

Abstract
The unprocessed gastrin precursor, progastrin (PG), is often overexpressed in colon cancer and other malignancies where it appears to stimulate colonic growth. Overexpression of progastrin also stimulates proliferation of normal colonic mucosa, but the receptors mediating these effects have not been identified. Here we report the development of a non-radioactive assay for assessment of PG binding to normal and transformed cells. Progastrin was labeled using biotinylation, and binding of biotinylated PG to cells was assessed using flow cytometry. Using this approach, we show strong and specific binding of PG to some cell lines (IEC-6, IEC-18, HT-29, COLO320) and minimal binding to others (HeLa, DC2.4, Jurkat). We also found PG binding to several non-gut epithelial lines, such as CHO-K1, COS-6 and HEK293 cells. The specificity of binding was confirmed by competition with cold, unlabeled PG but not with glycine-extended gastrin or amidated gastrin-17. Binding was not influenced by the presence of the classical CCK-2 receptor, but was partially dependent on the charged glycosaminoglycans (GAG). The analysis of primary colonic tissues isolated from wild type C57BL/6 mouse, revealed a small epithelial subpopulation of non-hematopoietic (CD45-negative) cells that strongly interacted with PG. Surprisingly, this population was greatly expanded in gastrin knockout mice. This nonradioactive, FACS-based assay should prove useful for further characterization of cells expressing the progastrin receptor.
Keywords: biotin, gastrin-related peptides, FASC assay, glycosaminoglycans, mouse, colonic mucosa
INTRODUCTION
Gastrin is a peptide hormone produced primarily by endocrine G cells in the gastric antrum, with lower levels found in the duodenum and pituitary. In the stomach, gastrin has a well-defined role in the maintenance and functioning of the gastric oxyntic mucosa involving both growth and acid secretion. Amidated gastrins (e.g. G-17 and G-34) act primarily on parietal and ECL cells through binding to the CCK-2/CCKB receptor, a high affinity G-protein coupled receptor [1]. Gastrin is initially generated as a 101 amino acid precursor peptide, preprogastrin that is converted to progastrin (PG) by cleavage of a signal peptide. The 80 amino acid precursor, progastrin (PG), is then converted to amidated gastrins via biosynthetic intermediates, glycine-extended gastrin (G-gly) [2]. In most cases, the levels of the nonamidated precursor forms of gastrin comprise <10% of the total secreted peptide, but increased levels are occasionally observed in disease states, such as in patients with colorectal cancer [3].
Work from a number of laboratories has suggested that nonamidated gastrins (e.g. PG and G-gly) contribute to the growth and development of colorectal cancer. First, the gastrin gene is expressed in the majority of colonic adenomas and cancers, and gastrin has been identified as an important downstream target of both Wnt and Ras signaling pathways. In these tumors, gastrin is present primarily in the nonamidated form due to the absence of processing enzymes in nonendocrine tissues [4-6]. The growth of colorectal cancers could be blocked by nonselective gastrin receptor antagonists or antisense gastrin mRNA [7, 8]. In vitro studies have demonstrated growth promoting effects for G-gly and progastrin on cultured cell lines [9-12]. Stronger evidence has come from in vivo studies with transgenic mice engineered to overexpress progastrin. hGAS mice express a human preprogastrin transgene in the liver, resulting in high circulating concentrations of progastrin (1-100 nM) and normal plasma concentrations of amidated gastrin [13]. These mice show colonic hyperproliferation and increased sensitivity to the induction of aberrant crypt foci (ACFs) and colonic adenomas after treatment with the carcinogen azoxymethane, AOM. More recent studies have shown that transgenic mice with elevated progastrin levels exhibit significantly higher levels of mitosis after DNA damage by gamma-irradiation, an effect that does not require other forms of gastrin. Indeed, infusion of progastrin 1-80 into gastrin deficient mice also significantly stimulated mitosis [14].
The effects of progastrin on colonic cell proliferation, as well as studies suggesting the activation of intracellular signaling pathways [15-17], would argue for the presence of specific receptors for progastrin. However, while binding sites for progastrin-derived peptides have been demonstrated in colonic crypts [18], the precise receptors mediating the trophic effects of progastrin and G-gly remain uncertain. Possibilities include a novel receptor or a modified version of the CCK-2 receptor. Some progress has been made in this area by studies showing high-affinity binding of 3H-labeled progastrin to IEC-6 and IEC-18 cells. This approach has led to the identification of a candidate progastrin-binding protein (Annexin II) [19]. Here, we report the development of an alternative approach combining biotinylated progastrin and flow cytometry. Using this approach, we confirm the presence of specific, high-affinity receptors for progastrin in epithelial cells that appears independent of the CCK-2 receptor.
EXPERIMENTAL PROCEDURES
Materials
Mice (described elsewhere) were housed in a barrier facility at the Irving Cancer Center of Columbia University. Mice were treated in accordance with the guidelines and procedures approved by the Institutional Animal Care and Use Committee. The cell lines - CHO-K1, CHO-pgsB 618, CHO-pgsD 677, IEC-6, IEC-18, HEK293, COLO-320, HT-29, COS-6, COS-7, Jurkat and HeLa - were obtained from the American Type Cell Collection (Manassas, VA). Progastrin (PG), C -terminally extended gastrin (Gas-CEP) and Gly-extended gastrin (G-gly) were synthesized by New England Peptides (Gardner, MA). Gastrin and heparin sulfate (HS), heparin, heparinase (EC 4.2.2.7), Biotin Labeling Kit were obtained from Sigma-Aldrich (St. Louis, MO). Streptavidin was obtained from Pierce (Rockford, IL). Phycoerythrin conjugated streptavidin (Str-PE) was obtained from Molecular Probes/Invitrogen (Carlsbad, CA).
Biotinylation procedure
Conjugation of biotin to progastrin was performed using Biotin Labeling Kit in accordance with the manufacturer’s instructions. Briefly, up to 50 μL of biotin-7-NHS solution (20mg/ml in DMSO) were drop added to the PG solution (2mg/ml in PBS) with continuous stirring for 2h at RT. The products of the reaction were separated by SephadexG-25 column chromatography. The efficiency of progastrin biotinylation was assessed by SDS-PAGE electrophoresis using 4-20% Tris-Tricine gels. Samples of the biotinylated or unmodified progastrin were pre-incubated with 1-5μg of streptavidin in 20μl PBS buffer supplemented by CaCl2 and MgSO4 (1mM each) at room temperature for 30 minutes. An equal volume of concentrated (× 2) loading buffer was then added to each sample. Samples were heated at 37°C for 10 minutes and loaded onto a gel that was run at 125V for 1h 30 minutes. The gel was then stained by SimplyBlue Safe Stain (Invitrogen) according to the manufacturer’s instructions.
Reporter assay
To access a possible growth promoting activity of PG (bio-PG) on epithelial cells we used a transient transfection assay with the growth-responsive reporter construct, pSREx5-LUC, containing five copies of the serum response element (SRE) fused to the firefly luciferase reporter gene (Stratagene, La Jolla, CA). IEC-18 epithelial cells (105 cells/well in 6 well plate) were transfected with 2μg of pSREx6-LUC and 0.1μg Renilla LUC reporter constructs, along with 6μl of Lipofectamine 2000 in 200μl of Opti-MEM medium. Twenty-four hours after transfection, cells were split into 6 smaller wells (in 12 well plates) in serum-free medium (DMEM+0.5% BSA) for an additional 24 hours. On the next day, the quiescent cells were stimulated with 1-100nM of PG (bio-PG) peptide for 6-20 hours. Following progastrin stimulation, cells extracts were prepared using dual-luciferase lysis buffer (Promega, Madison, WI). The firefly and renilla luciferase activities were measured with the “Dual luciferase reporter assay kit”(Promega, Madison, WI). Relative (firefly/renilla) luciferase activity (RLU) was calculated for each treatment and was performed in triplicate. Statistical significance of the PG (bio-PG) stimulating effect on RLU in treated and untreated IEC18 cells was assessed using a two-tail Student’ t-test.
