

Abstract
Background
O-Linked N-acetylglucosaminylation (O-GlcNAcylation) plays a role in many aspects of protein function. Whereas elevated O-GlcNAc levels contribute to diabetes related end-organ damage, O-GlcNAcylation is also physiologically important. Because proteins that play a role in vascular tone regulation can be O-GlcNAcylated, we hypothesized that O-GlcNAcylation increases vascular reactivity to constrictor stimuli.
Methods and Results
Aortas from male Sprague-Dawley rats and C57BL/6 mice were incubated for 24 h with vehicle or PugNAc (O-GlcNAcase inhibitor, 100μM). PugNAc incubation significantly increased O-GlcNAc-proteins, as determined by Western blot. PugNAc also increased vascular contractions to phenylephrine and serotonin, an effect not observed in the presence of L-NAME or in endothelium-denuded vessels. Acetylcholine-induced relaxation, but not that to sodium nitroprusside was decreased by PugNAc treatment, an effect accompanied by decreased levels of phosphorylated eNOSSer-1177 and AktSer-473.
Conclusion
Augmented O-GlcNAcylation increases vascular reactivity to constrictor stimuli, possibly due to its effects on eNOS expression and activity, reinforcing the concept that O-GlcNAcylation modulates vascular reactivity and may play a role in pathological conditions associated with abnormal vascular function.
Keywords: O-Linked N-acetylglucosaminylation (O-GlcNAc), PugNAc, eNOS, vascular reactivity
INTRODUCTION
O-linked attachment of N-acetyl-glucosamine (O-GlcNAc) on serine and threonine residues of nuclear and cytoplasmic proteins is a highly dynamic post-translational modification that plays a key role in signal transduction pathways. Numerous proteins, including kinases, phosphatases, transcription factors, and cytoskeletal proteins have been identified as targets of O-GlcNAc modification (1, 2). The cycling of O-GlcNAc on target proteins is controlled by two enzymes, O-GlcNAc transferase (OGT or uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase; UDP-NAc transferase) and β-N-acetylglucosaminidase (O-GlcNAcase). Whereas OGT catalyses the addition of O-GlcNAc to the hydroxyl group of serine or threonine residues of a target protein, O-GlcNAcase catalyses the hydrolytic cleavage of O-GlcNAc from post-translationally modified proteins (1, 2). The overall catalytic activity of OGT is controlled by the concentrations of its donor substrate, uridine 5′-diphosphate-N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is highly sensitive to fluxes in nutrients and energy, mainly to flux through the hexosamine biosynthetic pathway (HBP). It is found in high concentrations (is second only to that of ATP), and 2–5% of all cellular glucose is used to generate this sugar nucleotide (3, 4).
Almost every functional class of proteins, but mainly those involved in transcription or translation and in stress responses and energy metabolism, are subject to O-GlcNAc (1, 2). O-GlcNAcylation can either suppress or enhance transcription, depending on the promoter involved and other associated proteins. Proteins with an important role on vascular function, such as endothelial nitric oxide synthase (eNOS), sarcoplasmic reticulum Ca2+-ATPase (SERCA), phospholipase C (PLC), protein kinase C (PKC), and phosphoinositide-3 kinase (PI3K) are involved in cytoskeletal regulation. Further, these proteins are also targets for O-GlcNAcylation (1, 2), suggesting that this post-translational modification may play an important role in vascular reactivity.
Since the effects of increased O-GlcNAcylation on vascular proteins and vascular reactivity have not been addressed, the aim of the present study was to investigate whether changes in O-GlcNAcylation modifies vascular reactivity. We hypothesized that augmented vascular O-GlcNAc proteins increases reactivity to constrictor stimuli. To address our hypothesis we have used a pharmacological approach to increase O-GlcNAcylation: O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino- N-phenylcarbamate, or PugNAc, which blocks O-GlcNAcase activity by mimicking the enzyme-stabilized transition state (5–11). In addition, the effects of PugNAc on vascular reactivity were addressed in two species, rat and mouse.
METHODS
Animals
Eight week-old adult male Sprague-Dawley rats (230–250g; Harlan Laboratories, Indianapolis, IN) and C57BL/6 mice (12 weeks-old, 25–30 g; Harlan, Indianapolis, IN) were used in this study. All procedures were performed in accordance with the Guiding Principles in the Care and Use of Animals, approved by the Medical College of Georgia Committee on the Use of Animals in Research and Education. The animals were housed four per cage on a 12-h light/dark cycle and fed a standard chow diet with water ad libitum.
