

Abstract
Previous data has shown that adenylyl cyclase type 6 (AC6) is expressed principally in lipid rafts or caveolae of cardiac myocytes and other cell types while certain other isoforms of AC are excluded from these microdomains. The mechanism by which AC6 is localized to lipid rafts or caveolae is unknown. In this study, we show AC6 is localized in lipid rafts of COS-7 cells (expressing caveolin-1) and in HEK-293 cells or cardiac fibroblasts isolated from caveolin-1 knock-out mice (both of which lack prototypical caveolins). To determine the region of AC6 that confers raft localization, we independently expressed each of the major intracellular domains, the N-terminus, C1 and C2 domains, and examined their localization with various approaches. The N-terminus did not associate with lipid rafts or caveolae of either COS-7 or HEK-293 cells nor did it immunoprecipitate with caveolin-1 when expressed in COS-7 cells. By contrast, the C1 and C2 domains each associated with lipid rafts to varying degrees and was present in caveolin-1 immunoprecipitates. There were no differences in the pattern of localization of either the C1 or C2 domains between COS-7 and HEK-293 cells. Further dissection of the C1 domain into four individual proteins indicated that the N-terminal half of this domain is responsible for its raft localization. To probe for a role of a putative palmitoylation motif in the C-terminal portion of the C2 domain, we expressed various truncated forms of AC6 lacking most or all of the C-terminal 41 amino acids. These truncated AC6 proteins were not altered in terms of their localization in lipid rafts or their catalytic activity, implying that this C-terminal region is not required for lipid raft targeting of AC6. We conclude that while the C1 domain may be most important, both the C1 and C2 domains of AC6 play a role in targeting AC6 to lipid rafts.
Keywords: adenylyl cyclase, lipid rafts, caveolae, cyclic AMP, compartmentation
INTRODUCTION
It has recently been documented that GPCR’s and their associated signaling components such as AC isoforms are not randomly dispersed throughout the plasma membrane [1, 2]. Lipid rafts are rich in cholesterol and sphingolipids, and they display a high concentration of signaling proteins, including G protein-coupled receptors (GPCR’s), G-proteins, and effectors such as ion channels and adenylyl cyclases (AC’s)[1, 3, 4]. The caveolin/lipid raft signaling hypothesis proposes that compartmentation of signaling molecules in these microdomains provides a mechanism for temporal and spatial signal transduction and cross-talk among signaling pathways [5, 6]. However, by no means are all membrane-associated signaling proteins located in lipid rafts [3, 7]. Understanding how this differential plasma membrane localization occurs may illuminate the significance of raft versus non-raft targeting of GPCR signaling components.
Adenylyl cyclases are a key effector component of transmembrane signaling pathways that are both positively and negatively regulated by the activity of heterotrimeric G proteins and GPCR’s. There are nine different mammalian isoforms of AC’s and each isoform displays distinct lipid raft or non-raft localization [1, 8, 9]. AC3, AC5, AC6 and AC8 are expressed in lipid rafts of various cell types while the other isoforms of AC are not [10–15]. The distinct localization of the raft-associated AC isoforms facilitates regulation of theses enzymes by specific receptors and other regulatory influences such as calcium and contributes to the compartmentalization of cAMP signaling [9, 16, 17]. However, only relatively recently has it begun to be understood how a combination of protein–protein and protein–lipid interactions contributes to the manifestation GPCR signaling compartmentalization in lipid raft domains.
The nine membrane-bound AC’s share a common secondary structure comprising an intracellular N-terminus, two cassettes of six transmembrane domains in tandem separated by a cytoplasmic loop, termed the C1 domain, and a C-terminal cytoplasmic C2 domain. The ATP-binding C1a and C2a domains are the most conserved regions between AC isoforms, and their interaction forms the catalytic core [18, 19]. The transmembrane domains are not essential for catalytic activity because the C1a and C2a domains can be overexpressed separately in vitro to form a Gsα- and forskolin-stimulable enzyme [20]. However, by their association, the transmembrane domains have a crucial role in increasing the relative concentrations of their attached catalytic domains so that they readily associate to form the catalytic core [21]. The transmembrane spanning domains have also been suggested to be important in the regulation of protein assembly and membrane trafficking of AC6 [22]. Recent kinetic investigations have shown that dimerization is an important feature of G-protein regulation of AC. AC5 undergoes a cooperative activation by Gsα that is consistent with Gsα binding to an AC5 dimer [23] and AC2-AC5 heterodimers are more sensitive to G and forskolin than either isoform alone [24]. Other studies indicate that AC6, a homologue of AC5, also forms homodimers, as demonstrated by the co-immunoprecipitation of two differently epitope-tagged forms of AC6 [25]. In the case of AC6, dimerization was suggested to occur through intermolecular interactions between the first transmembrane cassettes since the co-expression of the first transmembrane cassette of AC6 not only co-immunoprecipitated but also reduced the plasma membrane expression and activity of full-length AC6.
The transmembrane regions of GPCR’s and other membrane proteins, which are embedded in the lipid bilayer, can dictate domain localization by interacting with the lipid and/or protein components of rafts/caveolae. In particular, cholesterol specifically enriched in lipid microdomains has attracted attention as a possible determinant of GPCR localization in lipid rafts. The intracellular loops and carboxyl- terminal tail may all be involved in receptor targeting to lipid rafts by means of different addressing signals: fatty acylation and protein-protein interactions. Crossthwaite et. al. investigated the regions of AC that dictate lipid raft localization by expressing various chimeric combinations of AC5 (a lipid raft-localized isoform) and AC7 (a non-raft localized isoform [26]. These investigators concluded that the C1 and the C2 domains were critical for lipid raft localization of AC5 and that the membrane-spanning domains have no role in this localization.
In the present study, we sought to identify the structural elements of AC6 that determine its localization to lipid rafts. We generated constructs corresponding to each intracellular segments of the raft-targeted AC6 and studied their localization properties when expressed in COS-7 cells. Detergent- and non detergent-based methods for raft isolation, combined with immunoprecipitations and confocal microscopic analysis, were used to confirm the cellular localization of the expressed proteins. Our results indicate that the targeting of AC6 to lipid raft depends only on the C1 and C2 cytosolic domains and is independent of the N terminal domain and the extreme C-terminal tail. Our studies also show that caveolins are not required for lipid raft localization of AC6. These studies will allow more detailed analysis of AC6 that can determine the mechanism by which this protein, and likely others, localize to lipid rafts.