Flow Cytometric Analysis of bio-PG Binding to Cells
Adherent cells were plated 2 days prior to binding experiments. If not otherwise specified, the cells were detached with 0.5 mM EDTA in PBS and washed twice with ice-cold binding buffer (RPMI 1640, 20 mM Hepes, 1% bovine serum albumin). Cells (4 × 105) were resuspended in the presence of the indicated concentration of peptide in a total volume of 200 μl and incubated for 90 min. at 4 °C under gentle agitation. Unbound ligand was removed by washing with binding buffer, and cell-bound PG was stained with phycoerythrin-conjugated streptavidin (1:200; Molecular Probes). Cells were then washed and resuspended in PBS-1%BSA. Labeled cells were analyzed by flow cytometry using FACScan or LSRII machines (Becton Dickinson, CA).
To remove cell-surface GAG, CHO-K1 and IEC-6 cells (106 cells) were washed twice and incubated for 90 min. at 37 °C with 1000 units/ml heparinase. For trypsin treatment, cells were washed once with PBS containing 2 mM EDTA and incubated for 5 min. at 37 °C with 0.25% trypsin in PBS. After enzyme treatment, cells were washed four times with 5 ml of binding buffer, detached with PBS/EDTA, and then assayed for bio-PG binding as described above.
Screening of murine colon by flow cytometry
To analyze bio-PG binding to mouse colonic cells, a single cell suspension was prepared from wild type (WT) or gastrin knockout C57BL/6 mice. The suspension was prepared as follows: colons were extracted and opened longitudinally, briefly rinsed, cut into ~3mm pieces and placed in 5ml/colon OPTI-MEM-10% FCS supplemented by 2U/ml Dispase (Roche, Palo Alto) in a rotary shaker at 37°C for 45 min. Single cell suspension was prepared by vigorous repeated pipetting, followed by filtration of resultant mix through a 70μM cell strainer. Cells were further washed twice by the above but dispase-free medium. The remaining cells were then placed in complete medium and incubated at 37°C for an additional 30-60 min. before labeling with bio-PG. This protocol provided ~50-70 % viable epithelial cells as judged by the DAPI (or 7AAD) exclusion test in flow cytometry.
One hundred microliters of each suspension (~107/ml) were incubated with bio-PG (1-500nM) in binding buffer for 30 min. at room temperature. Labeled cells were washed and processed for secondary labeling by streptavidin-phycoerythrin conjugate and fluorescein-conjugated antibodies to the respective cell surface marker. Finally, cells were washed to remove unconjugated dyes in PBS-2%FCS and processed for flow cytometry. 7-aminoactinomycin D (7-AAD; 5μl/probe) or 4,6-diamidino-2-phenylindol (DAPI; 1μg/ml) was added just before analysis to label dead cells and exclude them from the final analysis on FACScan or LSRII machine, respectively. Cell clumps were eliminated from analysis by using the corresponding (FSC-A/FCS-H) gates. Raw data were treated using Cell Quests or FlowJo software in order to generate the final analysis.
Analysis of Annexin II expression in epithelial cancer cells
Subconfluent (~80% confluent) cells were washed with phosphate-buffered saline (PBS; without Ca++/Mg++) and detached with 0.5mM EDTA for 5 minutes at room temperature. Cells were centrifuged, transferred into complete medium and incubated on a rotary shaker for 1 hour at 37°C. Cells were collected by centrifugation and resuspended to a final concentration ~1 × 107/ml in binding buffer (PBS/1mMCaCl2 +2% heat-inactivated mouse serum). Aliquots (100μl) of suspension were stained for surface Annexin II expression with FITC-conjugated mouse monoclonal (IgG1) anti-human Annexin II antibodies (5-10μg/ml; BD Biosciences) or an equal amount of control FITC-conjugated mouse IgG1 (eBiosciences) in binding buffer at 4°C for 45 minutes. Cells were washed twice with PBS and resuspended in binding buffer with DAPI (1μg/ml). The fluorescence of unstained or FITC-labeled cells was analyzed by flow cytometry. Viable cells were pre-selected using gates with the appropriate forward-side scattering characteristics and/or DAPI fluorescence. To stain cytosolic Annexin II, cells were fixed with 1.6% paraformaldehyde (PFA; for 15 min at room temperature) and then permeabilized with cold methanol for 20 minutes on ice. Fixed cells were ultimately stained as described for live cells above.
RESULTS
Quantification of progastrin biotinylation by gel electrophoresis
Human progastrin is a medium-sized protein (80 aa) that contains four possible sites for labeling with NHS-biotin, the ε-amino groups of 3 lysyl residues at positions 3, 53 and 54 and the α-amino group of N-terminal serine (Fig. 1A). Since progastrin’s biological activity is thought to be localized primarily to the carboxyl terminus of the peptide [14], the latter can not be additionally biotinylated without potential loss of binding and/or activity. Furthermore, preliminary western blot experiments demonstrated a high degree of heterogeneity in progastrin biotinylation when using commercially available kits (BK-200, Sigma or EZ-link Sulfo-biotin kit, Pierce). Due to the size of progastrin, it was not possible to effectively separate biotinylated and unmodified products using HPLC (data not shown). To overcome these difficulties, we first developed a relatively simple assay for quantification of the degree of progastrin biotinylation. We took an advantage of the different sizes of progastrin, biotin and streptavidin, and the strong interaction between biotin and streptavidin that is typically not disrupted even under the stringent reducing conditions of SDS gel-electrophoresis.
Fig. 1. Effective biotinylation of synthetic progastrin in vitro.


(A). Sequence of human progastrin. Amino acids with free amino groups available for biotinylation, are shown in bold and underlined. (B). The quality of PG biotinylation in samples was tested through analysis of bio-PG: strepavidin complexes in SDS-PAGE electrophoresis. Biotinylated PG (2μg; lanes 5-7) was incubated alone (lane 5) or with 1 or 2 μg of strepavidin (lanes 6, 7) in HBSS for 20 minutes at 20°C prior to electrophoresis. Unmodified PG (2.5μg) was processed in the same manner (lanes 1-3). The control lane (lane 4) contains streptavidin alone (2 μg). At the end of electrophoresis, the gel was stained with “SimplyBlue” reagent (Invitrogen). The migrating positions of the primary reagents are shown by arrows on the left. Filled arrowhead indicates the unique bio-PG: streptavidin complex. The positions of molecular weight markers (Mw; kD) are shown by arrows on the right. (C) Unmodified and biotinylated progastrin peptides stimulate the growth-responsive reporter (luciferase) activity in IEC-18 cells. The cells were transiently co-transfected with growth-factor responsive pSREx5-LUC and reference Renilla-Luciferase plasmids, serum-starved and treated or not with 0, 1, 10, 100nM of PG or bio-PG (empty, dotted, hatched and filled bars, respectively) for 20 hours. Relative (RLU) reporter-to-reference signals were determined for each sample and shown on the abscissa axis. Data are representative of two experiments that were performed.
