

Nearly 1 in 5 people will experience a major depressive episode at some point in their lives.1 In this review, we discuss data describing how genes, psychosocial adversity in childhood, and ongoing or recent psychosocial stress may impact multiple neurobiological systems relevant to major depressive disorder. Major depressive disorder may be caused by the cumulative effects of these 3 factors on the brain.2
A major depressive episode is characterized by a low mood or an inability to experience pleasure (anhedonia), or both, for more than 2 weeks, combined with several cognitive and vegetative symptoms and the occurrence of distress or impairment.3 A diagnosis of major depressive disorder can be made if a person suffers at least 1 such episode (without ever experiencing mania). However, most people with major depressive disorder have multiple episodes. Importantly, several medical illnesses such as diabetes, heart disease, autoimmune disorders and pain are common comorbid diagnoses.4,5 The relation between major depressive disorder and these chronic and disabling conditions appears to be bidirectional because one may influence the prognosis of the other.
Investigations into the neurobiology of major depressive disorder have traditionally focused on the monoamine neurotransmitters serotonin and norepinephrine. The monoamine hypothesis initially posited that depressed individuals are likely to have low levels of these neurotransmitters because various antidepressant drugs acutely increase their levels.6 However, even though monoaminergic antidepressants are generally used for first-line treatment, they do not exert their clinical benefit immediately and for some people they do not provide any benefit at all. We review the neurobiological research that may help explain this.
A revised monoamine hypothesis
Studies on the pathophysiology of major depressive disorder tend to focus on people who are currently depressed. Although informative, data derived from such studies often do not allow for a distinction to be made between cause and effect. These studies also do not allow researchers to distinguish between core mechanisms responsible for the disease and epiphenomena. For example, the finding that the rate of serotonin synthesis may be low in depressed patients can be explained in multiple ways.7 A reduction in serotonin synthesis may result in depression, depression may result in a reduction in serotonin synthesis, or a third factor may be responsible for both lowering serotonin synthesis rates and triggering depression.
Experimental studies involving patients whose depression is currently in remission may help clarify the role of serotonin in major depressive disorder. Indeed, one of the few areas of investigation with studies including such patients is concerned with the effects of experimental serotonin manipulations on mood. In most of these studies, patients ingest a tryptophan-deficient amino acid mixture that transiently decreases serotonin levels in the brain, because serotonin is derived from tryptophan (Figure 1).8 Patients taking medications at the time of study may experience a brief relapse, especially if they are taking drugs that affect the serotonin system. Patients not taking any medications are also likely to experience a brief relapse of symptoms during tryptophan depletion, especially if their depression has been in remission for only a few months. This suggests that a lowering of serotonin levels may result in depression.

Figure 1: The serotonin synapse. Serotonin is synthesized from tryptophan by the enzyme tryptophan hydroxylase. Serotonin is then packaged into vesicles for release into the synaptic cleft, which occurs when there is sufficient stimulation of the neuron. Serotonin released from the serotonin neuron into the synaptic cleft has multiple actions. (1) Serotonin binds to its receptors on other neurons. Activation of postsynaptic receptors results in transduction of the signal that initially stimulated the serotonin neuron. (2) Serotonin also binds to presynaptic serotonin receptors on the neuron from which it was released, which provides feedback and regulates plasticity of the neuron. (3) Serotonin is taken up back into the presynaptic serotonin neuron by the serotonin transporter. Serotonin is then recycled for future release or broken down by monoamine oxidase and excreted in urine. Photo by: Lianne Friesen and Nicholas Woolridge