EXPERIMENTAL
Materials and cell culture
Primary antibodies for AC5/6 were obtained from Santa Cruz Biotechnology. Primary antibody for FLAG was obtained from Sigma. Primary antibody for caveolin-1was obtained from BD/Transduction Laboratories. S-tag HRP-conjugated and anti-His antibody were obtained from Novagen. All other chemicals and reagents were obtained from Sigma Aldrich. COS-7 and HEK-293 cells were obtained from ATCC and were maintained in minimum essential medium (MEM) supplemented with 10% horse serum and penicillin/streptomycin in a 37°C incubator with 5% CO2. Cardiac fibroblasts were isolated from wild-type or caveolain-1 knockout mice using a method adapted from Liu et al [27]. Briefly, hearts were rapidly excised and then minced, pooled, and placed in a collagenase/pancreatin digestion solution. After five sequential digestions, the fibroblasts were pelleted and resuspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin, streptomycin, fungizone, and 10% fetal bovine serum (FBS, Atlanta Biologicals). After a 30-min period of attachment to uncoated culture plates, cells that were weakly attached or unattached were rinsed free and discarded. Cells were then cultured for two to four days. The purity of mouse cardiac fibroblast cultures was greater than 95% as determined by positive staining for vimentin and negative staining for smooth muscle actin and von Willebrand factor, as previously described [13]. All animals were treated according to the principles of laboratory animal care (NIH publication No. 85-23, revised 1985) and applicable US law.
Construction and expression of AC6 wild-type and truncation mutants
We generated WT Flag-AC6 using pCDNA3.1-AC6 as a initial template. PCR fragments were and then cloned into pEGFP-N1 vector, which had been digested with identical enzymes. Positive clones were sequenced to verify the right open reading frame and the desired mutation. Truncation mutants were fused with the FLAG epitope. We constructed the following protein fragments of human AC6: N-terminus: amino acids 1–150, C1: amino acids 306–672 and C2: amino acids 991–1165. Each construct was cloned into the pTriEX-4 vector (EMD Biosciences) with fused S-tags and His-tags. Other fragments of human AC6 were constructed and fused with GFP. These proteins are C1-306 (composed of amino acids 306–387 of AC6), C1-388 (composed of amino acids 388–490 of AC6), C1-491 (composed of amino acids 491–589 of AC6) and C1-590 (composed of amino acids 590–672 of AC6. Each construct was cloned into the pEGFP vector (Invitrogen) with fused EGFP. All of the above constructs are represented schematically in Figure 3. Transient transfections were performed using Lipofectimine 2000 (Invitrogen) for 36–48 hr.
Figure 3. Schematic diagram of AC6 fragments and truncated proteins constructed for these studies.

Wild type (full length) AC6 is represented by the top bar, with each of the two cassettes of 6-transmembrane domains represented by boxes for simplicity. Each bar below represents the individual proteins constructed and expressed in this study, with their designation and various epitope tags shown. The scale of these bars is representative only
Triton X-100 insolubility
Cos-7 cells were grown to confluence in 35-mm dishes and extracted with 1 ml of lysis buffer 25 mM Mes, pH 6.5,0.15 M NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride. After 30 min on ice without agitation, the Triton-soluble extract was gently decanted, and the remaining Triton-insoluble material was solubilized in 1 ml of 1% SDS. This latter fraction, termed the triton-insoluble extract, was homogenized by passing through a 26- gauge needle ten times. Equal proportions of each fraction were separated by SDS-polyacrylamide gel electrophoresis and analyzed by immunoblotting.
Non-detergent isolation of caveolar and non-caveolar membranes
Transfected cells were fractionated using a detergent-free method described previously [28]. Two 10-cm plates containing 70 to 80% confluent Cos-7 or HEK-293 cells were washed twice in ice-cold PBS and scraped into a total of 2 ml of 500 mM sodium carbonate, pH 11. Cells were homogenized with a tissue grinder with three 10 sec bursts and then a sonicator with three 20 sec bursts. A full 1 min rest period was included in between each burst. 1 ml of homogenate was brought to 45% sucrose by addition of an equal volume of 90% sucrose in MBS (25 mM MES and 150 mM NaCl, pH 6.5) and loaded in an ultracentrifuge tube. A discontinuous sucrose gradient was layered on top of the sample by placing 2 ml of 35% sucrose prepared in MBS with 250 mM sodium carbonate then 1 ml of 5% sucrose (also in MBS/Na2CO3). The gradient was centrifuged at 46,000 rpm on a SW55Ti rotor (Beckman Coulter, Fullerton, CA) for 16 to 18 h at 4°C. Fractions (10 × 0.5 ml) were collected from the top of the gradient, separated by SDS-PAGE using proportional loading, and analyzed by immunoblotting. In some studies, the faint light-scattering band was collected from the 5 to 35% sucrose interface (caveolin-enriched membranes) while the bottom of the gradient (45% sucrose) was collected as noncaveolar membranes.
Immunofluorescent confocal microscopy
Intracellular localization of was visualized in living Cos-7 cells. Microscopy was performed using a Zeiss LSM 410 confocal microscope equipped with a Krypton/Argon laser. EGFP fluorescence was examined using a fluorescein isothiocyanate filter under a 40x oil immersion lens. For each experimental condition, fluorescence distribution patterns similar to the images shown were observed in the majority (~90%) of cells inspected. GFP-fusion protein expression was determined by quantifying total fluorescence (excitation filter 475/20 nm, emission filter 515/30 nm), which was measured as relative fluorescence units (RFU). Upon addition of coelenterazine total protein expression was determined by the light passed by the 450/58 filter, and was measured in relative luciferase units (RLU). Cos-7 cells (1 × 105 cells) were plated onto poly-L-lysine-coated glass coverslips and cultured for 24 h. Cells were then transfected with 0.4 μg of cDNA using the Lipofectamine 2000 (Invitrogen). Cells were then washed with phosphate-buffered saline (PBS; 12.1 mM Na2HPO4, 4 mM KH2PO4, and 130 mM NaCl, pH 7.4) and fixed using 4% paraformaldehyde (1 h, 20 °C). After fixing, cells were washed further in PBS and permeabilized (1 h, 20 °C) with PBS containing 0.2% Triton X-100 and 1% goat serum (blocking solution) and incubated (12 h, 20 °C) with anti-His TRITC conjugated antibody or anti-caveolin-1 monoclonal antibody. After further washes in PBS (3 × 10 min), cells were incubated (1 h, 20 °C) with Alexa Fluor 488 goat anti-mouse antibody. Coverslips were mounted in Slowfade® light antifade (Molecular Probes) according to the manufacturer’s procedures, and the cells were visualized on a Zeiss Axiovert LSM510 confocal microscope, using a x63 oil immersion objective and a slice depth of 1 μm.