Thus, we sought to optimize progastrin biotinylation using commercially available reagents (see Materials and Methods), monitoring the degree of biotinylation through electrophoresis after pre-incubation with streptavidin. We reasoned that biotinylated progastrin would interact avidly with streptavidin, resulting in a shift into a high molecular weight zone. Indeed, pre-incubation of unmodified progastrin with streptavidin did not change the electrophoretic mobility of either protein (Fig. 1B, lanes 2, 3). However, biotinylated progastrin was entirely shifted by the excess of streptavidin into a high molecular weight zone to form a major band of ~70kD. This matches well the size of a complex between one streptavidin molecule (52kD) and two bio-PG molecules (19kD) (Fig. 1B, lanes 6, 7; arrowhead). Curiously, tetrameric streptavidin migrated significantly slower (~160kD, lane 4) than may be expected from its known molecular weight (52 kD). This apparently reflects the formation of streptavidin aggregates [20] which are not easily disrupted by SDS in electrophoresis. Overall, these data rather suggest a biotinylation of each PG molecule with at least one biotin moiety under these experimental conditions.
Progastrin is known to stimulate some degree of proliferation in responsive epithelial cell lines under serum-free conditions [17]. To demonstrate that biotinylation of progastrin did not attenuate its biological function, we tested both biotinlated and unmodified PG in a reporter gene assay in PG-responsive IEC-18 cells. IEC-18 cells were transfected with the pSREx5-LUC reporter plasmid, which contains the luciferase gene under the control of a growth factor-responsive promoter containing five serum-response elements (SRE). Transfected cells were serum starved and stimulated with 1-100nM PG (or bio-PG) for 20 hours. We detected a prominent and similar stimulation of reporter activity in response to both peptides (Fig. 1C). Thus, the growth-factor signaling effect of original PG appears to be preserved in the biotinylated PG.
Optimization and analysis of biotinylated progastrin (bio-PG) binding to epithelial cells
Previous reports by our group and others have shown that transgenic mice over-expressing human progastrin exhibit markedly increased proliferation in their colonic epithelium [13, 21]. A growth-promoting effect by progastrin has also been demonstrated in vitro using a variety of colon cancer cell lines and immortalized intestinal epithelial cell lines. In order to investigate the potential utility of bio-PG for analysis of progastrin receptor expression, we first studied its specific binding to a panel of epithelial cell lines under different conditions. We initially selected a protocol that had previously been employed successfully for binding of recombinant iodinated progastrin (I125-PG) to intestinal epithelial cells [17]. Bio-PG (1-500nM) was incubated with 106 cells in 100μL HBSS buffer for 1h at 37°C, followed by washing in buffer and additional incubation with PE-labeled streptavidin (1:200) for 15 min. After two final washings and fixation (1% PFA), cells were analyzed by flow cytometry. However, in initial studies we found some nonspecific binding of bio-PG to many cell lines tested under these conditions (not shown). In addition, we found that the trypsin treatment, typically needed to detach adherent cells, strongly decreased the ability of IEC-6 and CHO-K1 cells to bind bio-PG (Fig. 2). Finally, binding of bio-PG to IEC-6 cells was strongly influenced by the confluency of the cells, with greater binding observed for cells grown to high confluency (data not shown).
Fig. 2. Bio-PG (100nM) binding is sensitive to protease treatment.

IEC-6 (top) or CHO-K1 (bottom) epithelial cells were detached from plastic support by either combined Trypsin-EDTA (Trypsin) treatment or EDTA treatment alone. Cells were processed for bio-PG/Streptavidin-PE labeling for 1 hour at 4°C. The stained cells were analyzed by flow cytometry as described in Material and Methods. The controls represent the fluorescent profiles of the same cells pre-incubated with streptavidin-PE reagent alone under the same conditions.
In order to increase the specificity and sensitivity of FACS-based assay for bio-PG binding, we introduced several modifications to our procedures. First, cells were grown to 90% confluency prior to harvest. Second, rather than using trypsin, adherent cells were gently detached by HyQtase (“Hyclone”) and incubated in complete medium with agitation for 1h before carrying out the binding assay. HyQtase was superior to EDTA in the generation of isolated, single cells that could be analyzed by flow cytometry. Third, 10% fetal calf serum was added to the binding buffer. Fourth, an additional signal amplification step was introduced after primary labeling, when the biotinylated goat anti-streptavidin antibodies (1:100) were added to cells, followed by secondary labeling with PE-conjugated streptavidin. As a result of these modifications, we were able to detect strong binding of bio-PG to the intestinal epithelial cell lines IEC-6 and IEC-18 (Fig. 3 and data not shown). The specificity of this binding was confirmed by vigorous competition with 30-fold excess of unmodified progastrin (Fig. 3, top panel). However, as noted in the figure, there was still some degree of residual bio-PG binding to IEC-6 cells, which may reflect incomplete competition or the existence of non-specific bio-PG interaction with additional cell surface molecules.
Fig. 3. Specific binding of bio-PG to the IEC-6 intestinal epithelial cell line.

(A). Competition of bio-PG binding to IEC-6 cells by an excess of unlabeled ligand. Cells were pre-incubated with 100 nM bio-PG alone (grey line) or in presence of 30- fold excess of PG (black heavy line) prior to labeling by phycoerythrin-conjugated streptavidin (Str-PE) followed by flow cytometry analysis. Control labeling by Str-PE alone is shown as a fine black line. (B). The presence of amidated gastrin (G-17) in the binding reaction does not compete for bio-PG binding to IEC-6 cells. Cells were processed as in (A) but G-17 was used instead of PG in competition experiments (heavy black line). (C). Glycine extended gastrin (G-gly) does not compete for bio-PG binding to IEC-6 cells. Cells were processed as in (A) but G-Gly was used instead of PG in competition experiment (heavy black line).
We applied our bio-PG binding assay to a panel of cancer lines (Fig. 4). As expected, we detected bio-PG interaction with the colonic cancer lines, HT-29 and COLO320. Unexpectedly, we also detected some degree of specific binding to non-intestinal epithelial cells, (COS-6, COS-7 > HEK293 > CHO-K1>AGS) suggesting the existence of an abundant low affinity receptor(s) on the surface of epithelial cells of varied origin (Fig. 4). Minimal or low levels of binding were detected for HeLa and DC2.4 cells under these conditions, and no binding could be detected for any hematopoietic cell line. Thus, progastrin binding appeared largely confined to permanent cell lines of presumed epithelial origin.
Fig. 4. Comparative binding of biotinylated progastrin to a panel of epithelial and hematopoietic (DC2.4) cells.
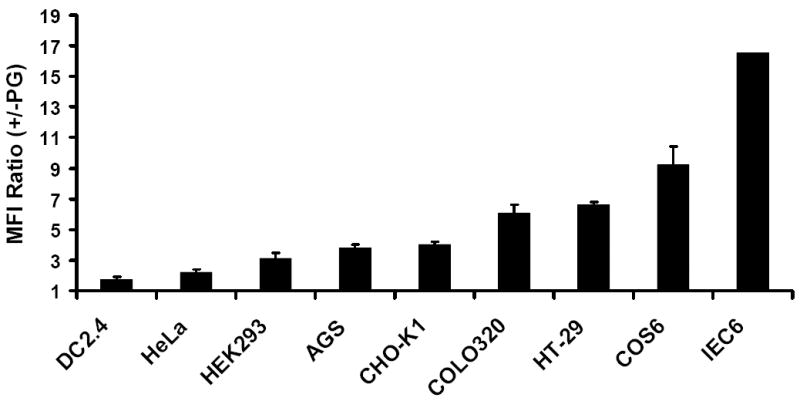
The indicated cells were pre-incubated with 100 nM of bio-PG for 1h followed by labeling with phycoerythrin –conjugated streptavidin (Str-PE). The stained cells were then analyzed by flow cytometry. Mean fluorescent intensities (relative to staining by Str-PE alone), which were calculated basing on five independent experiments, are shown.