Vascular functional studies
After euthanasia, the thoracic aorta was removed and cleaned from fat tissue in an ice-cold physiological salt solution (PSS), containing (mM): NaCl, 130; NaHCO3, 14.9; KCl, 4.7; KH2PO4, 1.18; MgSO4·7H2O 1.18; CaCl2·2H2O, 1.56, EDTA, 0.026, glucose 5.5. Segments of thoracic aorta were incubated in Eagle’s Minimum Essential Medium (EMEM) containing L-glutamine (1%), fetal bovine serum (10%), penicillin and streptomycin (0,5%), and incubated with vehicle (methanol) or PugNAc (100μM) during 24h. Following incubation, thoracic aortas (4 mm in length) were carefully mounted as ring preparations on standard organ chambers for isometric tension recordings by a PowerLab 8/SP data acquisition system (ADInstruments Pty Ltd., Castle Hill, Australia). The tissue was continuously bubbled with 95% O2 and 5% CO2 and maintained at 37°C, under a resting tension (30mN for rats; 5mN for mice). After a 45 min equilibration period aorta integrity was assessed first by stimulation of vessels with potassium chloride (KCl - 120 mM) and, after washing and a new stabilization period, by contracting the segments with phenylephrine (PE; 1μM) followed by stimulation with acetylcholine (ACh; 10μM).
Concentration-response curves to PE (1nM to 100μM) and serotonin [5-hydroxytriptamine (5-HT), 1nM to 100μM] were performed to evaluate vascular contractility, both in the presence and absence of a NO synthase inhibitor, Nω-nitro-L-arginine methyl ester (L-NAME 100μM) for 40 minutes. Endothelium-dependent relaxation was assessed by measuring the relaxation response to ACh (1nM to 100μM) and endothelium-independent relaxation was assessed by measuring the relaxation response to sodium nitroprusside (SNP 0.1nM to 10μM) in PE-contracted vessels (1μM).
Western Blot Analysis
Proteins (60 μg) extracted from aorta were separated by electrophoresis on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% skim milk in Tris-buffered saline solution with Tween (10%) for 1 hour at 24°C. Membranes were then incubated with antibodies overnight at 4°C. Antibodies were as follows: anti-O-GlcNAc antibody, CTD 110.6 (1:2000; Pierce Biotechnology, USA), total eNOS, Akt and PI3K [(1:1000) Cell Signaling Technology, Inc], beta-actin [(1:10000), Sigma-Aldrich, Inc]. Immunoblots for nonphosphoproteins were carried out in the same membranes used to evaluate their phosphorylated forms: p-eNOS (Ser1177), eNOS (Thr495), p-Akt (Ser473) and p-PI3K (Tyr458) [(1:400); Cell Signaling Technology, Inc]. O-GlcNAc transferase (OGT) protein expression (1:400, Santa Cruz antibodies) was also evaluated.
After incubation with secondary antibodies, signals were developed with chemiluminescence, visualized by autoradiography, and quantified densitometrically. Results are normalized to beta-actin protein and expressed as arbitrary units.
Data Analysis
The results are shown as mean ± SEM (n), where n represents the number of animals used in the experiments. Contractions were recorded as changes in the displacement (mN) from baseline. Relaxation is expressed as percent change from the PE contracted levels. Concentration–response curves were fitted using a nonlinear interactive fitting program (Graph Pad Prism 3.0; GraphPad Software Inc., San Diego, CA, U.S.A.) and two pharmacological parameters were obtained: the maximal effect generated by the agonist (or Emax) and -log EC50 (or pD2). Statistical analyses of Emax and pD2 values were performed using one-way ANOVA or Student’s t-test. Post hoc comparisons were performed using Newman-Keuls’s test. Western blot data were analyzed by one-sample t test and the P value was computed from the t ratio and the numbers of degrees of freedom. Values of P<0.05 were considered statistically significant.
RESULTS
In order to understand the role of O-GlcNAcylation in relation to vascular function, we used PugNAc, a potent inhibitor of O-GlcNAcase (5–11), to increase O-GlcNAcylation levels. Rat thoracic aortas incubated with PugNAc (100μM) for 24 hours displayed increased vascular content of O-GlcNAc-proteins (Fig. 1). OGT expression was decreased after treatment with PugNAc (Fig. 2), suggesting a compensatory mechanism for the increased vascular O-GlcNAcylation. Our next aim was to investigate if increased amounts O-GlcNAc-proteins lead to changes in vascular reactivity.
Figure 1. PugNAc increases the content of O-GlcNAc-proteins in rat aorta.

On the top, representative Western blot image of O-GlcNAc-proteins; on the bottom, corresponding bar graphs showing the relative O-GlcNAc-proteins after normalization to β-actin expression. Results are presented as mean ± SEM for n=4 in each experimental group. *, p<0.05 vs. vehicle (methanol).
Figure 2. PugNAc decreases OGT expression in rat aorta.

On the top, representative Western blot image of OGT; on the bottom, corresponding bar graphs showing the relative OGT after normalization to β-actin expression. Results are presented as mean ± SEM for n=4 in each experimental group. *, p<0.05 vs. vehicle (methanol).
Vascular reactivity to the alpha1-adrenergic agonist PE was evaluated. PE-induced contraction was augmented in rat aortas incubated with PugNAc (Fig. 3A), when compared with vehicle incubation. Treatment with L-NAME (100μM) augmented vascular responses to PE and abolished the differences in PE-response between the groups (Fig. 3B). Similarly, no differences in PE reactivity were observed between endothelium-denuded aortas treated with PugNAc or vehicle (Fig. 3C). PugNAc incubation did not change contraction induced by 120 mM KCl [Emax (mN) = 24.3±1.2 vehicle vs. 28.5±2.2 PugNAc; n=6].