Immunoprecipitation
Transfected cells were lysed in buffer A (20 mM Tris, pH 8.0, 1% NP-40, 0.1% SDS, 140 mM NaCl, 1 mM phenylmetanesulfonyl fluoride. For co-immunoprecipitation experiments, cell lysates were incubated with anti-caveolin-1 monoclonal antibody for 1 hour at 4°C followed by the addition of protein G agarose beads (Santa Cruz Biotechnology, CA) and further incubated overnight at 4°C on a rocker platform. The resulting immunoprecipitates were separated by centrifugation and both the precipitate and supernatant were collected for analysis. Precipitates underwent washing at three levels of stringency before being separated by SDS-PAGE and analyzed by immunoblotting.
Immunoblot analysis
Whole cell lysates were obtained from COS-7 by scraping cells in modified RIPA lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Igepal CA-630, plus mammalian protease inhibitor cocktail, Sigma cat# P-8340) and homogenizing by sonication. Equal protein amounts of the lysates were separated by SDS-polyacrylamide gel electrophoresis (made) and transferred to PVDF membranes (Millipore) by electroblotting. Membranes were blocked in 20mM phosphate buffered saline (PBS) with 3% nonfat dry milk and incubated with primary antibody (see Materials) overnight at 4°C. Bound primary antibodies were visualized using appropriate secondary antibody with conjugated horseradish peroxidase (Santa Cruz Biotechnology) and ECL reagent (Pierce). In some experiments, proteins were detected using an S-Tag-HRP detection kit (Novagen). Most primary antibodies recognized multiple non-specific species. Only the band representing the appropriately sized immunoreactive band is shown. The amount of protein per sample was determined using a dye-binding protein assay (Bio-Rad).
Measurement of cAMP accumulation
Cells plated at 80% confluency on 24-well plates were washed three times with serum and NaHCO3-free DMEM supplemented with 20 mM HEPES, pH 7.4 (DMEH) and equilibrated for 30 min. Assay for cAMP accumulation was performed by incubation with drugs of interest plus 0.2 mM isobutylmethylxanthine, a PDE inhibitor, for 10 min. To terminate reactions, assay medium was aspirated and 200μL lysis buffer (GE Healthcare) was added to each well. cAMP content of the extract was quantified using the Biotrak EIA Kit (GE Healthcare) using the manufacturer’s non-acetylation protocol. Data were normalized to the amount of protein per sample, as determined using a dye-binding protein assay (Bio-Rad).
RESULTS
Expression of AC6 and caveolins in HEK-293 and COS-7cells
We first sought to characterize the native expression of AC6 in two cell types, COS-7 and HEK-293 cells. We isolated buoyant lipid raft fractions from both these cell types using non-detergent fractionation followed by sucrose gradient centrifugation. COS-7 cells expressed readily detectible levels of AC5 and AC6 (using an antibody that can not distinguish between these two isoforms) almost exclusively in buoyant fractions also expressing caveolin-1 (Figure 1, top). These buoyant fractions were devoid of immunorectivity for either β-adaptin (a clathrin-coated pit marker) or mannosidase II (a Golgi marker) while these proteins were readily detectible in fractions 7 through 10 (data not shown). Transient transfection of the wild-type human AC6 led to increased AC5/6 immunoreactivity in these same buoyant fractions. By contrast, HEK-293 cells express much lower levels of AC5/6 immunoreactivity, but the detectible protein is also predominantly localized in the buoyant fractions (Figure 1, bottom). The HEK-293 cell line used in our studies expresses little or no caveolin-1 or caveolin-2 (latter data not shown). Transient transfection of human AC6 led to readily detectible AC5/6 immunoreactivity in buoyant fractions from HEK-293 cells. The non-raft protein adaptin-β, a component of clathrin coated pits, is excluded from buoyant fractions in all experiments from both cell types (data not shown). Thus, COS-7 and HEK-293 cells differ in their expression levels of AC5/6 but the localization of AC6 in these two cells is consistent.
Figure 1. Endogenous and overexpressed AC6 are co-localized in lipid rafts independent of caveolin-1 expression.

Untreated COS-7 or HEK-293 cells (control) or cells incubated with an adenovirus expressing human AC6 for 24 h (Adv-AC6) were fractionated using a non-detergent method to isolate lipid rafts and caveolae (see Methods). Following sucrose density centrifugation, 10 fractions were collected and fractions 2–10 were separated by SDS-PAGE and analyzed by immunoblot using either anti-caveolin-1 or anti-his tag and S-protein. Images shown are representative of 4 experiments.
To more definitively understand the role of caveolins in the localization of AC6, we isolated cardiac fibroblasts from wild-type and caveolin-1 knock-out mice. Mouse cardiac fibroblasts (MCF) express AC2, AC3 and AC5/6 along with caveolin-1 (Figure 2). Caveolin-2 expression was not detected in MCF. AC3 and AC5/6 immunoreactivity was detected primarily in buoyant lipid raft fractions that also contained caveolin-1 expression. By contrast, AC2 was detected only in non-buoyant fractions, consistent with our previous findings from other cell types [1, 13]. Parallel studies of MCF isolated from caveolin-1 knockout mice yielded identical results except for the fact that no caveolin-1 expression was detected (Figure 2, bottom). As with studies of HEK-293 and COS-7 cells, β-adaptin and mannosidase II immunoreactivity were detected exclusively in non-buoyant fractions (data not shown). Taken together, these data imply that caveolin expression is not required for the lipid raft localization of AC6 or AC3.