Recently, a progastrin–interacting molecule, Annexin II, was identified by others in a biochemical screen of a PG-responsive cancer cell line [19]. Consequently, we wished to determine whether the same interaction of bio-PG with Annexin II could be detected on the surface of epithelial cells in our assays. Indeed, real-time qRT-PCR analysis revealed abundant expression of Annexin II mRNA in the tested epithelial cell lines and in GI tissues, comparable to the level of expression of a housekeeping gene, GAPDH (data not shown). Therefore, next we tested for expression of Annexin II protein on the surface of epithelial IEC-6 and IEC-18 cells by flow cytometry assay using the anti-human Annexin II antibody (hAnxa2) which had previously been used to detect surface Annexin II expression in macrophages [22]. We did not detect any significant binding of the antibody to the surface of epithelial cells under a variety of conditions (Fig. 5A and data not shown). In contrast, the same antibody did detect the presence of intracellular Annexin II in the same cells that have been fixed and permeabilized (Fig. 5B). Importantly, fixation alone did not stimulate anti-hAnxa2 antibody binding to the epithelial cell surface. Additionally, the presence of Annexin II antibodies (25 μg/ml) in the binding reaction did not affect bio-PG interaction with IEC-6 and IEC-18 cells (data not shown). These observations indicate that Annexin II protein expression is primarily localized to the cytosol in most epithelial cell lines examined, and thus is unlikely to mediate the binding of bio-PG in our assay.
Fig. 5. Detection of the surface (A) and cytosolic (B) Annexin II expression in IEC-18 cells by flow cytometry.
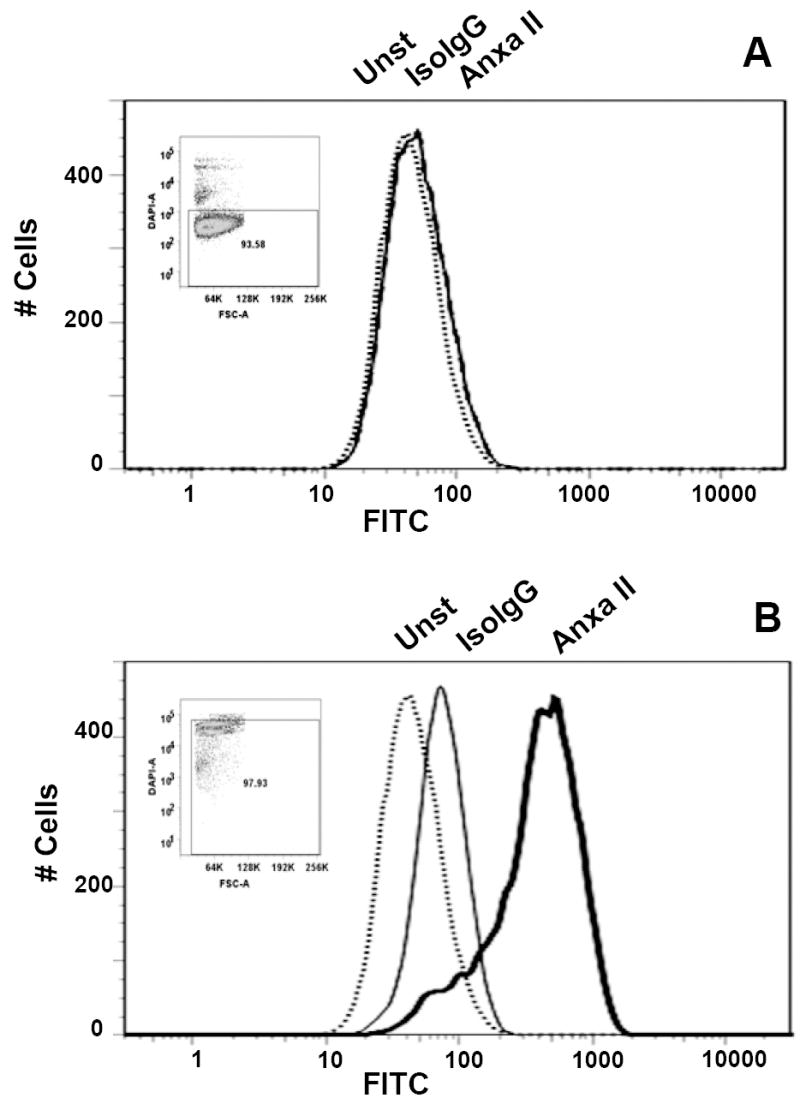
Cells were detached with 0.5 mM EDTA, and the cell suspension was directly used for labeling (A), or initially fixed and permeabilized by PFA (1.6%) and cold methanol, respectively, (B). Aliquots (106) of the cell suspensions were left untreated (Unst; dotted line) or incubated with the fluorescein-conjugated anti-Annexin II (ANXA II; heavy line) or isotype-matched (IsoIgG; fine line) antibodies. Cells were washed and resuspended in DAPI-containing buffer prior to analysis. Inserts show DAPI fluorescence (Y- axis) versus forward light scattering characteristics of the analyzed populations.
Bio-PG does not interact with the CCK-2 receptor
In theory, circulating progastrin could undergo proteolytic cleavage to give rise to more fully processed gastrin isoforms, such as amidated gastrin or glycine extended gastrin. In addition, due to the absence of cysteine bonds, progastrin may exist in several different conformational states in solution. This could expose the gastrin pentapeptide sequence known to interact with the amidated gastrin receptor, CCK-2, to the surface of cells. In order to examine the possibility that overexpressed progastrin may function through the CCK-2 receptor or through more processed isoforms, we first tested competition for bio-PG binding by G-gly and G-17. While a 30-fold excess of progastrin successfully competed for binding by bio-PG to IEC-6 cells (Fig. 3, top panel), no competition was observed with a 30-fold excess of amidated gastrin-17 or a 30-fold excess of G-gly (Fig. 3, middle and bottom panels). Curiously, in the latter case, we observed a slightly better interaction of bio-PG with IEC-6 cells, the maximum of which, however, did not exceed one of bio-PG binding alone. Overall these data suggest that the progastrin-interacting surface molecule(s) does not avidly bind G-gly or G-17.
To address the possible role of the CCK-2 receptor more directly, we employed AGS cells that stably express the CCK-2 receptor (AGS-E cells) [23]. Importantly, we found that both AGS-E and the parent AGS cell line bind bio-PG with equal efficiency (data not shown). Since FACS binding studies cannot exclude PG binding to CCK-B bearing AGS-E cells with extremely low affinity, we applied a sensitive Ca++- flux assay to AGS-E cells treated with either progastrin, amidated G-17, G-gly or a peptide corresponding to the carboxy-terminal 26 amino acid residues of human preprogastrin (carboxy-extended gastrin or Gas-CEP). As shown in Fig. 6, a low concentration (20nM) of amidated G-17 stimulated a robust Ca++ flux in AGS-E cells, similar in range to that observed with ionomycin. Only a very weak Ca++ flux was seen with 10μM of G-gly (Fig. 6A), while no Ca++ response could be observed with 10- 20μM concentrations of progastrin (or bio-PG; data not shown). Curiously, Gas-CEP stimulated a detectable Ca++- flux, albeit at concentrations ~10 fold higher than required for amidated gastrin (Fig. 6A, B). Importantly, none of the peptides induced any detectable Ca++ flux in the parent AGS cell line not expressing the CCK-2 receptor (data not shown). These data strongly suggest that unprocessed progastrin does not bind or signal through the CCK-B receptor. Thus, the biologic activity of PG is likely determined by a distinct receptor.