However, individuals without a personal or family history of major depressive disorder tend to not show any mood changes following tryptophan depletion,8 despite the fact that tryptophan depletion alters the activity of mood-regulating regions of the brain, such as the amygdala, in these individuals as it does in patients with major depressive disorder.9 Thus, lowering serotonin levels does not induce depression in all people. It is possible that a depressive episode alters the serotonin system such that a person becomes more vulnerable to the effects of future changes in serotonin levels. However, even in the absence of a personal history of major depressive disorder, people with a family history may report mood-worsening following tryptophan depletion.8 Clearly, there are several possible factors that contribute to a person's vulnerability to the effects of altered levels of serotonin on mood.10
Genes influencing serotonin metabolism moderate the impact of stress
Scientists have not identified a gene or a series of genes that cause depression. Rather, certain variations in genes, called polymorphisms, may increase risk for depression. Genes can predispose individuals to major depressive disorder in many ways. For example, genes help control the metabolism of neurotransmitters and their receptors, the numbers of particular types of neurons and their synaptic connections, the intracellular transduction of neuronal signals, and the speed with which all of these can change in response to environmental stressors.11 The serotonin transporter gene is the most studied in major depressive disorder (Figure 1).12 This gene is of interest because it contains a polymorphism that gives rise to 2 different alleles (long and short). People usually have 2 copies of each gene in their DNA; therefore, a person can be homozygous for the long allele, homozygous for the short allele or heterozygous (1 long and 1 short allele). The short allele slows down the synthesis of the serotonin transporter. This is thought to reduce the speed with which serotonin neurons can adapt to changes in their stimulation.13 Given that an acute stressor increases serotonin release, the polymorphism may influence a person's sensitivity to stress. Indeed, healthy people with the short allele display exaggerated amygdala activation when exposed to stress-evoking stimuli.14 These people may also have a greater likelihood of mood-worsening following tryptophan depletion.15
Stress is a common precipitating factor for depression.16,17 However, not everyone exposed to stress becomes depressed. Rather, stress interacts with a person's genetic makeup to influence his or her risk for developing major depressive disorder (Figure 2). A gene–environment interaction suggesting that carriers of the short allele of the serotonin transporter may be especially vulnerable to depression when under stress was first suggested in 2003 in a prospective study involving a birth cohort. In a group of 26-year-old men and women, having had a major depressive episode in the past year was best predicted by the combination of having the short allele of the serotonin transporter and having had multiple stressful life events in the past 5 years.18 The interactive effects of the serotonin transporter polymorphism and stress on a person's risk for major depressive disorder have been reported by various groups.19–22 Nevertheless, controversy remains. Not all studies have observed this gene–environment interaction. In a study including patients in remission from a major depressive episode, tryptophan depletion had less pronounced effects on mood in carriers of the short allele than in people homozygous for the long allele, not more as might have been expected.23 In any case, the impact of individual genes on the risk of major depressive disorder is thought to be small. The interactive effects of multiple genes and psychosocial stress on the risk of depression are only starting to be explored.24

Figure 2: Schematic representation of gene–environment interaction. Genetic polymorphisms may influence a person's risk for major depressive disorder in certain psychosocial environments. Genes may contribute to a person's risk for major depressive disorder through polymorphisms or variations in their DNA sequence that may influence the expression or activity of the gene product. A person may have 2 identical alleles of a gene (homozygous) or 2 different alleles (heterozygous). Although carrying a certain allele may in itself not affect a person's risk for developing major depressive disorder, several genetic polymorphisms have been shown to interact with the environment in terms of their effect on disease vulnerability. Please note that this figure is a schematic representation. Individual polymorphisms and individual studies may differ in terms of how a person's risk for major depressive disorder is affected by carrying 1 or 2 vulnerability alleles. Limitations of studies to date include the method used to assess disease vulnerability (e.g., some studies used a person's score on a depression questionnaire rather than a formal diagnosis of major depressive disorder as the outcome variable) and the retrospective nature of most studies' assessment of psychosocial stress exposure.5–7,9–13