Figure 2. Endogenous AC3 and AC6 are localized in lipid rafts in cardiac fibroblasts from both wild-type and caveolin-1 knock-out mice.

Cardiac fibroblasts were isolated from wild-type and caveolin-1 knock-out mice and then fractionated using a non-detergent method to isolate lipid rafts and caveolae (see Methods). Following sucrose density centrifugation, 10 fractions were collected and fractions 2–10 were separated by SDS-PAGE and analyzed by immunoblot using either anti-caveolin-1 or anti-his tag and S-protein. Images shown are representative of 4–5 experiments.
Expression of intracellular domains of AC6
A recent study by Crossthwaite et. al. showed that the transmembrane spanning domains were not required for targeting AC5 to lipid rafts[26]. These investigators constructed chimeric AC’s from raft (AC5) and non-raft (AC7) localized isoforms and found that the C1 and C2 domains determined the lipid raft localization of AC5. Taking a different approach, we expressed individual recombinant proteins corresponding to each of the three large cytosolic domains of AC6 (N-terminus, C1 and C2 domains) to determine if they localized to lipid rafts (see schematic representation of these protein constructs in Figure 3). Each protein was fused with a His epitope and an S-protein tag for simplified isolation and detection. Confocal microscopy confirmed that each protein is expressed largely in the cytosol, with only a minor proportion of the C1 or C2 domains localized to the plasma membrane where the bulk of caveolin-1 staining was found (Figure 4). Thus, while expression of the individual cytosolic domains of AC6 does not lead to a large percentage of expression in the plasma membrane, the approach of expressing these individual domains was deemed useful for probing for the regions of AC6 responsible for its characteristic lipid raft localization.
Figure 4. C1 and C2 domains, but not the N-terminus, of AC6 are partially associated with the plasma membrane.
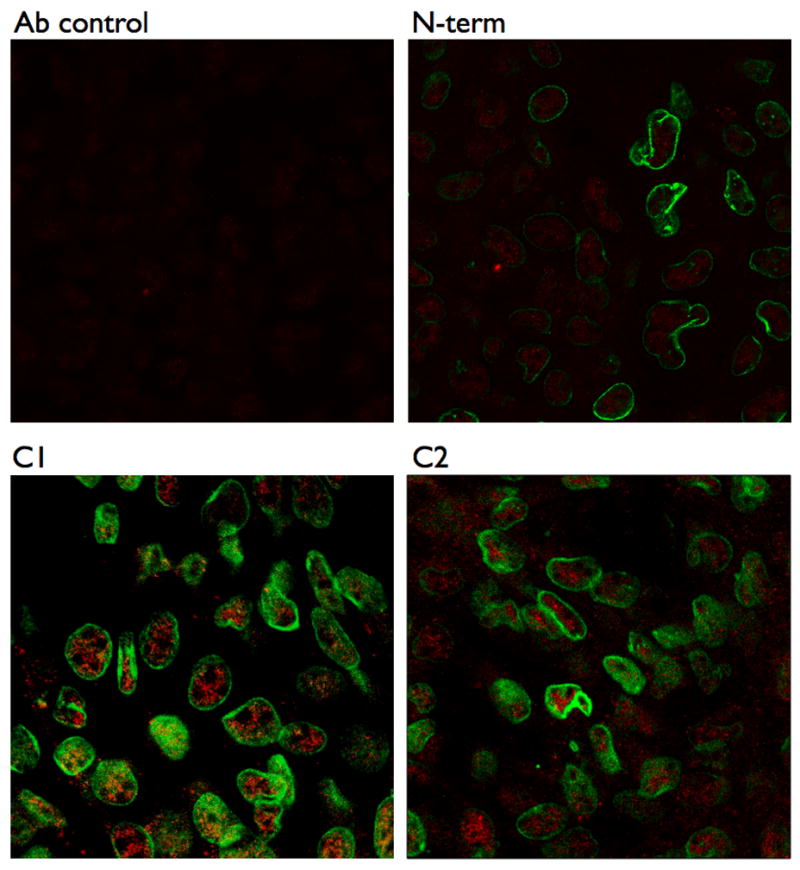
COS-7 cells were transfected with each intracellular domain construct from AC6 then were fixed in 2% paraformaldehyde, washed and permeablized (see Methods). Cells were then incubated with an anti-His TRITC-conjugated antibody to detect the expressed proteins (red) and a caveolin-1 monoclonal antibody (followed by Alexa Fluor 488 secondary antibody, green) before being analyzed by confocal microscopy. Ab control (top, left) shows mock-transfected cell incubated with anti-His TRITC antibody. Images are representative of 3 experiments.
Thus, we expressed each protein in COS-7 and HEK-293 cells and performed lipid raft fractionation studies. Detection of the proteins via the fused S-protein tag revealed that while the bulk of the expressed proteins were in the heavy fractions, as expected based upon their appearance in cytosolic portions of the cell, only C1 and, to a slightly lesser extent, C2 could be detected in the buoyant lipid raft fractions from either cell type (Figure 5). As in previous studies, HEK-293 cells did not contain caveolin-1 immunoreactivity. As a complementary approach, we used detergent-based solubilization of cells (1% Triton X-100) to determine the association of the N-terminus, C1 and C2 domains with lipid rafts and also found that the C1 domain most readily associated with detergent-insoluble membranes (data not shown). Expression of both C1 and C2 domains in the same cells did not alter the localization of either construct in lipid rafts, indicating that there is limited or no cooperativity between these two regions of AC6 (data not shown).
Figure 5. The C1 domain of AC6 most strongly localizes in lipid raft/caveolar fractions.