Fig. 6. Calcium flux stimulated by various progastrin-derived peptides in AGS-E cells expressing the CCK-2 (gastrin) receptor.
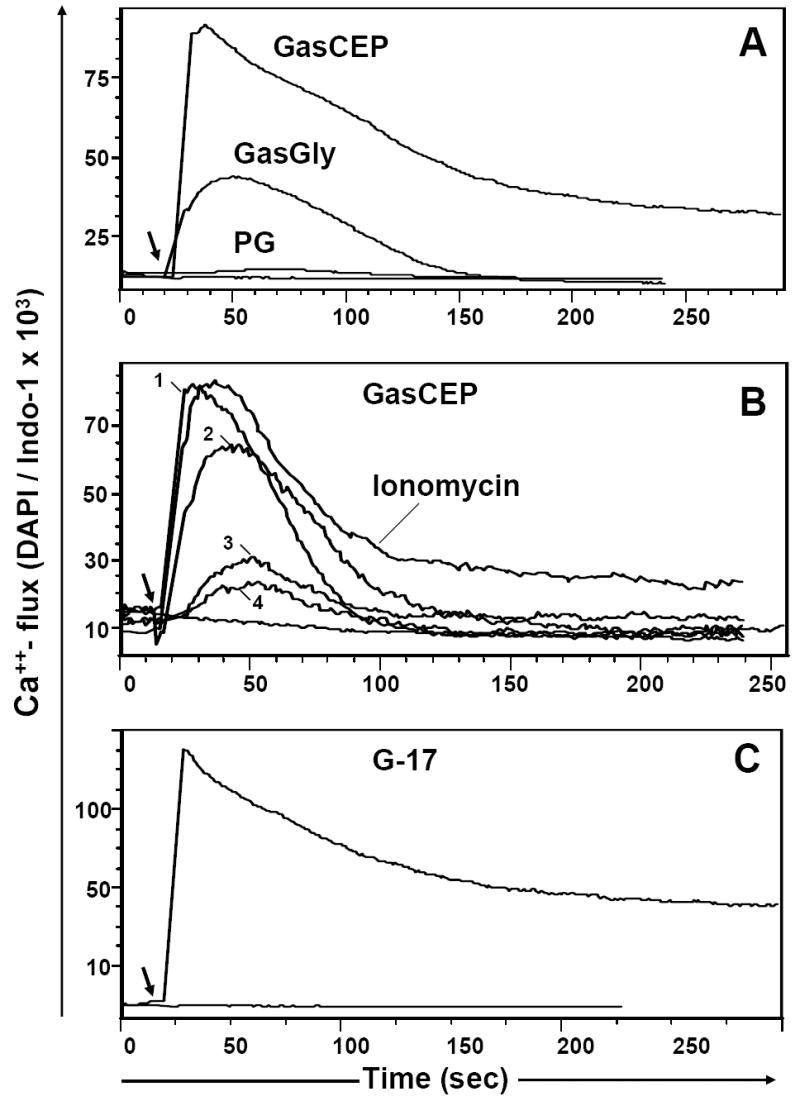
(A). Ca++ - flux in response to 10 μM of the following peptides: C-terminally extended gastrin (GasCEP), Gly-extended gastrin (G-gly) or progastrin (PG); (B). CCK-2R- dependent Ca++ flux in AGS/CCK-B cells treated with various concentrations of Gas-CEP peptide (nM): 5000(1), 500(2), 50(3) or 20(4) and Ca++ ionophore, ionomycin (1μg/ml); (C). Effective Ca++ - flux in response to the completely processed hormone, amidated gastrin (20 nM). The baseline Ca++ level in loaded AGSE cells is shown on each graph. The stimulant was added at the time indicated by the arrow. Representative data for one of three performed experiments is shown.
Progastrin interacts with surface glycosaminoglycans (GAG)
During the process of optimizing the conditions for bio-PG binding to cultured cell lines, we observed that binding could be markedly inhibited by certain agents, such as high salt (300nM NaCl), spermidine (5μM) or polybrene (10μg/ml), that are highly positively charged and typically exhibit strong hydrophilic interactions (not shown). This suggested the possibility of an electrostatic interaction between the positively charged region(s) of bio-PG and a negatively-charged cellular membrane component, which could then be competed by cationic molecules.
Recently, important electrostatic interactions between several chemokine receptor ligands and sulfated (and thus negatively charged) glycosaminoglycans (GAGs) has been demonstrated [24]. The biologic functions of chemokines are thought to be influenced by their association with cellular or extracellular matrix GAGs. Usually attached to a core protein to form proteoglycans, the common GAGs (heparin, heparan sulfate (HS), dermantan sulfate, and chondroitin sulfate (CS)) are highly sulfated oligosaccharides. With the exception of CS, all these GAGs are characterized by a high degree of structural heterogeneity. Another common GAG is hyaluronic acid, but it is never attached to a core protein and is not a GAG sulfate [24].
A number of reports have suggested that GAGs, through enhancing local concentrations of chemokines and growth factors, promote the interaction of these ligands with surface receptors. Thus, we hypothesized that similar electrostatic interactions may exist for progastrin with surface/extracellular matrix GAG’s. To address this issue, we tested whether bio-PG binds to epithelial CHO-K1 cells, and found robust binding (Fig. 7A, left column). The capacity of bio-PG to interact with proteoglycans was then investigated using CHO-K1-derived pgsB618 and pgsD677 cells which lack galactosyltransferase I (pgsB618) or N-acetylglucosaminyltransferase and glucuronyltransferase I (pgsD677) activities. Consequently, they are deficient for the synthesis of any GAG or HS, respectively. In sharp contrast to the parental CHO-K1 cells, little or no bio-PG binding could be detected with these cells, even when high concentrations of the bio-PG peptide (up to 500nM) were used (Fig. 7A, two right bars). In addition, to confirm the above findings, we treated CHO-K1, IEC-6, IEC-18 and colonic HT-29 cells with heparinase. Indeed, this treatment resulted in a decrease in bio-PG binding to these cells (Fig. 7B). However, heparinase treatment had a minimal effect on the-PG stimulating effect on the reporter activity of the growth responsive reporter plasmid, pSREx5-LUC in IEC-18 cells (data not shown). Thus, the interaction of PG with GAGs does not appear to play a major role in PG signaling.
Fig. 7. Bio-PG binding is dependent on surface proteoglycans.