Brain-derived neurotrophic factor
One polymorphism that may moderate the interactive effect of the serotonin transporter polymorphism and psychosocial stress is located in the gene that codes for brain-derived neurotrophic factor. This growth factor plays a major role in the birth, survival and maturation of brain cells during development. Brain-derived growth factor is important for cell growth and for allowing changes in the synapses between neurons (synaptic plasticity) throughout life. Brain-derived neurotrophic factor contributes to these processes primarily by activating DNA-binding factors that stimulate gene transcription. For example, in the raphe nuclei located in the brain stem, brain-derived neurotrophic factor stimulates transcription of genes involved in serotonin function, such as the serotonin transporter and tryptophan hydrolase (the serotonin synthesizing enzyme). In turn, activation of serotonin receptors by serotonin released from the raphe nuclei stimulates expression of the brain-derived neurotrophic factor gene. During brain development, this cyclic process promotes outgrowth, synapse formation and survival of serotonin neurons, and the eventual innervation of multiple brain regions. The ability of the serotonin system to adapt and change in response to various stimuli continues to be influenced by brain-derived neurotrophic factor throughout life.25
A common polymorphism in the gene that codes for brain-derived neurotrophic factor produces alleles called “Val” and “Met.” This polymorphism affects the intracellular transport and secretion of brain-derived neurotrophic factor.26 People with the Met allele have been found to have a relatively small hippocampus at birth and to display hippocampal hypoactivity in a resting state, hippocampal hyperactivation during learning, and relatively poor hippocampus-dependent memory function.27,28 This may contribute to a hippocampal hypersensitivity to stress. It could also explain why studies have found that having the Met allele, in addition to having the short allele of the serotonin transporter and psychosocial stress, increases vulnerability to depression more than having the short allele of the serotonin transporter and psychosocial stress alone.29 These 3 factors combined may increase depression vulnerability even if the stress took place in childhood.30,31
Further evidence for a role of brain-derived neurotrophic factor in the pathophysiology of major depressive disorder comes from postmortem studies, which have found low levels of brain-derived neurotrophic factor in the hippocampus and prefrontal cortex of symptomatic depressed patients.27,28 In addition, a recent review reported that serum levels of brain-derived neurotrophic factor in patients with major depressive disorder are abnormally low.32 In healthy people, serum levels of brain-derived neurotrophic factor correlate negatively with sensitivity to stress and positively with brain levels of N-acetyl-aspartate, a putative marker of neuronal integrity that can be measured by neuroimaging.33
Psychosocial adversity in childhood and the hypothalamic–pituitary–adrenal axis
Stressful events often do not occur at random. This may be especially true of childhood stressors. A child's environment is influenced by his or her own behaviour as well as by the behaviour of his or her parents. The same genes influence the behaviour of both generations (provided the child grows up with his or her biological parents). In studies of the impact of psychosocial adversity during childhood on the risk of adult depression, it is often difficult to separate the effects of genes from those of the environment.2,11
Experimental studies involving nonhuman primates and other mammals can be more informative. For example, monkeys temporarily reared by peers rather than by their mothers develop exaggerated stress responses. These responses have been associated with abnormalities in serotonin activity as well as in the hypothalamic–pituitary–adrenal axis (Figure 3).34 Data from studies in rats also suggest that childhood experiences can alter the reactivity of the hypothalamic–pituitary–adrenal axis and show that these alterations are at least partially mediated by modifications in genes that do not involve any actual changes in the underlying DNA (epigenetic changes).35 Specifically, the hippocampi of adult rats deprived of maternal care during infancy display these epigenetic modifications in the gene for the glucocorticoid receptor, which helps mediate the effects of cortisol released from the adrenal glands in response to stress (Figure 3). The process that leads to epigenetic changes, which affect gene transcription, is known to be influenced by serotonin.36 The resulting changes in glucocorticoid receptor expression in the hippocampus increase the reactivity of the hypothalamic– pituitary–adrenal axis.37 This epigenetic process may even occur in utero38 and is thought to be extremely long lasting. This might help explain why people with major depressive disorder often show abnormalities in this neuroendocrine system.39