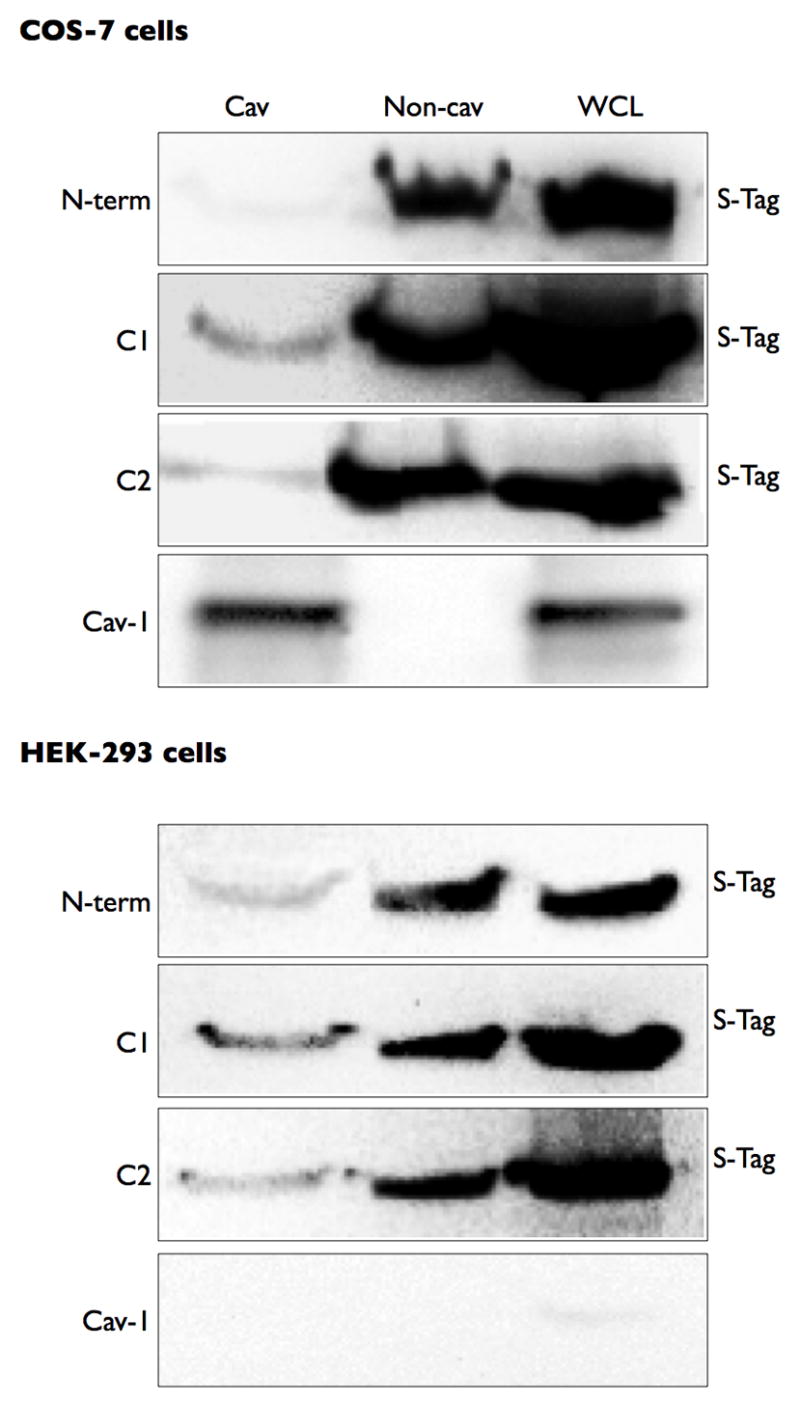
COS-7 or HEK-293 cells were transfected with each intracellular domain construct. Cells were then fractionated using a non-detergent method to isolate lipid rafts and caveolae (see Methods). Following sucrose density centrifugation, fractions were 2–4 were combined as the Cav fraction and fractions 7–10 were combined as the non-cav fraction. Each pooled fraction and a whole cell lysate control were separated by SDS-PAGE and analyzed by immunoblot using an anti-His antibody to detect the expressed protein construct. Images shown are representative of 4–5 experiments.
We have previously shown that AC6 co-immunoprecipitates with caveolins [28]. Thus, we immunoprecipitated caveolin-1 from plasma membrane fractions of COS-7 cells expressing each of the three intracellular domains and probed for the S-tag fused to each construct. We observed that virtually no N-terminal protein could be detected in caveolin-1 complexes, indicating that this portion of AC6 did not associate with plasma membrane caveolin complexes (Figure 6). However, virtually all the C1 protein and approximately half the C2 protein that was present in plasma membranes from COS-7 cells was detected in caveolin-1 immunoprecipitated complexes. These data support the idea that the C1, and to a lesser extent C2, form key interactions with caveolin-1 complexes.
Figure 6. The C1 and C2 domains are localized in Triton X-100 insoluble fractions and immunoprecipitate with caveolin-1.
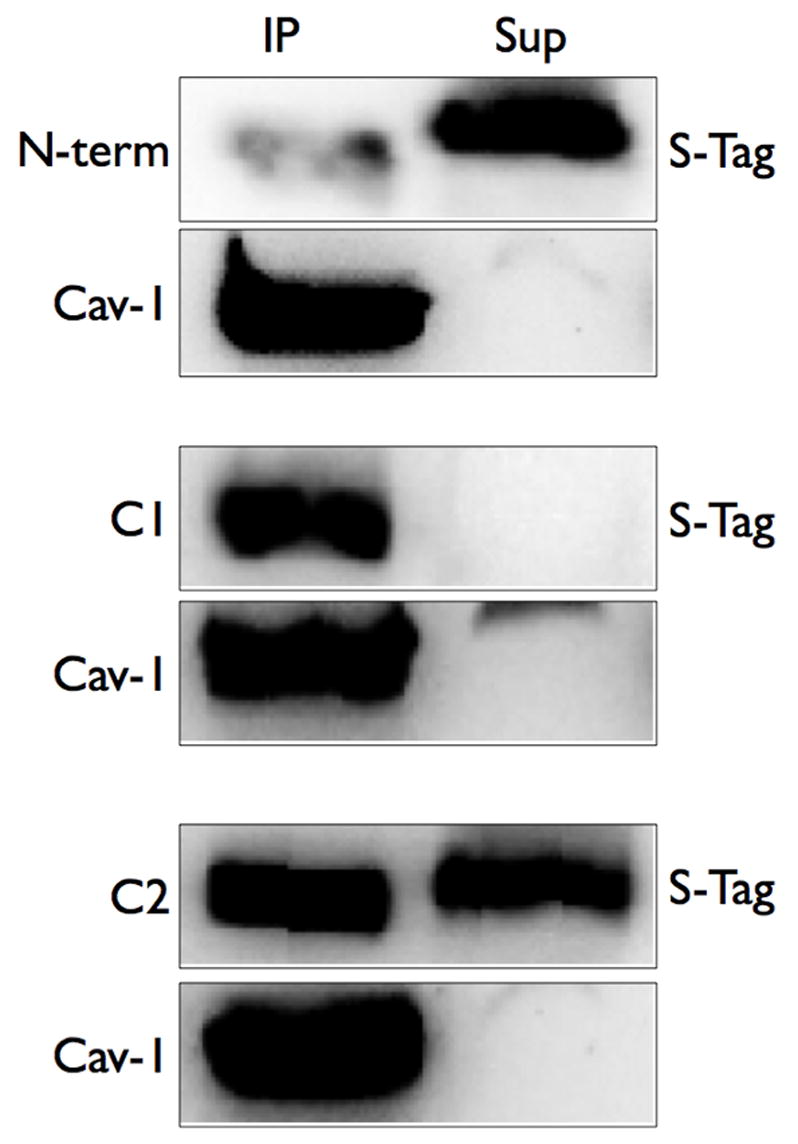
COS-7 cells were transfected with each intracellular domain construct. Cells were then separated into soluble and insoluble fractions in Triton-X 100 (left panels) or were lysed and immunoprecipitation of caveolin-1 was performed (right panels, see Methods). Soluble and insoluble fractions or caveolin-1 immunoprecipitates (IP) or supernatants (Sup) were separated by SDS-PAGE and analyzed by S-Tag detection (to detect AC6 domain proteins) or immunoblotting (to detect caveolin-1). Images shown are representative of 5 experiments.
