and restenosis; p. 1665. [Google Scholar]
- 165.Mitsakakis K, Lousinian S, Logothetidis S. Early stages of human plasma proteins adsorption probed by Atomic Force Microscope. Biomolecular Engineering. 2007;24:119. doi: 10.1016/j.bioeng.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 166.Boukari A, Francius G, Hemmerle J. AFM force spectroscopy of the fibrinogen adsorption process onto dental implants. Journal of Biomedical Materials Research - Part A. 2006;78:466. doi: 10.1002/jbm.a.30778. [DOI] [PubMed] [Google Scholar]
- 167.Bremmell KE, Kingshott P, Ademovic Z, Winther-Jensen B, Griesser HJ. Colloid probe AFM investigation of interactions between fibrinogen and PEG-like plasma polymer surfaces. Langmuir. 2006;22:313. doi: 10.1021/la052143a. [DOI] [PubMed] [Google Scholar]
- 168.Chen X, Davies MC, Roberts CJ, Tendler SJB, Williams PM, Davies J, Dawkes AC, Edwards JC. Recognition of protein adsorption onto polymer surfaces by scanning force microscopy and probe-surface adhesion measurements with protein-coated probes. Langmuir. 1997;13:4106. [Google Scholar]
- 169.Conti M, Donati G, Cianciolo G, Stefoni S, Samori B. Force spectroscopy study of the adhesion of plasma proteins to the surface of a dialysis membrane: Role of the nanoscale surface hydrophobicity and topography. Journal of Biomedical Materials Research. 2002;61:370. doi: 10.1002/jbm.10168. [DOI] [PubMed] [Google Scholar]
- 170λ.Siedlecki CA, Agnihotri A, Rice Z, Soman P. Detection of plasma proteins in multicomponent protein films by atomic force microscopy. Microscopy and Microanalysis. 2006;12:34. Surface adhesion of plasma proteins was monitored by AFM in multicomponent protein films created by rounds of stamping and filling - a useful procedure for testing of biocompatible surfaces. [Google Scholar]
- 171.Berglin M, Andersson M, Sellborn A, Elwing H. The effect of substrate molecular mobility on surface induced immune complement activation and blood plasma coagulation. Biomaterials. 2004;25:4581. doi: 10.1016/j.biomaterials.2003.11.050. [DOI] [PubMed] [Google Scholar]
- 172.Chu SH, Choi SH, Watt GD, Kim JW, Park Y, Davis RC, Harb JN, King GC, Lillehei PH. Proceedings of SPIE - The International Society for Optical Engineering. 2004. Fabrication of cell structures for bio-nanobattery; p. 443. [Google Scholar]
- 173.Francois P, Vaudaux P, Nurdin N, Mathieu HJ, Descouts P, Lew DP. Physical and biological effects of a surface coating procedure on polyurethane catheters. Biomaterials. 1996;17:667. doi: 10.1016/0142-9612(96)86736-6. [DOI] [PubMed] [Google Scholar]
- 174.Hayama M, Yamamoto KI, Kohori F, Uesaka T, Ueno Y, Sugaya H, Itagaki I, Sakai K. Nanoscopic behavior of polyvinylpyrrolidone particles on polysulfone/polyvinylpyrrolidone film. Biomaterials. 2004;25:1019. doi: 10.1016/s0142-9612(03)00629-x. [DOI] [PubMed] [Google Scholar]
- 175.Zhou C, Khlestkin VK, Bracken D, De Keersmaecker K, Laureyn W, Engelborghs Y, Borghs G. Solvent-controlled organization of self-assembled polymeric monolayers on gold: An easy approach for the construction of protein resistant surfaces. Langmuir. 2005;21:5988. doi: 10.1021/la0471590. [DOI] [PubMed] [Google Scholar]
- 176λ.Yamashita I. Fabrication of a two-dimensional array of nano-particles using ferritin molecule. Thin Solid Films. 2001;393:12. Ferritin used as a nanobiotechnolohical tool to deposit metallic arrays. [Google Scholar]
- 177.Choi SH, Kim JW, Chu SH, Park Y, King GC, Lillehei PT, Kim SJ, Elliott JR. Proceedings of SPIE - The International Society for Optical Engineering. 2005. Ferritin-templated quantum-dots for quantum logic gates; p. 213. [Google Scholar]