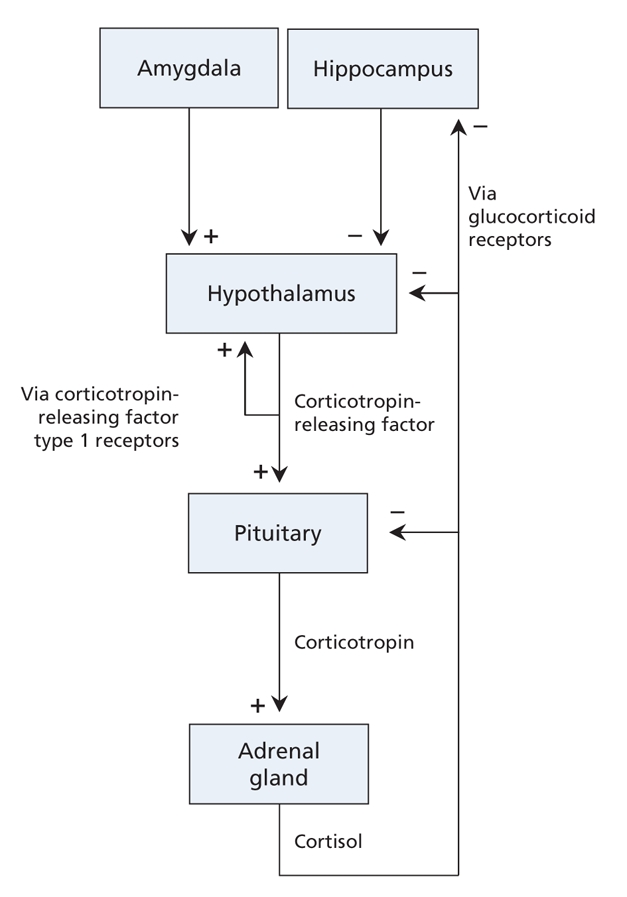
Figure 3: The hypothalamic–pituitary–adrenal axis. This system is activated by stress directly at the level of the hypothalamus or indirectly at the level of the amygdala. The hypothalamus produces and releases corticotropin-releasing factor. Local stimulation of corticotropin-releasing factor type 1 receptors results in additional release of corticotropin-releasing factor. This creates a feed-forward loop, which facilitates a rapid response to the stressor. In the pituitary, stimulation of corticotropin-releasing factor type 1 receptors results in release of corticotropin (also known as adrenocorticotropic hormone). The adrenal glands are stimulated by corticotropin to produce the stress hormone cortisol, which affects many organs, including the brain. The hippocampus is an important target of cortisol. Local activation of glucocorticoid receptors helps the hippocampus control the hypothalamic–pituitary–adrenal axis. Glucocorticoid receptors are also found in the hypothalamus and pituitary. Chronic stress increases the level of corticotropin-releasing factor and cortisol and decreases expression of corticotropin-releasing factor type 1 receptors and glucocorticoid receptors. Similar changes have been found in some patients with major depressive disorder. Stress-and depression-associated changes at the level of the hippocampus in particular are thought to underlie the structural changes seen in this brain region, which in turn may contribute to chronic disinhibition of the hypothalamic– pituitary– adrenal axis.
The role of corticotropin-releasing factor in determining sensitivity to stress
Stress may activate the hypothalamus and therefore may activate the hypothalamic–pituitary–adrenal axis directly, by stimulating local synthesis and release of corticotropin-releasing factor (Figure 3).37 Stress may also activate this axis indirectly by releasing corticotropin-releasing factor from neurons in other regions of the brain, including the amygdala.40 These neurons may also contribute to activation of the serotonin and norepinephrine systems.41,42 Reciprocal connections between the norepinephrine system and the hypothalamus create a feed-forward cascade in which stress progressively activates corticotropin-releasing factor and norepinephrine signalling. Activation of this system is thought to increase vigilance and fear.43,44
The combined dysregulation of the hypothalamic and extrahypothalamic corticotropin-releasing factor systems may help explain why patients with major depressive disorder often have inappropriately high levels of corticotropin-releasing factor and elevated levels of norepinephrine in their blood plasma and cerebrospinal fluid, and why they display faulty processing of environmental threats and exaggerated stress reactions.39 Childhood adversity might also contribute to the abnormalities seen in these systems.43 A recent study suggests that the impact of childhood abuse on a person's vulnerability for depression may be moderated by polymorphisms in the corticotropin-releasing factor type 1 receptor gene.45 Evidence for gene–stress interactions affecting the risk of major depressive disorder can be found across neurotransmitter systems (Figure 2).
Stress-induced changes in the dopamine system
Persistent changes in the body's stress response during infancy and childhood are thought to increase responses to even relatively low-threat negative events experienced later in life. Sensitization to negative events may help explain why recurrent depressive episodes are more likely than the first episode to occur independently of stress.46 A complex relation between serotonin dysfunction, hypersensitivity to stress, and vulnerability to developing major depressive disorder has been suggested.47
In addition, dopamine is increasingly thought to play an important role in the pathophysiology of major depressive disorder. Environmental threats perceived by the amygdala increase the levels of dopamine in the prefrontal cortex and the ventral striatum.48 Local inhibitory feedback ensures a return to homeostasis. However, a severe stressor may disrupt this feedback system by altering striatal levels of brain-derived neurotrophic factor.