To further dissect the regions of the C1 domain that may be responsible for the lipid raft localization that we observed, we constructed four individual proteins of roughly equal size that comprise the C1 domain of AC6 and fused each to GFP. These proteins are C1-306 (composed of amino acids 306–387 of AC6), C1-388 (composed of amino acids 388–490 of AC6), C1-491 (composed of amino acids 491–589 of AC6) and C1-590 (composed of amino acids 590–672 of AC6. These constructs are each represented schematically in Figure 3. We expressed each of these C1 domain fragments into COS-7 cells and probed for their localization in buoyant lipid raft fractions and their formation of immunprecipitable complexes with caveolin-1. We found that only C1-306 and C1-388 partitioned to buoyant fractions from sucrose gradient centrifugation (Figure 7, left panel). In addition, these same proteins were detected in caveolin-1 immunoprecipitates while C1-491 and C1-561 were not (Figure 7, right panel). Immunoprecipitation of each expressed C1 fragment was also performed, utilizing the antibodies to the fused GFP. These studies confirm that caveolin-1 is readily detectible in immunoprecipitated complexes with C1-306 and C1-388 and to a lesser extent C1-491, but not with C1-561. These data are consistent with the idea that elements of the N-terminal half of the C1 domain are responsible for lipid raft localization.
Figure 7. Segments in the N-terminal half of the C1 domain localize to lipid rafts and immunoprecipitate with caveolin-1.

COS-7 cells were transfected with one of four proteins corresponding to a segment of the AC6 C1 domain. A: Cells were then fractionated using a non-detergent method to isolate lipid rafts and caveolae, then fractions 2–10 were separated by SDS-PAGE and analyzed by immunoblot using either anti-caveolin-1 (top image) or anti-GFP. B: Caveolin-1 (Cav-1) or GFP was immunoprecipitated (IP, see Methods) and precipitates were immunoblotted (IB) for the indicated protein. Images shown are representative of 3–5 experiments.
Many proteins that localize in lipid rafts possess post-translational modifications, such as palmitoylation, that drive their association with these lipid domains. Examination of the AC6 amino acid sequence reveals a putative palmitoylation site in the carboxy terminus at cysteine 1145[29]. Thus, we expressed epitope-tagged full length human AC6 and three different C-terminally truncated AC6 proteins. AC6-1127 is a protein truncated at Q1127, a total of 41 amino acids short of the wild-type AC6 and devoid of the C1145 residue that is the sole putative palmitoylation site in the C-terminal region (see Figure 3). AC6-1144 is a protein truncated at G1144, thus possesses the C1145 residue but would not be palmitoylated due to the lack of a full palmitoylation motif (cys-aliphatic-aliphatic-any AA). AC6-1148 is a protein truncated at V1148 and containing the full palmitoylation motif. We expressed each of these truncated AC6 proteins and wild-type AC6 in COS-7 cells and performed both lipid raft fractionations and caveolin-1 immunoprecipitations to detect the localization of these recombinant proteins. Each of these truncated protein retains full catalytic activity, as evidenced by the increase forskolin-stimulated cAMP production following transfection of COS-7 cells with each construct (Figure 8c). Each of the truncated AC6 proteins localized to caveolin-rich, lipid raft fractions in a manner indistinguishable from full length AC6 (Figure 8a). Furthermore, immunoreactivity for each truncated protein was also readily detectible in caveolin-1 immunoprecipitates from COS-7 cells (Figure 8b). Thus, deletion of the putative palmitoylation site in the C-terminal tail of AC6 does not alter the characteristic lipid rat localization of this protein. These data re consistent with the idea that none of the final 41 amino acids in the C-terminal play a critical role in AC6 localization.
Figure 8. Truncation of the C-terminal 41 amino acids of AC6 does not alter localization in lipid rafts or cAMP production.

COS-7 cells were transfected with either full length AC6 (wtAC6) or three different C-terminally truncated versions of AC6 (AC6-1127, AC6-1144 and AC6-1148). A: Cells were fractionated using a non-detergent method to isolate lipid rafts and caveolae and the fractions were separated by SDS-PAGE and expressed AC6 proteins detected by immunoblot for their fused FLAG epitopes. B: Caveolin-1 was immunoprecipitated and precipitates (IP) and supernatants (Sup) were separated by SDS-PAGE and expressed AC6 proteins detected by immunoblot for their fused FLAG epitopes. Images shown are representative of 3–5 experiments (A and B). cAMP production was also measured in cells expressing wtAC6 or truncated versions. Cells were incubated with (control) or without 10 μM forskolin for 10 min and accumulated cAMP detected by EIA (see Methods). # denotes p > 0.05 by paired t-test as compared to control. * denotes p > 0.05 by paired t-test as compared to vector only.