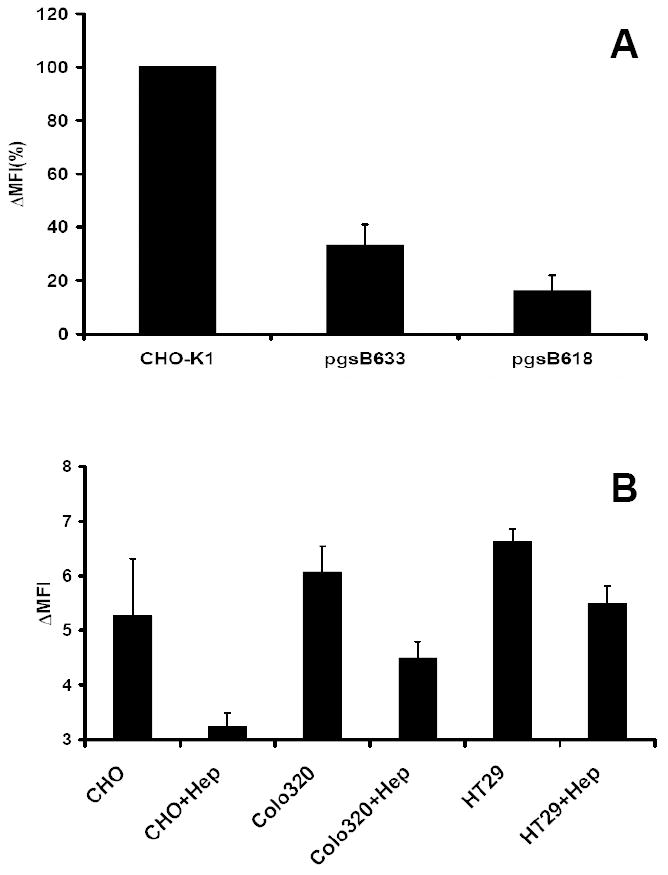
(A). The wild type and proteoglycan-deficient strains of CHO-K1 cells were analyzed for bio-PG (100 nM) binding by flow cytometry as above, except that the binding reaction was performed in HBSS- 1% BSA. The Mean Fluorescence Intensity was calculated and presented as % of MFI for wild type CHO-K1 cells (MFI=100%). The figure shows combined data from three separate experiments. (B). Heparinase pre-treatment attenuates bio-PG to epithelial cells. The indicated cells were treated (+Hep) or not with heparinase (1000U/ml) for 1.5 hours at 37°C before testing bio-PG binding as above. Data (in triplicate) for one of two performed experiments. Statistically significant (P<0.05) decrease of bio-PG binding was found in treated (+Hep) versus untreated samples.
Binding of bio-PG to primary mouse gut cells
While a number of studies have indicated that progastrin is upregulated in colon cancer cells where it may stimulate autocrine growth, evidence from transgenic animals suggests that progastrin also stimulates proliferation of normal intestinal epithelial cells. Indeed, proliferative effects of progastrin were first recognized in progastrin-overexpressing human gastrin (hGAS) transgenic mice that showed increased circulating levels of PG [25]. However, these in vivo observations cannot be used to conclude that progastrin’s target in the colon is necessarily an epithelial cell, since indirect or paracrine effects remain a possibility. In order to characterize further the PG-binding cells in the normal mouse gut, we tested whether a specific bio-PG binding could be detected in colonic epithelial cells isolated from WT (C57BL/6) mice. We used a gentle treatment of freshly isolated mouse colonic mucosa with dispase to dissociate cells while avoiding damage to surface membrane receptors. As an internal control for receptor functionality, we monitored the labeling of lymphocyte receptors (CD3, CD4, B220, CD45) following tissue dissociation, assuming that effective lymphocyte labeling would indicate integrity of the putative PG receptor (data not shown). Analysis of bio-PG binding in a single cell suspension of unchallenged wild type mouse colonic cells revealed the presence of ~9 % labeled cells (Fig. 8, left panel, heavy line). Importantly, no detectable binding of the secondary reagent - streptavidin coupled to phycoerythrin - was observed (Fig. 8, left panel, thin line). Bio-PG binding to normal mouse colonic epithelial cells could be effectively competed by an excess of unlabeled PG (Fig. 8, left panel, dotted line), consistent with specific binding.
Fig. 8. FACS analysis of bio-PG binding to viable mouse colonic cells isolated from wild type (WT, left panel) and gastrin knockout (Gas-KO, right panel) C57BL/6 mice.
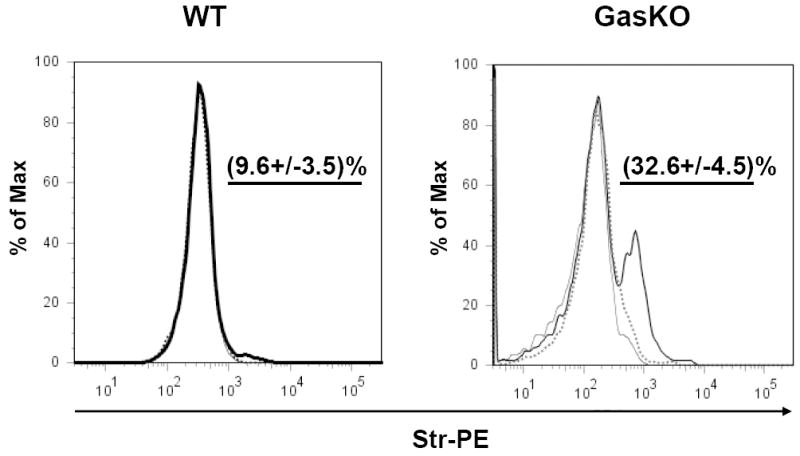
Colons were dissociated and the resultant single cell suspensions were pre-incubated with 100 nM of bio-PG alone (heavy line) or in the presence of 50-fold excess of unmodified PG (dotted line) followed by incubation with the secondary reagent, Str-PE. Cells stained with Str-PE alone are shown as a thin line. Labeled cells were analyzed with flow cytometry. The mean percentage (and standard deviation) of the bio-PG-interacting (PE-positive) viable cells for three separate experiments, is shown numerically in each panel above the gated subpopulation (straight line). Dead (DAPI-stained) cells were excluded from this analysis by appropriate gating (not shown).
Several recent studies have suggested that gastrin deficient mice are highly responsive to exogenously administered progastrin [14, 21, 26]. Thus, we wondered whether gastrin deficiency might enhance PG binding due to upregulation of the putative PG receptor. To address this question, we prepared a single cell suspension from the colon of gastrin knockout mice (GAS-KO mice in a C57BL/6 background), and treated these cells with bio-PG and streptavidin-PE followed by FACS. Dead or necrotic cells were excluded from the analysis by DAPI or 7AAD stains. We observed a significant increase (up to 30%) in the proportion of bio-PG labeled cells from the colons of GAS-KO mice (Fig. 8, right panel). Importantly, sorting of colonic mucosal cells using the hematopoietic specific marker CD45 revealed no binding of progastrin by CD45+ cells, consistent with the overall conclusion that bio-PG binding was specific for colonic epithelial cells (Fig. 9, bottom right panel).
Fig. 9. FACS analysis of bio-PG binding to colonic lymphocytes isolated from gastrin knockout mice.