Abnormal feedback in the striatal dopamine system may help explain why depressed patients often attribute inappropriate salience to even mildly negative stimuli. Further, changes in the striatal dopamine system are thought to underlie the anhedonia reported by many patients.48,49 Evidence for dopamine abnormalities in major depressive disorder is mixed but does exist.50 A polymorphism in the dopamine type 2 receptor gene was recently found to influence the effect of past stressful life events on current mood.51 The genetic makeup of the dopamine system may help influence vulnerability to major depressive disorder via interaction with the environment (Figure 2).
Structural changes in the depressed brain
Major depressive disorder has traditionally been viewed as an illness in which depressive episodes are followed by periods of euthymic mood. Nonetheless, patients showing clinical signs of remission may present with persistent neurobiological abnormalities. These abnormalities may worsen over time, and some patients may become chronically depressed. Similar to multiple sclerosis, there may be variability in the course of the disease over time. In multiple sclerosis, there are 2 common patterns of disease progression: relapsing–remitting and primary–progressive. The existing data for major depressive disorder suggest that although many patients may have a relapsing–remitting variant of the disease, some may have a primary–progressive variant.
Most relevant to this idea are structural neuroimaging studies which indicate that individuals with recurrent major depressive episodes may have relatively small hippocampi even during periods of clinical remission. Recurring or enduring illness and lack of antidepressant treatment are thought to contribute to progressive volume reductions of the hippocampus, which in turn may explain the memory problems of some patients, as well as several other symptoms of the disorder.52–54 Patients may also present with volumetric abnormalities in other subcortical brain regions, including the amygdala and ventral striatum, and in cortical regions, including the anterior cingulate cortex, orbitofrontal cortex and prefrontal cortex (Figure 4).53–55 These too may persist during remission and might help explain why patients in remission continue to overreact to threatening stimuli.56 This cognitive reactivity contributes to the risk of future relapse.57

Figure 4: Structural and functional brain abnormalities in patients with major depressive disorder, and the site of action of novel neurostimulation techniques with antidepressant potential. Structural abnormalities in the brains of patients with major depressive disorder have been observed in the cortical and subcortical regions. The anterior cingulate cortex, especially the subgenual cingulate, may show volume reduction. This has also been observed in other subregions of the prefrontal cortex as well as in the orbitofrontal cortex. Subcortical regions in which volume reduction has been observed include the amygdala, hippocampus and ventral striatum. (A) Transcranial magnetic stimulation of the dorsolateral prefrontal cortex and (B) deep-brain stimulation of the subgenual cingulate have been shown to have antidepressant effects in some patients. The rationale for deep-brain stimulation of the subgenual cingulate or the ventral striatum is largely based on neuroimaging findings of functional dysregulation in this region. (C) Vagus nerve stimulation might have antidepressant properties via its effects on the locus coeruleus, an area in the brain stem from which norepinephrine neurons originate. Image by: Lianne Friesen and Nicholas Woolridge
Neurobiological abnormalities underlying brain atrophy in major depressive disorder
Structural brain changes in patients with major depressive disorder have been attributed to abnormal function of the hypothalamic–pituitary–adrenal axis. Chronic hypercortisolemia results in downregulation of glucocorticoid receptors in the hippocampus (Figure 3). Subsequent reductions in the transcription of brain-derived neurotrophic factor and other target genes may contribute to hippocampal atrophy.2,27 This may further compromise the function of the hypothalamic–pituitary–adrenal axis.58 Similar cortisol-mediated effects may be responsible for structural changes in the amygdala, prefrontal cortex and other brain regions. All or some of the structural changes may contribute to the risk for relapse following a period of apparent homeostasis.59,60
The growth and survival of brain cells also involves multiple actions of glutamate, an amino acid neurotransmitter.2 Normal glutamate signalling includes activation of multiple types of receptors.61 Acute stress moderately increases synaptic glutamate neurotransmission in, for example, the hippocampus and amygdala, thereby increasing the levels of brain-derived neurotrophic factor and stimulating neuroplasticity. Termination of the stressor usually results in a return to the steady state. In times of chronic stress, however, excessive glutamate levels may lead to activation of N-methyl-D-aspartic acid type glutamate receptors outside the synapse. Hyperactivation of these receptors, located on both neurons and glial cells, increases intracellular calcium to a level that decreases rather than increases levels of brain-derived neurotrophic factor, induces atrophy and causes cell death.61 The degeneration of glial cells, which normally help avoid the toxic buildup of glutamate released by neurons, may accelerate this process. Continued disruptions in the glutamate system may ultimately result in hypoactivity of cortical brain regions. Patients with depression may have reduced levels of glutamate and associated proteins in the prefrontal cortex, as indicated by neuroimaging studies.61,62 This provides evidence that glutamate toxicity plays a role in the pathophysiology of major depressive disorder.