DISCUSSION
There has been growing appreciation in recent years that signaling proteins form complexes with other key partners or can be compartmentized within the cell. Our lab and others have demonstrated that AC’s have discreet localization properties within the plasma membrane of mammalian cells and that this distinct localization can impart selective coupling between AC’s and specific GPCR, allow tight regulation of downstream effectors by cAMP, or establish highly efficient regulation of AC activity by other cellular signals [1, 15, 28, 30]. One well described AC isoform in regards to localization and function is AC6. This isoform is highly enriched in lipid rafts of all mammalian cells in which it is expressed and forms complexes with numerous other signaling molecules, including β-adrenergic receptors, caveolins and other AC’s [11, 12]. In this report, we describe data from our efforts to define how AC6 forms protein–lipid and protein–protein interactions that allow the enzyme to partake in higher-order signaling complexes and contribute to cAMP compartmentalization. We conclude that the cytosolic C1 and C2 domains play a central role but that these domains do not appear to rely upon known mechanisms for lipid raft localization. While much more information needs to be gathered to fully understand the mechanisms of AC localization in lipid rafts, these data are critical initial steps to unraveling the complex puzzle of GPCR-AC signaling.
Three different, but perhaps complimentary, mechanisms have emerged that can cause the localization of proteins in lipid rafts or caveolae. First, proteins can bind to caveolin oligomers that form on the inner leaflet of lipid rafts to drive formation of caveolae [31]. Endothelial nitric oxide synthase, insulin receptors, epidermal growth factor receptors are examples of proteins that depend upon caveolin binding for their distinct localization. Second, proteins can partition to lipid rafts via acylation, particularly palmitoylation or myristoylation of two or more closely spaced residues[32, 33]. Proteins such as caveolins, G proteins and certain GPCR’s depend upon such post-translational modification to achieve their specific localization [34, 35]. Finally, there are sequence-dependant features of some transmembrane proteins that drive interactions with lipid moieties to form “lipid shells” [36].
None of these mechanisms for localizing proteins to lipid rafts and caveolae appear sufficient to explain the distinct localization of AC6 to these lipid microdomains. As we report here, AC6 is localized to buoyant lipid raft fractions (as defined using sucrose density centrifugation) in both cells that express caveolins and cells that lack caveolin expression (either naturally or via knock-out, Figures 1 and 2). Thus, the caveolin binding hypothesis can’t explain AC6 localization. Crosthwaite et. al. recently reported that the chimeric substitution of the two 6 transmembrane spanning domains of AC5 (a lipid raft localized isoform) with those sequences from AC7 (a non-raft localized isoform) did not alter the characteristic localization in lipid rafts [26]. These data are inconsistent with the hypothesis transmembrane segments participate in lipid shell formation to drive lipid raft localization of AC’s. Instead, other data from Crossthwaite and coworkers implicate the intracellular segments of the enzyme as the elements driving lipid raft localization. Our data confirm and extend these findings. By using reductionist approaches (e.g. truncation, expression of individual intracellular domains) we show that the C1 and C2 domains, but not the C-terminal 44 amino acids of C2 containing a putative palmitoylation site, are critical for lipid raft localization of AC6.
Several caveats of the reductionist approach we have utilized are evident. First, the individually expressed cytosolic domains of AC6 do not have membrane targeting sequences or hydrophobic transmembrane domains. Thus, expression of these bait proteins yields a sizable proportion of protein that does not associate with membranes. This lack of targeting likely limits the protein’s ability to find natural partners or form avid lipid associations. However, the epitopes fused to each of these bait proteins allow sensitive detection via immunoblot and thereby allow interpretable results despite a low abundance of bait protein finding its target. Second, these individual proteins may not fully form their native tertiary structure since they lack the rest of the complementary portions contained in the full length AC6. This lack of native structure may limit their association with normal binding partners and reduce their native localization. However, our data suggest that an appreciable proportion of the C1 domain, and to a slightly lesser extent the C2 domain, do form associations that cause them to appear in lipid raft fractions (Figures 5 and 6). Furthermore, the N-terminal half of the C1 domain appears to contain sequences that confer this lipid raft localization, as evidenced by the partitioning of C1-306 And C1-388 into lipid rafts (Figure 7). One may be tempted to interpret this data as meaning several interactions are responsible for lipid raft association of AC6. However, more studies are needed to determine the mechanisms for the observed localization.
The approaches used to characterize lipid rafts and caveolae can be fraught with methodological pitfalls and carry high probability of experimental artifact. Cell fractionation and sucrose density centrifugation destroys the cell architecture and has potential for contamination of fractions with other cellular domains. Immunoprecipitation studies carry significant probability that proteins form complexes after extraction, yielding false positives, or that weaker protein-protein interactions are lost in the process, yielding false negatives. Light microscopy is limited in resolution and falls short of being capable of defining a 50–100 nm lipid raft. Electron microscopy depends upon reliable and specific antibodies and a high degree of epitope exposure in the protein of interest. Thus, no single method for defining lipid raft or caveolar localization of a protein of interest is sufficient to reach reliable conclusions. In the current studies, we performed immunoprecipitations, non-detergent cell fractionation with sucrose density centrifugation and detergent-based extraction of membranes in an effort to alleviate artifactual concerns. We also used fluorescent microscopy to confirm plasma membrane association of our expressed proteins. Nonetheless, defining how (and in some cases when) proteins associate with lipid rafts will require development of improved methods [32].
GPCR-AC signaling is beginning to be understood in terms of internal intermolecular associations, molecular dimerization and hetero-oligomerization with other signaling partners. These conformations and associations likely influence the formation of the AC catalytic core, regulate the trafficking of AC’s to the plasma membrane and dictate both the control of AC activity by various signals and the physiological impact of the cAMP generated by the enzyme. Complex formation of signaling molecules also allows rapid and specific signal transduction and processing. The present findings make inroads to understanding the mechanism(s) driving lipid raft localization of AC’s by focusing on smaller components of these large proteins that are responsible. It is likely that these same forces are at work targeting many other transmembrane signaling proteins, including receptors, cell adhesion molecules and others, to lipid rafts and caveolae. Thus, characterizing these mechanisms are key to understanding how signaling complexes form, how second messengers are compartmentized and how these aspects of cellular signaling might be altered in pathophysiology.