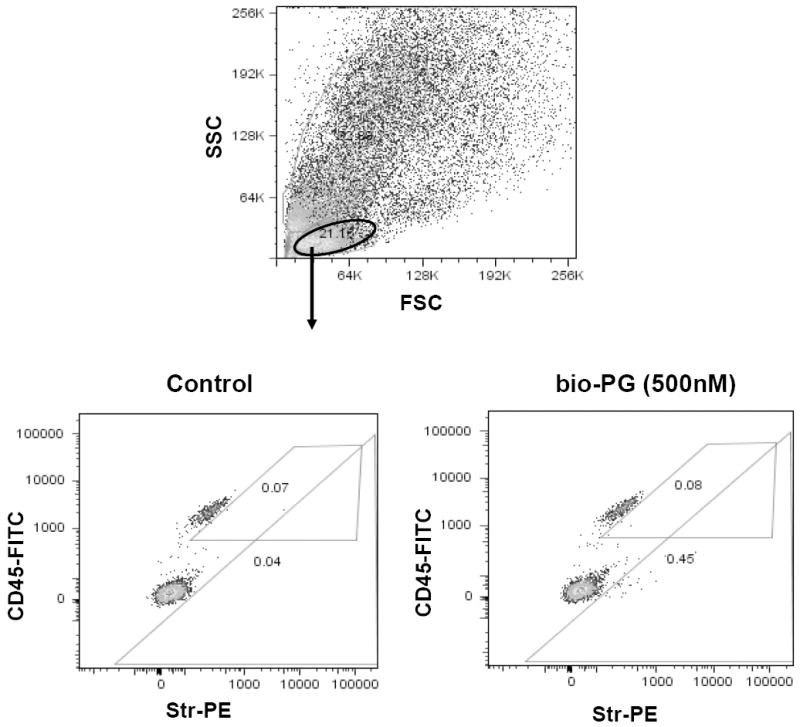
Single cell suspension was prepared from colons of two Gas-KO mice as described in Material and Methods. Experimental sample (106 cells) was successively incubated with bio-PG (100nM), str-PE (1:100) and CD45-FITC (0.2μg) in 100μl binding buffer for 30-60 minutes at room temperature in the dark. Control sample was stained in the same way but incubation with bio-PG was omitted. Stained samples were washed, resuspended in DAPI containing buffer (PBS) and processed by flow cytometry. Dead (DAPI stained) cells (~30%) were excluded from this analysis by appropriate gating (not shown). (Top) Representative forward/side (light) scattering characteristics of dissociated colonic cells in control sample. Similar data were obtained for experimental sample (not shown). The oval gate shows the lymphocyte-containing cell fraction which was analyzed in the bottom panels. (Bottom) The lymphocyte fractions for the control (left) or experimental (right) sample, were concomitantly analyzed for CD45 expression (FITC fluorescence; ordinate) and bioPG/str-PE binding (PE fluorescence; abscissa). The numbers in respective gates indicate a proportion of PE-positive cells in the CD45+ or CD45- subpopulations. No significant binding of bio-PG/Str-PE to CD45+ cells was detected (0.08% on the right experimental panel versus 0.07% on left control panel).
DISCUSSION
Here we report the characterization of progastrin binding to permanent cell lines and primary colonic cells using a novel non-radioactive, flow cytometry-based assay. Studies with permanent cell lines showed strong binding to several epithelial cell lines (IEC-6 > COS-6 > AGS) but not to two hematopoietic cell lines in good agreement with previous reports [17, 27]. Progastrin binding occurred independent of the gastrin/CCK-2 receptor and could not be competed away by either amidated gastrin-17 or glycine-extended gastrin. In addition, progastrin binding appeared to be partially dependent on charged glycosaminoglycans. Using the same approach, we identified a subpopulation of mouse colonic epithelial cells that interacted avidly with biotinylated PG ex vivo. Interestingly, this colonic subpopulation is significantly enriched (3-7 fold) in gastrin knockout mice, suggesting upregulation in the absence of the progastrin ligand. Taken together, these data indicate that colonic epithelial cells are one of the primary targets of PG binding.
Several reports have previously suggested the possibility that the incompletely processed forms of gastrin, such as G-gly, may be able to bind or stimulate the CCK-2 receptor [28-30], although most studies have supported the existence of a novel receptor. Nevertheless, in order to address the possible role of the CCK-2/gastrin receptor in progastrin binding, we compared binding of bio-PG to AGS cells with or without CCK-2 receptor expression. Binding of bio-PG occurred independent of CCK-2 receptor expression, in contrast to amidated gastrin that could bind only to the CCK-2-expressing AGS cells. Additionally, in contrast to amidated gastrin, PG did not stimulate Ca++ flux in AGS-E cells expressing the CCK-2 receptor, even at supra physiological (μM) concentrations. Neither amidated G-17 nor glycine-extended gastrin (G-Gly) was able to compete for binding with bio-PG suggesting the involvement of a distinct, non-CCK-2 receptor. Similar, small molecule antagonists (such as L365,270) were not able to compete for this binding (data not shown). Overall, these data indicate quite clearly that PG binding in these cell lines is mediated via a distinct, non-CCK-2 receptor.
Our study reveals for the first time that the PG molecules can effectively interact with negatively-charged proteoglycans modified by heparan sulfate. CHO-K1-derived cell lines that lack galactosyltransferase I or N-acetylglucosaminyltransferase and glucuronyltransferase I showed decreased binding of bio-PG compared to the parent CHO-K1 cell line. In addition, treatment of epithelial cells with heparinase also reduced binding by bio-PG. The interaction between PG and the GAGs is likely an electrostatic interaction involving a stretch of several positively charged amino acids (#34-37, HHRR) in the PG peptide, similar to that which has been demonstrated for several chemokines and growth factors [24]. While the exact identity and function of these heparin sulfate–modified molecules remains to be established, it is pertinent to speculate that syndecans and/or glypicans, well known co-receptors for several growth factors and chemokines, are reasonable candidate for mediating these interactions [25].
Although physiologic effects of progastrin have been reproducibly demonstrated through in vivo models, and binding of PG to cell lines confirm in vitro, the precise identity of the progastrin receptor(s) has remained elusive. Recently, the cloning of one potential PG receptor was reported by Singh et al by using colon cancer cells lines [19]. Using a straightforward biochemical approach involving the crosslinking of radiolabeled gastrins to membrane proteins followed by mass spectrometry, the protein Annexin II (ANXA II) was identified. While Annexin II is classically a cytosolic or secreted molecule rather than surface receptor, the data to date supports a role for Annexin II in the growth response to progastrin and additional investigation is clearly warranted. However, our FACS-based assays with bio-PG did not reveal any significant binding of PG to the surface of HeLa cells expressing Annexin II [31]. In addition, Annexin II is not upregulated in the colon of Gas-KO mice (unpublished data). Finally, we did not detect Annexin II expression on the surface of IEC-6 or IEC-18 epithelial cells by flow cytometry. The FACS-based assay, however, strongly depends on the quality of antibodies used which potentially may not recognize a specific conformation of Annexin II on the surface of epithelial cells. Thus, keeping in mind the latter reservation, our data taken together strongly suggest the existence of another cell surface molecule(s) on colonic cells that mediates the initial interactions with progastrin.
Recent studies by Ottewell et al [14] suggest that gastrin deficient (GAS-KO) mice are highly responsive to the infusion of synthetic progastrin. Thus, we hypothesized that gastrin deficient mice might show increased expression of the progastrin receptor. Indeed, we detected increased numbers of colonic epithelial cells that bind bio-PG in gastrin knockout mice. The increased detection of the PG receptor could be due to an increased level of receptor expression or an actual expansion of a colonic lineage expressing the receptor. Given that several GPCR ligands have been shown to induce receptor internalization and degradation even in the absence of downstream signaling ([32] and references therein), the simplest explanation is that ligand deficiency (in the GAS-KO animals) results in the stabilization of surface receptor expression. Alternatively, progastrin (or other forms of gastrin) could potentially stimulate differentiation of the small population of PG binding cells present in the normal colon. Further studies will be needed to characterize the PG-binding molecules in the colon and their potential regulation by progastrin-derived peptides.