Abnormal brain activation patterns may help explain mood lability
Structural brain abnormalities in patients with major depressive disorder have been linked to changes in metabolic activity (Figure 4).2 These functional abnormalities have been directly associated with disease severity.63 Overall, neuroimaging studies indicate that major depressive episodes may be characterized by multiple abnormalities in the interconnectivity of various subcortical (especially limbic) and cortical brain regions.64 A relative lack of cortical regulation of the limbic system in the face of psychosocial adversity may help explain the stress sensitivity, emotional lability, irritability and suicidality often seen in people with depression.42,65 Various brain regions continue to show functional abnormalities upon clinical remission.63 Inducing a brief depressive relapse by lowering serotonin levels (via tryptophan depletion) or by lowering norepinephrine and dopamine levels (via administration of the drug α-methyl-para-tyrosine) activates both overlapping and distinct brain regions.66,67 Thus, different neurotransmitter systems may contribute in different ways to the cortical regulation of mood.
Treatment strategies
Antidepressant medications
It is clear that even a revised monoamine hypothesis does not do justice to the multitude of interconnected systems involved in the pathophysiology of major depressive disorder. This explains why treatment with existing antidepressant drugs, most of which target monoamines, frequently does not lead to clinical remission.68 The time lag between the start of and response to antidepressants in patients whose symptoms improve can be explained by the effects of these drugs on brain-derived neurotrophic factor and other growth-regulating systems.25,69 The effects of these drugs on neuroplasticity are particularly relevant given that treatment may reverse or even prevent structural brain abnormalities. Among the new drugs currently under investigation are those that target the corticotropin-releasing hormone, dopamine and glutamate systems.70 However, a recent study found no significant antidepressive effect of a drug with selective action on the corticotropin-releasing factor type 1 receptor.71 In contrast, in a study that used the anesthetic ketamine, which has actions on both dopamine and glutamate, a single intravenous administration of a subanesthetic dose was found to have robust and rapid antidepressant effects even in patients whose depression is considered resistant to conventional drug treatment strategies.72 However, most patients experienced a relapse within 1 week. Other glutamate-modulating agents currently under investigation for treatment of major depressive disorder include memantine and riluzole, and there are others in development.61,73,74 Finally, there has been a recent interest in drugs targeting other neurotransmitters such as gamma-aminobutyric acid, melatonin and substance P.70
Studies are underway to advance personalized medicine based on pharmacogenetics. A recent study found no evidence for an effect of the polymorphism in the serotonin transporter gene on the clinical response to citalopram (a selective serotonin reuptake inhibitor). Yet, patients with the short allele reported more adverse outcomes.75 A polymorphism in the gene for the serotonin type 2A receptor, however, has been found to influence the outcome of treatment with citalopram.76
Other polymorphisms with possible effects on patients' responses to antidepressant drugs include those resulting from allelic variations in other genes with roles in the serotonin system. Genes involved in the function of the hypothalamic– pituitary–adrenal axis and the corticotropin-releasing factor and norepinephrine systems have also been implicated.77 In addition, polymorphisms in genes for certain liver enzymes involved in drug metabolism may influence plasma drug levels and may therefore impact efficacy.77 A screening kit to identify how a person will process certain medicines based on his or her genetic makeup and thus help guide doctors to prescribe effective and tolerable medications has recently been developed but is not currently commercially available in Canada.