Figure 3. PugNAc enhances contraction to PE in rat aorta.
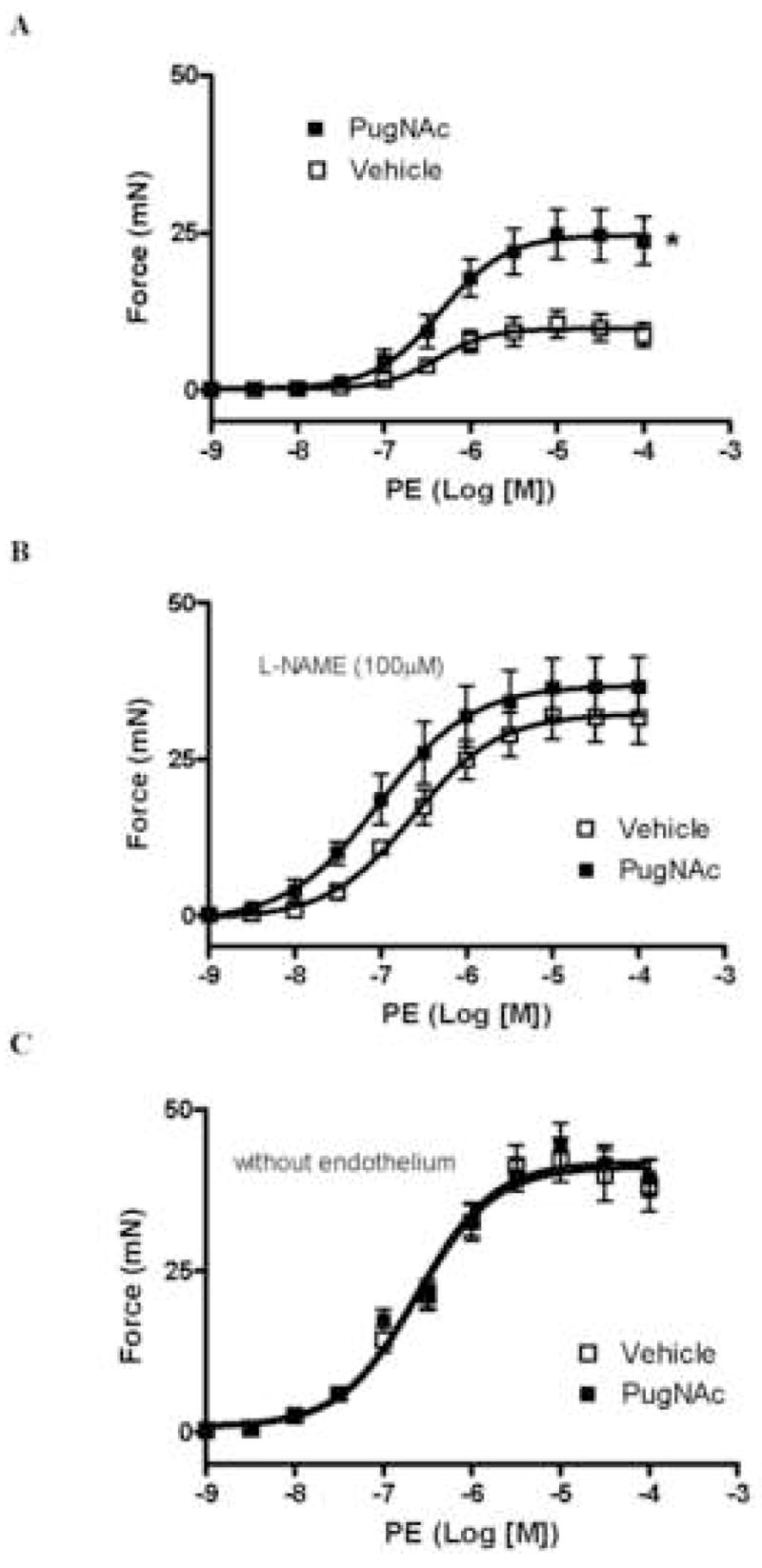
A – Incubation with PugNAc increases vascular contraction to PE in rat thoracic aorta (n=6), when compared to methanol incubation (n=6). B – L-NAME incubation and C – endothelium removal (n=6) abolish the differences between the groups. Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (methanol).
To determine if increased levels of O-GlcNAcylation interfere specifically with alpha1-adrenergic-mediated responses, we performed concentration-response curves to 5-HT. Serotonin-induced contraction was increased in aortas after PugNAc incubation, when compared with control arteries [pD2 = 5.4±0.03 vehicle vs. 5.2±0.02 PugNAc; Emax (mN) = 3.95±0.53 vehicle vs. 7.48±0.51 PugNAc; n=6].
Endothelium-dependent and -independent relaxation was determined by assessing responses to ACh and SNP, respectively. PugNAc impaired ACh-induced relaxation in rat aorta (Fig. 4A), but did not modify SNP-induced relaxation (Fig. 4B).
Figure 4. PugNAc decreases responses to ACh, but not to SNP, in rat aorta.