Acknowledgments
Authors are grateful to Dmitry Kolpashchikov for HPLC analysis of progastrin. This work was supported by the NIH through the grant # 5 R01 DK052778 to Timothy C. Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kopin AS, et al. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc Natl Acad Sci U S A. 1992;89(8):3605–9. doi: 10.1073/pnas.89.8.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dockray GJ, et al. The gastrins: their production and biological activities. Annu Rev Physiol. 2001;63:119–39. doi: 10.1146/annurev.physiol.63.1.119. [DOI] [PubMed] [Google Scholar]
- 3.Ferrand A, Wang TC. Gastrin and cancer: a review. Cancer Lett. 2006;238(1):15–29. doi: 10.1016/j.canlet.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Kochman ML, et al. Post-translational processing of gastrin in neoplastic human colonic tissues. Biochem Biophys Res Commun. 1992;189(2):1165–9. doi: 10.1016/0006-291x(92)92326-s. [DOI] [PubMed] [Google Scholar]
- 5.Nemeth J, et al. Identification of progastrin derived peptides in colorectal carcinoma extracts. Gut. 1993;34(1):90–5. doi: 10.1136/gut.34.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Solinge WW, et al. Expression but incomplete maturation of progastrin in colorectal carcinomas. Gastroenterology. 1993;104(4):1099–107. doi: 10.1016/0016-5085(93)90279-l. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin GS, Shulkes A. Gastrin as an autocrine growth factor in colorectal carcinoma: implications for therapy. World J Gastroenterol. 1998;4(6):461–463. doi: 10.3748/wjg.v4.i6.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh P, et al. Gastrin gene expression is required for the proliferation and tumorigenicity of human colon cancer cells. Cancer Res. 1996;56(18):4111–5. [PubMed] [Google Scholar]
- 9.Baldwin GS, et al. Biologically active recombinant human progastrin(6-80) contains a tightly bound calcium ion. J Biol Chem. 2001;276(11):7791–6. doi: 10.1074/jbc.M009985200. [DOI] [PubMed] [Google Scholar]
- 10.Hollande F, et al. Glycine-extended gastrin acts as an autocrine growth factor in a nontransformed colon cell line. Gastroenterology. 1997;113(5):1576–88. doi: 10.1053/gast.1997.v113.pm9352860. [DOI] [PubMed] [Google Scholar]
- 11.Seva C, Dickinson CJ, Yamada T. Growth-promoting effects of glycine-extended progastrin. Science. 1994;265(5170):410–2. doi: 10.1126/science.8023165. [DOI] [PubMed] [Google Scholar]
- 12.Singh P, et al. Incomplete processing of progastrin expressed by human colon cancer cells: role of noncarboxyamidated gastrins. Am J Physiol. 1994;266(3 Pt 1):G459–68. doi: 10.1152/ajpgi.1994.266.3.G459. [DOI] [PubMed] [Google Scholar]
- 13.Wang TC, et al. Processing and proliferative effects of human progastrin in transgenic mice. J Clin Invest. 1996;98(8):1918–29. doi: 10.1172/JCI118993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ottewell PD, et al. COOH-terminal 26-amino acid residues of progastrin are sufficient for stimulation of mitosis in murine colonic epithelium in vivo. Am J Physiol Gastrointest Liver Physiol. 2005;288(3):G541–9. doi: 10.1152/ajpgi.00268.2004. [DOI] [PubMed] [Google Scholar]
- 15.Brown D, et al. pp60c-Src Kinase mediates growth effects of the full-length precursor progastrin1-80 peptide on rat intestinal epithelial cells, in vitro. Endocrinology. 2003;144(1):201–11. doi: 10.1210/en.2002-220501. [DOI] [PubMed] [Google Scholar]
- 16.Hollande F, et al. Adherens junctions and tight junctions are regulated via different pathways by progastrin in epithelial cells. J Cell Sci. 2003;116(Pt 7):1187–97. doi: 10.1242/jcs.00321. [DOI] [PubMed] [Google Scholar]
- 17.Singh P, et al. Progastrin1-80 stimulates growth of intestinal epithelial cells in vitro via high-affinity binding sites. Am J Physiol Gastrointest Liver Physiol. 2003;284(2):G328–39. doi: 10.1152/ajpgi.00351.2002. [DOI] [PubMed] [Google Scholar]
- 18.Karelina Y, Baldwin GS. Binding sites for progastrin-derived peptides in colonic crypts. J Gastroenterol Hepatol. 2001;16(2):169–75. doi: 10.1046/j.1440-1746.2001.02429.x. [DOI] [PubMed] [Google Scholar]
- 19.Singh P, et al. Annexin II binds progastrin and gastrin-like peptides, and mediates growth factor effects of autocrine and exogenous gastrins on colon cancer and intestinal epithelial cells. Oncogene. 2007;26(3):425–40. doi: 10.1038/sj.onc.1209798. [DOI] [PubMed] [Google Scholar]
- 20.Bayer EA, et al. Postsecretory modifications of streptavidin. Biochem J. 1989;259(2):369–76. doi: 10.1042/bj2590369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ottewell PD, et al. Progastrin stimulates murine colonic epithelial mitosis after DNA damage. Gastroenterology. 2003;124(5):1348–57. doi: 10.1016/s0016-5085(03)00288-9. [DOI] [PubMed] [Google Scholar]
- 22.Fan X, et al. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol Biol Cell. 2004;15(6):2863–72. doi: 10.1091/mbc.E03-09-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noble PJ, et al. Stimulation of gastrin-CCKB receptor promotes migration of gastric AGS cells via multiple paracrine pathways. Am J Physiol Gastrointest Liver Physiol. 2003;284(1):G75–84. doi: 10.1152/ajpgi.00300.2002. [DOI] [PubMed] [Google Scholar]
- 24.Amara A, et al. Stromal cell-derived factor-1alpha associates with heparan sulfates through the first beta-strand of the chemokine. J Biol Chem. 1999;274(34):23916–25. doi: 10.1074/jbc.274.34.23916. [DOI] [PubMed] [Google Scholar]
- 25.Alexopoulou AN, Multhaupt HA, Couchman JR. Syndecans in wound healing, inflammation and vascular biology. Int J Biochem Cell Biol. 2007;39(3):505–28. doi: 10.1016/j.biocel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 26.Cobb S, et al. Deletion of functional gastrin gene markedly increases colon carcinogenesis in response to azoxymethane in mice. Gastroenterology. 2002;123(2):516–30. doi: 10.1053/gast.2002.34754. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Owlia A, Singh P. Precursor peptide progastrin(1-80) reduces apoptosis of intestinal epithelial cells and upregulates cytochrome c oxidase Vb levels and synthesis of ATP. Am J Physiol Gastrointest Liver Physiol. 2003;285(6):G1097–110. doi: 10.1152/ajpgi.00216.2003. [DOI] [PubMed] [Google Scholar]
- 28.Stubbs M, et al. CCK2 gastrin receptor as a potential target for therapy in leukaemia cell lines. Oncol Rep. 2005;14(4):1055–8. doi: 10.3892/or.14.4.1055. [DOI] [PubMed] [Google Scholar]
- 29.Artru P, et al. Gastrin-17 and G17-gly induce proliferation of LoVo cells through the CCK B/gastrin receptor. Gastroenterol Clin Biol. 1998;22(67):607–12. [PubMed] [Google Scholar]
- 30.Iwase K, et al. Regulation of growth of human gastric cancer by gastrin and glycine-extended progastrin. Gastroenterology. 1997;113(3):782–90. doi: 10.1016/s0016-5085(97)70172-0. [DOI] [PubMed] [Google Scholar]
- 31.Sullivan DM, et al. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39(36):11121–8. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 32.Feng Z, et al. Cutting edge: human beta defensin 3--a novel antagonist of the HIV-1 coreceptor CXCR4. J Immunol. 2006;177(2):782–6. doi: 10.4049/jimmunol.177.2.782. [DOI] [PubMed] [Google Scholar]