Nonpharmacologic approaches
The most traditional approach for treating major depressive disorder by nonpharmacologic means is psychotherapy. This approach may be especially helpful for patients with a history of childhood adversity or recent stress.78 Another approach that has been available for decades is electroshock or electroconvulsive therapy; however, its use is limited by its invasive nature, which includes the requirement of general anesthesia and the risk of retrograde amnesia, which may be irreversible in some patients.79
A small number of other neurostimulation techniques have recently been developed based on current insights into the neurobiology of major depressive disorder. These include electrical stimulation of the vagus nerve, transcranial magnetic stimulation of the prefrontal cortex, and electrical stimulation of the subgenual cingulate or the ventral striatum (Figure 4).68 The development of transcranial magnetic stimulation has been guided by findings of low activity in the prefrontal cortex of patients with depression (Figure 4). The rationale for deep-brain stimulation procedure is based on findings of functional impairments in the subgenual cingulate and ventral striatum and abnormalities in corticolimbic interconnectivity.64
Deep brain stimulation is still under experimental investigation. Transcranial magnetic stimulation and vagus nerve stimulation have been approved in Canada for treatment of major depressive disorder. However, because of their relatively invasive nature, techniques such as electroconvulsive therapy are currently only indicated for patients whose depression is resistant to conventional treatments.
Future directions
Major depressive disorder is thought to result from the complex interplay of multiple inherited genetic factors and subsequent exposure to a wide range of environmental variables throughout life. The exact roles of the monoamine and other neurotransmitter systems as well as their extracellular, intracellular, local and regional targets continue to be defined. The reciprocal connections between various brain regions and the neurochemical messengers involved in these connections are being increasingly studied from a systems perspective. Furthermore, researchers are increasingly considering the impact of an individual's psychosocial environment on brain chemistry, activity and anatomy, and vice versa. The impact of early childhood events in particular may have long-lasting effects,80 especially if epigenetic changes are involved.28,35 Clearly, researchers as well as clinicians must pay careful attention to the possible contribution of childhood adversity to adulthood psychopathology, for example, by incorporating family history and childhood background into diagnostic interviews with patients.
This review focused mainly on psychosocial adversity as an environmental risk factor, which largely reflects the current interest of this field. Much remains to be explored in terms of how genes interact with other environmental variables to influence the risk of major depressive disorder. For example, we have yet to discover why some people become depressed when they develop inflammatory disease or spend a winter at higher latitude, while others do not. The genetic factors influencing this differential susceptibility are unknown. Major depressive disorder is likely to have a large number of causes, both genetic and environmental. We have attempted to provide a framework for a complex disease that requires a multifaceted approach in research, diagnosis and treatment.
Key points.
Major depressive disorder is caused by the cumulative impact of genetics, adverse events in childhood and ongoing or recent stress.
Gene–environment interactions seem to predict a person's risk for major depressive disorder better than genes or environment alone.
Structural and functional brain abnormalities in patients with major depressive disorder may be associated with low levels of brain-derived neurotrophic factor, abnormal function of the hypothalamic–pituitary–adrenal axis and glutamate-mediated toxicity.
These abnormalities are thought to contribute to recurrent episodes of major depressive disorder and chronic illness.
Existing options for antidepressant treatment are limited by their delayed onset of action, lack of efficacy and adverse outcomes.
Future developments include the advancement of personalized medicine by means of genotyping for interindividual variability in drug action and metabolism.
Supplementary Material
Acknowledgments
We thank Heidi L. Fitterling and Katherine A. Collins for their feedback on an early draft of this manuscript.
Footnotes
A list of terms used in this article is available at www.cmaj.ca/cgi/content/full/180/3/305/DC1
Contributors: All of the authors contributed substantially to the content and revision of this article and approved the version submitted for publication.
Competing interests: Sanjay Mathew and Dennis Charney have been named as inventors on a use-patent of ketamine for the treatment of depression. Dennis Charney has received consulting fees from Unilever UK Central Resources Limited. Sanjay Mathew has received grant support from Alexza Pharmaceuticals, GlaxoSmithKline, Novartis and NARSAD, and he has received consulting or lecture fees from AstraZeneca and Jazz Pharmaceuticals. None declared for Marije aan het Rot.
Correspondence to: Dr. Marije aan het Rot, Mount Sinai School of Medicine, Department of Psychiatry, One Gustave L. Levy Place, Box 1217, New York NY, USA; fax 212 241-7973; Marije.aanhetRot@MSSM.edu
REFERENCES
- 1.Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003;289:3095-105. [DOI] [PubMed]
- 2. Charney DS, Manji HK. Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE 2004:re5. [DOI] [PubMed]
- 3.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington (DC): The Association; 1994.