A - Incubation with PugNAc decreases vascular relaxation to ACh in rat thoracic aorta (n=8), when compared to methanol incubation (n=8). B – PugNAc incubation does not change SNP-induced relaxation (n=8). Experimental values of the relaxation induced by ACh or SNP were calculated relative to the maximal changes from the contraction produced by PE, which was taken as 100%. Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (methanol).
The effects of PugNAc on contractile activity were also checked in mouse thoracic aorta. Similar results were obtained, i.e. increased vascular content of O-GlcNAc proteins augmented PE-induced responses [pD2 = 6.8±0.03 vehicle vs. 6.9±0.03 PugNAc; Emax (mN) = 6.9±0.3 vehicle vs. 11.1±0.6 PugNAc; n=6], and decreased ACh-induced relaxation [pD2 = 7.0±0.02 vehicle vs. 6.2±0.01 PugNAc; Emax (%) = 88±2.4 vehicle vs. 76±3 PugNAc; n=6], indicating that the effects of PugNAc, or increased O-GlcNAc levels, on vascular reactivity are not species-specific.
Considering that 1) vascular responses to ACh, but not SNP, were decreased by PugNAc; 2) PugNAc does not change vascular reactivity to PE in the presence of L-NAME (inhibitor of NOS activity); 3) under high glucose conditions and increased O-GlcNAcylation, phosphorylation of eNOS at Ser1177 (eNOS-Ser1177) is decreased both in vascular and penile tissue from diabetic animals (37–38); we determined whether PugNAc changes the expression and activity (indicated by phosphorylation levels) of enzymes in the eNOS/Akt/PI3K pathway.
Our data show that increased vascular O-GlcNAcylation is associated with decreased phosphorylation of eNOS at Ser1177 (eNOS-Ser1177), but not at Thr495 (eNOS-Thr495) (Fig. 6). In addition, vessels with increased O-GlcNAcylation displayed decreased phosphorylation of Akt at Ser473 (Fig. 7), but not PI3K at Tyr458/199 (Fig. 8). Total expression of eNOS, Akt or PI3K was not modified (Figs. 6, 7 and 8, respectively).
Figure 6. Effect of PugNAc on Akt proteins in rat thoracic aorta.
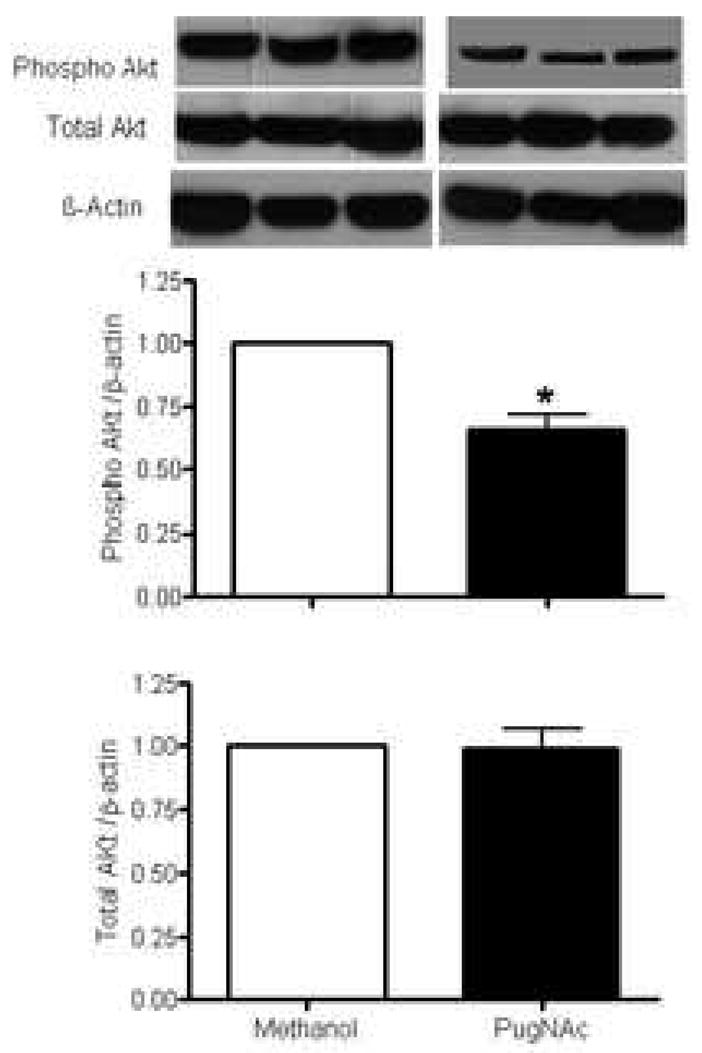
On the top, representative Western blot image of Akt total, Akt phosphorylated (Ser473) and β-actin; on the bottom, bar graphs showing the relative Akt total and phosphorylated proteins after normalization to β-actin expression (n=4). Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (methanol).
Figure 7. Effect of PugNAc on PI3K proteins in rat thoracic aorta.