Acknowledgments
The authors thank Drs. Anjaparavanda P. Naren and Chunying Li for the generation of the N-terminal, C1 and C2 domain proteins. We are also grateful to Ms. Fengying Li for her technical assistance. This work was supported by a grant-in-aid from the American Heart Association, Southeast Affiliate (grant number 0555291B).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostrom RS, Insel PA. Br J Pharmacol. 2004;143(2):235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, Tang CM, Roth DM. Ann N Y Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 3.Foster LJ, De Hoog CL, Mann M. Proc Natl Acad Sci U S A. 2003;100(10):5813–5818. doi: 10.1073/pnas.0631608100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel HH, Murray F, Insel PA. Annu Rev Pharmacol Toxicol. 2008;48:359–391. doi: 10.1146/annurev.pharmtox.48.121506.124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlegel A, Volonté D, Engelman JA, Galbiati F, Mehta P, Zhang XL, Scherer PE, Lisanti MP. Cellular Signalling. 1998;10(7):457–463. doi: 10.1016/s0898-6568(98)00007-2. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Journal of Biological Chemistry. 1998;273(10):5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 7.Cooper DM, Crossthwaite AJ. Trends Pharmacol Sci. 2006;27(8):426–431. doi: 10.1016/j.tips.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Hanoune J, Defer N. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 9.Cooper DM. Biochem J. 2003;375(Pt 3):517–529. doi: 10.1042/BJ20031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Head BP, Patel HH, Roth DM, Lai NC, Niesman IR, Farquhar MG, Insel PA. J Biol Chem. 2005;280(35):31036–31044. doi: 10.1074/jbc.M502540200. [DOI] [PubMed] [Google Scholar]
- 11.Ostrom RS, Violin JD, Coleman S, Insel PA. Molecular Pharmacology. 2000;57(5):1075–1079. [PubMed] [Google Scholar]
- 12.Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. Mol Pharmacol. 2002;62(5):983–992. doi: 10.1124/mol.62.5.983. [DOI] [PubMed] [Google Scholar]
- 13.Ostrom RS, Naugle JE, Hase M, Gregorian C, Swaney JS, Insel PA, Brunton LL, Meszaros JG. J Biol Chem. 2003;278(27):24461–24468. doi: 10.1074/jbc.M212659200. [DOI] [PubMed] [Google Scholar]
- 14.Smith KE, Gu C, Fagan KA, Hu B, Cooper DM. J Biol Chem. 2002;277(8):6025–6031. doi: 10.1074/jbc.M109615200. [DOI] [PubMed] [Google Scholar]
- 15.Fagan KA, Smith KE, Cooper DM. Journal of Biological Chemistry. 2000;275(34):26530–26537. doi: 10.1074/jbc.M001369200. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg SF, Brunton LL. Annu Rev Pharmacol Toxicol. 2001;41:751–773. doi: 10.1146/annurev.pharmtox.41.1.751. [DOI] [PubMed] [Google Scholar]
- 17.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW. Science. 2001;293(5527):98–101. doi: 10.1126/science.293.5527.98. [DOI] [PubMed] [Google Scholar]
- 18.Ludwig MG, Seuwen K. J Recept Signal Transduct Res. 2002;22(1–4):79–110. doi: 10.1081/rrs-120014589. [DOI] [PubMed] [Google Scholar]
- 19.Tang WJ, Hurley JH. Mol Pharmacol. 1998;54(2):231–240. doi: 10.1124/mol.54.2.231. [DOI] [PubMed] [Google Scholar]
- 20.Tang WJ, Gilman AG. Science. 1995;268(5218):1769–1772. doi: 10.1126/science.7792604. [DOI] [PubMed] [Google Scholar]
- 21.Sunahara RK, Dessauer CW, Gilman AG. Annual Review of Pharmacology and Toxicology. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 22.Gu C, Cali JJ, Cooper DM. Eur J Biochem. 2002;269(2):413–421. doi: 10.1046/j.0014-2956.2001.02708.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen-Goodspeed M, Lukan AN, Dessauer CW. J Biol Chem. 2005;280(3):1808–1816. doi: 10.1074/jbc.M409172200. [DOI] [PubMed] [Google Scholar]
- 24.Baragli A, Grieco ML, Trieu P, Villeneuve LR, Hebert TE. Cell Signal. 2008;20(3):480–492. doi: 10.1016/j.cellsig.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 25.Ding Q, Gros R, Chorazyczewski J, Ferguson SS, Feldman RD. Mol Pharmacol. 2005;67(2):564–571. doi: 10.1124/mol.104.006817. [DOI] [PubMed] [Google Scholar]
- 26.Crossthwaite AJ, Seebacher T, Masada N, Ciruela A, Dufraux K, Schultz JE, Cooper DM. J Biol Chem. 2005;280(8):6380–6391. doi: 10.1074/jbc.M411987200. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Thangavel M, Sun SQ, Kaminsky J, Mahautmr P, Stitham J, Hwa J, Ostrom RS. Naunyn Schmiedebergs Arch Pharmacol. 2008;377(4–6):359–369. doi: 10.1007/s00210-007-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. J Biol Chem. 2001;276(45):42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- 29.Mumby SM. Curr Opin Cell Biol. 1997;9(2):148–154. doi: 10.1016/s0955-0674(97)80056-7. [DOI] [PubMed] [Google Scholar]
- 30.Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. Journal of General Physiology. 2000;116(2):147–161. doi: 10.1085/jgp.116.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Journal of Biological Chemistry. 1997;272(10):6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 32.Brown DA. Physiology (Bethesda) 2006;21:430–439. doi: 10.1152/physiol.00032.2006. [DOI] [PubMed] [Google Scholar]
- 33.Zacharias DA, Violin JD, Newton AC, Tsien RY. Science. 2002;296(5569):913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 34.Uittenbogaard A, Smart EJ. J Biol Chem. 2000;275(33):25595–25599. doi: 10.1074/jbc.M003401200. [DOI] [PubMed] [Google Scholar]
- 35.Resh MD. Sci STKE. 2006;2006(359):re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 36.Anderson RG, Jacobson K. Science. 2002;296(5574):1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]