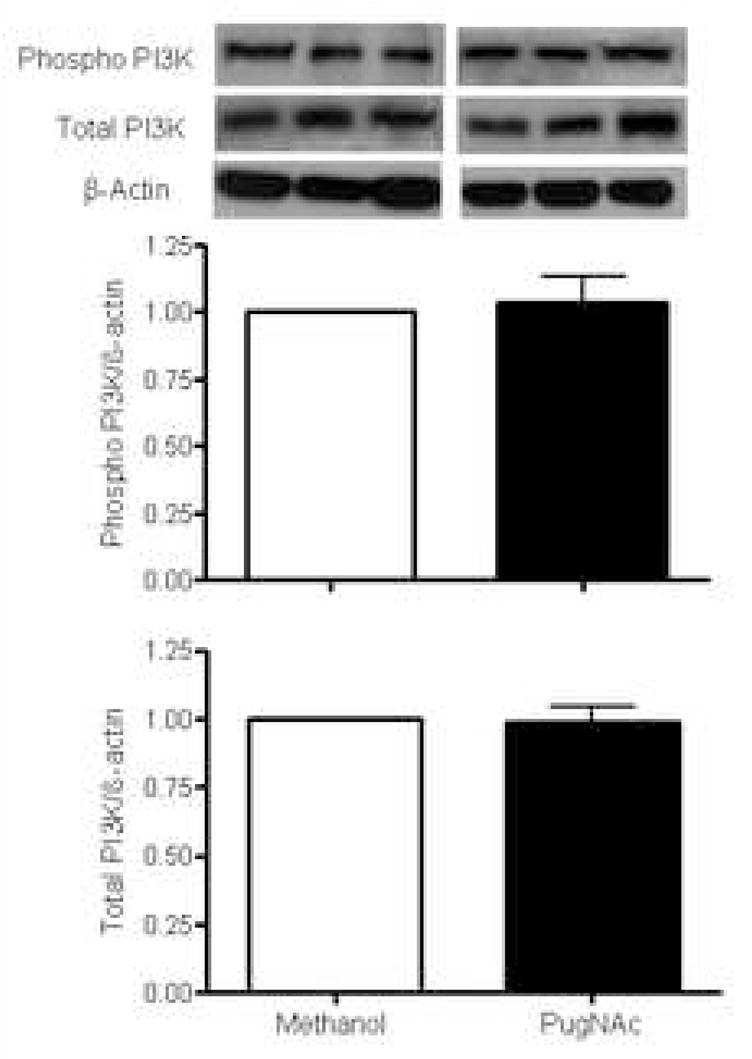
On the top, representative Western blot image of PI3K total, PI3K phosphorylated (Tyr458) and β-actin; on the bottom, bar graphs showing the relative PI3K total and phosphorylated proteins after normalization to β-actin expression (n=4). Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (methanol).
DISCUSSION
While much evidence suggests that O-GlcNAcylation may interfere with many aspects of cardiovascular function (12–27), the effects of abnormal or augmented O-GlcNAcylation on vascular reactivity have been virtually unstudied.
Abnormal vascular reactivity, including impaired endothelium-dependent relaxation and enhanced sensitivity to vasoconstrictors, is a hallmark of several diseases, including hypertension and diabetes. However, our current understanding of the cellular and molecular mechanisms leading to vascular dysfunction are still incomplete. Many proteins important to cardiovascular function have been identified as targets for O-GlcNAcylation, a post-translational modification that influences protein expression, degradation and trafficking. Although preliminary evidence suggests that increased O-GlcNAc levels may be associated with impairment of endothelial and smooth muscle cell function, the effects of O-GlcNAcylation on vascular reactivity are still unknown.
Several proteins can either be directly modified by O-GlcNAc or their activity is modified in response to increased levels of O-GlcNAc. Targets for these modifications include: cytokines, transcription factors, proteins involved in calcium handling, insulin responses, glucose metabolism and signaling pathways (24). Considering that vascular responses to ACh, but not to SNP, are decreased by PugNAc, and that PugNAc does not change vascular reactivity to PE in the presence of L-NAME (inhibitor of NOS activity), we decided to investigate if increased levels of O-GlcNAcylation modifies the PI3K/Akt/eNOS pathway. Indeed, our data strongly suggest that increased O-GlcNAcylation augments vascular reactivity via changes in eNOS/NO pathway. The discrepancy in IP3K and Akt/eNOS phosphorylation indicate that a specific set of proteins in a signaling pathway are subject to O-GlcNAc modification.
Other reports have shown that O-GlcNAcylation interferes with the eNOS pathway. Under high glucose conditions and increased O-GlcNAcylation, phosphorylation of eNOS at Ser1177 (eNOSSer1177) is decreased both in vascular and penile tissue from diabetic animals (6, 7). In addition, increased platelet aggregation induced by advanced glycation end-products (AGEs) is associated with decreased eNOSSer1177 and increased O-glycosylation of eNOS (9). PugNAc decreased phosphorylation of eNOS-Ser1177 as well as phosphorylation of Akt-Ser473, suggesting that a reduction of Akt-mediated eNOS phosphorylation at Ser1177 contributes to the enhanced vascular reactivity to constrictor stimuli. It is possible that increased O-GlcNAcylation may change other phosphorylation sites on eNOS, such as Ser615 and Ser633, which causes activation of eNOS function, and Thr495 and Ser114, which reduces eNOS activity (28). In contrast to Ser1177, PugNAc did not change phosphorylation of eNOS-Thr495, in agreement with previous reports (6, 7).
Du and collaborators (2001) showed a possible mechanism underlying impaired endothelium-dependent vasodilatation in patients with diabetes, a pathology where O-GlcNAc levels are increased. They showed that in cultured cells, hyperglycemia is able to inhibit eNOS activity and increase O-GlcNAc modification of the enzyme with a reciprocal decrease in phosphorylation. This was blocked by inhibition of glutamine:fructose-6-phosphate transferase (GFAT), suggesting that flux through HBP was a contributing factor (6).
Increased flux through the HBP, either through increased glucose uptake or glucosamine treatment, increases the production of UDP-GlcNAc and stimulates O-GlcNAc modification of proteins. GFAT is the rate-limiting enzyme of the pathway and converts fructose 6-phosphate to glucosamine 6-phosphate, with glutamine as the amine donor. However, the effects seen with glucosamine treatment may potentially be mediated via a number of other pathways, since this amino sugar, in addition to increasing O-GlcNAc levels, also increases UDP-GlcNAc levels, which is used for multiple N-glycosylation reactions that are involved in protein synthesis. N-glycosylation (29), glucosamine-6-phosphate accumulation (30), or ganglioside synthesis (31), all have the potential to alter gene expression through changes in intracellular signaling. In addition, high levels of glucosamine-6-phosphate, generated in response to glucosamine, inhibit glucose-6-phosphate dehydrogenase, thereby depleting NADPH and enhancing oxidative stress (30). A potent inhibitor of O-GlcNAcase, [O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino- N-phenylcarbamate (PugNAc), by mimicking the O-GlcNAcase-stabilized transition state, also increases O-GlcNAc-proteins and represents a more direct approach, with which to evaluate the effects of increased O-GlcNAcylation (32).
O-GlcNAc is considered to be similar to protein phosphorylation (O-phosphate attachment) in that both modifications occur on serine and threonine residues, both are dynamically added and removed from proteins in response to cellular signals, and both alter the function and association of the modified protein. Many phosphorylation sites are also known glycosylation sites, and this reciprocal occupancy may produce different activities or alter stability in the protein (8). Therefore, one potential mechanism by which O-GlcNAcylation may change vascular reactivity includes the complex interplay between O-GlcNAcylation and phosphorylation.
We cannot discharge that other pathways may contribute to this altered vascular response. It is also likely that glycosylation of other proteins, in addition to eNOS, contributes to changes in vascular reactivity induced by augmented O-GlcNAc levels. Accordingly, a recent report showed that the inositol 1,4,5-trisphosphate (InsP3) receptor activity is decreased by the addition of O-GlcNAc (10). Since InsP3 receptors represent the main channel for intracellular calcium (Ca+2) release in many cell types, including endothelial and smooth muscle cells, this mechanism may also contribute to vascular dysfunction under conditions in which O-GlcNAc is high. In addition, activation of the hexosamine pathway, which leads to increase O-GlcNAc modification and decrease O-linked phosphorylation of eNOS, is probably only one of the various mechanisms by which hyperglycemia leads to vascular dysfunction. Increased hexosamine pathway flux, altered cellular redox state, increased formation of diacylglycerol and subsequent activation of protein kinase C isoforms as well as accelerated non-enzymatic formation of advanced glycated end products (AGEs), all play a role in the deleterious effects of hyperglycemia. It is very likely that proteins modified by O-GlcNAc are different in different pathological conditions such as diabetes, hypertension (and treatment with PugNAc) and, consequently, one should consider that changes in vascular reactivity induced by hyperglycemia and pharmacologically-induced increased O-GlcNAcylation are not exactly the same.
In conclusion, our data suggest that increased O-GlcNAcylation augments vascular reactivity to constrictor stimuli via changes in the Akt/eNOS pathway and strongly support our hypothesis that augmented O-GlcNAcylation increases vasoconstriction. These important data drive our attention to a new mechanism for vascular response control, adding a novelty to this research area and suggest that abnormal O-GlcNAcylation may be associated with vascular dysfunction in pathological conditions, such as arterial hypertension.
Figure 5. Effect of PugNAc on eNOS proteins in rat thoracic aorta.
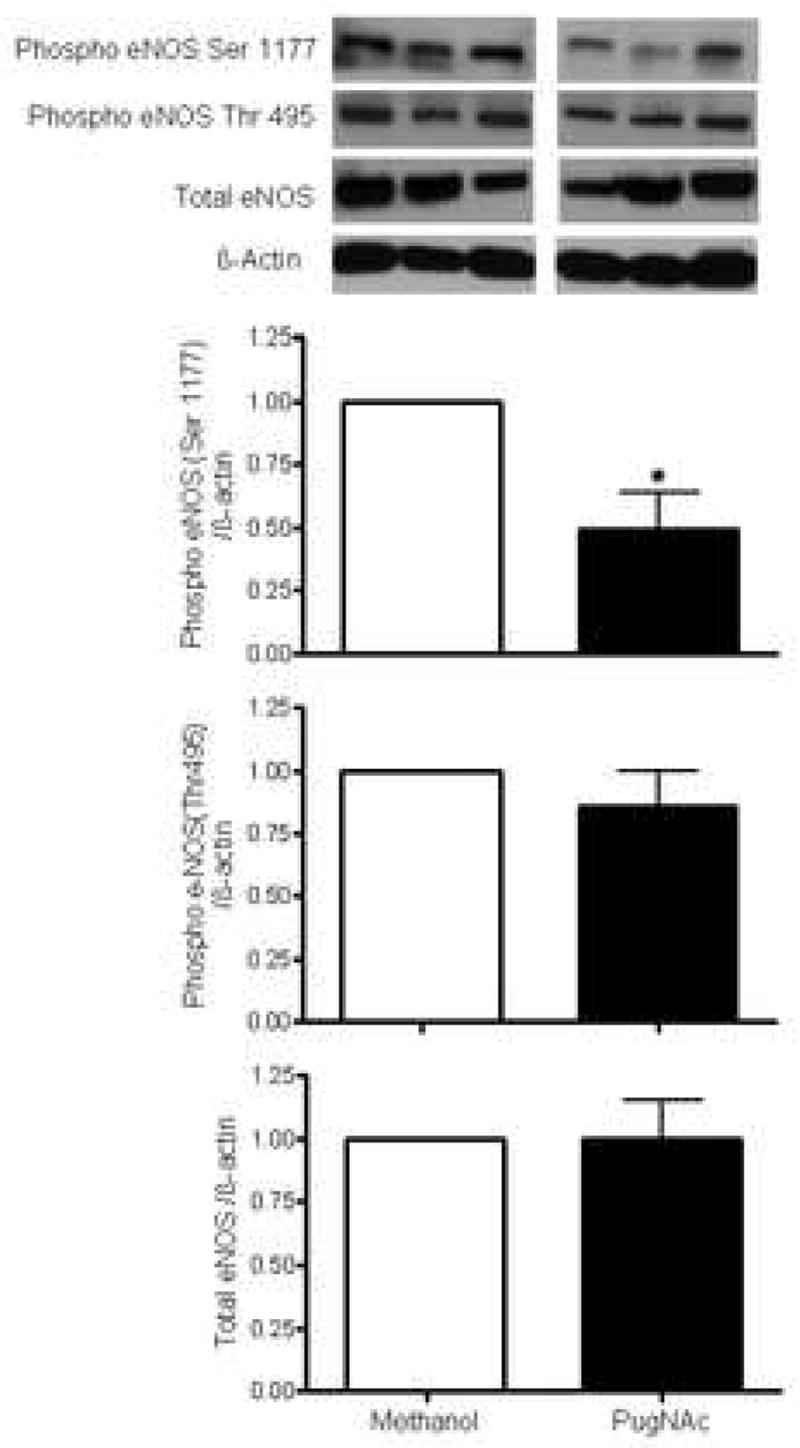
On the top, representative Western blot image of eNOS total, eNOS phosphorylated (Ser1177 and Thr 495) and β-actin; on the bottom, bar graphs showing the relative eNOS total and phosphorylated proteins after normalization to β-actin expression (n=4). Results are presented as mean ± SEM in each experimental group. *, p<0.05 vs. vehicle (methanol).
Acknowledgments
National Institutes of Health (HL74167), USA and CAPES, Brazil.
ABBREVIATIONS
- ACh
acetylcholine
- eNOS
endothelial nitric oxide synthase
- GFAT
glutamine:fructose-6-phosphate transferase
- HBP
hexosamine biosynthetic pathway
- KCl
potassium chloride
- L-NAME
Nω-nitro-L-arginine methyl ester
- O-GlcNAc
O-linked attachment of N-acetyl-glucosamine
- O-GlcNAcase
β-N-acetylglucosaminidase
- OGT
O-GlcNAc transferase
- PE
phenylephrine
- PI3K
phosphoinositide-3 kinase
- PKC
protein kinase C
- PLC
phospholipase C
- PSS
physiological salt solution
- PugNAc
O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino- N-phenylcarbamate
- SERCA
sarcoplasmic reticulum Ca2+-ATPase
- SNP
sodium nitroprusside
- UDP-GlcNAc
uridine 5′-diphosphate-N-acetylglucosamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–22. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 2.Fülöp N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res. 2007;73:288–97. doi: 10.1016/j.cardiores.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–8. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- 4.Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem. 1991;266:4706–12. [PubMed] [Google Scholar]
- 5.Goldberg HJ, Whiteside CI, Hart GW, Fantus IG. Posttranslational, reversible O-Glycosylation is stimulated by high glucose and mediates plasminogen activator inhibitor-1 gene expression and Sp1 transcriptional activity in glomerular mesangial cells. Endocrinology. 2006;147:222–231. doi: 10.1210/en.2005-0523. [DOI] [PubMed] [Google Scholar]
- 6.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–8. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musicki B, Kramer MF, Becker RE, Burnett AL. nactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. PNAS. 2005;102:11870–5. doi: 10.1073/pnas.0502488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamemura K, Hayes BK, Comer FI, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of Thr-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J Biol Chem. 2002;277:19229–35. doi: 10.1074/jbc.M201729200. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Liu Y, Cui B, Mi Q, Huang Y, Fan L, et al. 17β-oestradiol partially attenuates the inhibition of nitric oxide synthase-3 by advanced glycation end-products in human platelets. Clin Exp Pharmacol Physiol. 2007;34:972–8. doi: 10.1111/j.1440-1681.2007.04680.x. [DOI] [PubMed] [Google Scholar]
- 10.Rengifo J, Gibson CJ, Winkler E, Collin T, Ehrlich BE. Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J Neurosci. 2007;27:13813–21. doi: 10.1523/JNEUROSCI.2069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitworth CE, Macauley MS, Stubbs KA, Dennis RJ, Taylor EJ, Vocadlo DJ, et al. Analysis of PugNag and NAG-thiazoline as transition state analogues for human O-GlcNAcase: Mechanistic and structural insights into inhibitor selectivity and transition state poise. J Am Chem Soc. 2007;129:635–644. doi: 10.1021/ja065697o. [DOI] [PubMed] [Google Scholar]
- 12.Patti ME, Virkamaki A, Landaker EJ, Kahn CR, Yki-Jarvinen H. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal muscle. Diabetes. 1999;48:1562–71. doi: 10.2337/diabetes.48.8.1562. [DOI] [PubMed] [Google Scholar]
- 13.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA. 2002;99:5313–8. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, et al. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106:466–72. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 15.Hebert LF, Daniels MCJZ, Crook ED, Turner RL, Simmons S. Tea Overexpression of glutamine:fructose-6-phosphate amidotransferase in transgenic mice leads to insulin resistance. J Clin Invest. 1996;98:930–6. doi: 10.1172/JCI118876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Virkamaki A, Yki-Jarvinen H. Allosteric regulation of glycogen synthase and hexokinase by glucosamine-6-phosphate during glucosamine-induced insulin resistance in skeletal muscle and heart. Diabetes. 1999;48:1101–7. doi: 10.2337/diabetes.48.5.1101. [DOI] [PubMed] [Google Scholar]
- 17.Parker G, Taylor R, Jones D, McClain D. Hyperglycemia and inhibition of glycogen synthase in streptozotocin-treated mice: role of O-linked N-acetylglucosamine. J Biol Chem. 2004;279:20636–42. doi: 10.1074/jbc.M312139200. [DOI] [PubMed] [Google Scholar]
- 18.Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, et al. A single nucleotide polymorphism in MGEA5 encoding O-GlcNAcselective N-acetyl-beta-d glucosaminidase is associated with type 2 diabetes in Mexican Americans. Diabetes. 2005;54:1214–21. doi: 10.2337/diabetes.54.4.1214. [DOI] [PubMed] [Google Scholar]
- 19.Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H148–H58. doi: 10.1152/ajpheart.1997.272.1.H148. [DOI] [PubMed] [Google Scholar]
- 20.Dutta K, Carmody MW, Cala SE, Davidoff AJ. Depressed PKA activity contributes to impaired SERCA function and is linked to the pathogenesis of glucose-induced cardiomyopathy. J Mol Cell Cardiol. 2002;34:985–96. doi: 10.1006/jmcc.2002.2035. [DOI] [PubMed] [Google Scholar]
- 21.Ren J, Gintant GA, Miller RE, Davidoff AJ. High extracellular glucose impairs cardiac E–C coupling in a glycosylation-dependent manner. Am J Physiol Heart Circ Physiol. 1997;273:H2876–H83. doi: 10.1152/ajpheart.1997.273.6.H2876. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, et al. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–13. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- 23.Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, et al. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278:44230–44237. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- 24.Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279(29):30133–42. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 25.Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sc. 2007;112:375–84. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- 26.Kneass ZT, Marchase RB. Neutrophils exhibit rapid agonist-induced increases in protein-associated O-GlcNAc. J Biol Chem. 2004;279:45759–65. doi: 10.1074/jbc.M407911200. [DOI] [PubMed] [Google Scholar]
- 27.Daniels MC, McClain DA, Crook ED. Transcriptional regulation of transforming growth factor beta1 by glucose: investigation into the role of the hexosamine biosynthesis pathway. Am J Med Sci. 2000;319:138–42. doi: 10.1097/00000441-200003000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–9. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 29.Li SY, Sigmon VK, Babcock SA, Ren J. Advanced glycation end-product induces ROS accumulation, apoptosis, MAP kinase activation and nuclear O-GlcNAcylation in human cardiac myocytes. Life Sc. 2007;80:1051–6. doi: 10.1016/j.lfs.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 30.Guinez C, Mir AM, Leroy Y, Cacan R, Michalski JC, Lefebvre T. Hsp70-GlcNAc-binding activity is released by stress, proteasome inhibition, and protein misfolding. Biochem Biophys Res Commun. 2007;361:414–20. doi: 10.1016/j.bbrc.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 31.Minamino T, Komuro I. Vascular Cell Senescence. Contribution to Atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 32.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part III: Cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–7. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]