- 4.Demyttenaere K, Bonnewyn A, Bruffaerts R, et al. Comorbid painful physical symptoms and depression: prevalence, work loss, and help seeking. J Affect Disord 2006;92:185-93. [DOI] [PubMed]
- 5.Barnes DE, Alexopoulos GS, Lopez OL, et al. Depressive symptoms, vascular disease, and mild cognitive impairment: findings from the Cardiovascular Health Study. Arch Gen Psychiatry 2006;63:273-9. [DOI] [PubMed]
- 6.Hirschfeld RMA. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry 2000;61(Suppl 6):4-6. [PubMed]
- 7.Rosa-Neto P, Diksic M, Okazawa H, et al. Measurement of brain regional alpha-[11C]methyl-L-tryptophan trapping as a measure of serotonin synthesis in medication-free patients with major depression. Arch Gen Psychiatry 2004;61:556-63. [DOI] [PubMed]
- 8.Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry 2007;12:331-59. [DOI] [PubMed]
- 9.Cools R, Calder AJ, Lawrence AD, et al. Individual differences in threat sensitivity predict serotonergic modulation of amygdala response to fearful faces. Psychopharmacology (Berl) 2005;180:670-9. [DOI] [PubMed]
- 10.Jans LAW, Riedel WJ, Markus CR, et al. Serotonergic vulnerability and depression: assumptions, experimental evidence and implications. Mol Psychiatry 2006;12:522-43. [DOI] [PubMed]
- 11.Wurtman RJ. Genes, stress, and depression. Metabolism 2005;54(Suppl 1):16-9. [DOI] [PubMed]
- 12.Levinson DF. The genetics of depression: a review. Biol Psychiatry 2006;60:84-92. [DOI] [PubMed]
- 13.Lesch KP, Bengel D, Heils A, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996; 274: 1527-31. [DOI] [PubMed]
- 14.Munafo MR, Brown SM, Hariri AR. Serotonin transporter (5-HTTLPR) genotype and amygdala activation: a meta-analysis. Biol Psychiatry 2008;63:852-7. [DOI] [PMC free article] [PubMed]
- 15.Neumeister A, Konstantinidis A, Stastny J, et al. Association between serotonin transporter gene promoter polymorphism (5-HTTLPR) and behavioral responses to tryptophan depletion in healthy women with and without family history of depression. Arch Gen Psychiatry 2002;59:613-20. [DOI] [PubMed]
- 16.Kendler KS, Gardner CO, Prescott CA. Toward a comprehensive developmental model for major depression in women. Am J Psychiatry 2002;159:1133-45. [DOI] [PubMed]
- 17.Kendler KS, Gardner CO, Prescott CA. Toward a comprehensive developmental model for major depression in men. Am J Psychiatry 2006;163:115-24. [DOI] [PubMed]
- 18.Caspi A, Sugden K, Moffitt TE, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003;301:386-9. [DOI] [PubMed]
- 19.Kendler KS, Kuhn JW, Vittum J, et al. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry 2005;62:529-35. [DOI] [PubMed]
- 20.Zalsman G, Huang Y-y, Oquendo MA, et al. Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. Am J Psychiatry 2006;163:1588-93. [DOI] [PubMed]
- 21.Cervilla JA, Molina E, Rivera M, et al. The risk for depression conferred by stressful life events is modified by variation at the serotonin transporter 5HTTLPR genotype: evidence from the Spanish PREDICT-Gene cohort. Mol Psychiatry 2007; 12:748-55. [DOI] [PubMed]
- 22.Kilpatrick DG, Koenen KC, Ruggiero KJ, et al. The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry 2007;164:1693-9. [DOI] [PubMed]
- 23.Neumeister A, Hu X-Z, Luckenbaugh DA, et al. Differential effects of 5-HTTLPR genotypes on the behavioral and neural responses to tryptophan depletion in patients with major depression and controls. Arch Gen Psychiatry 2006;63:978-86. [DOI] [PubMed]
- 24.Cicchetti D, Rogosch FA, Sturge-Apple ML. Interactions of child maltreatment and serotonin transporter and monoamine oxidase A polymorphisms: depressive symptomatology among adolescents from low socioeconomic status backgrounds. Dev Psychopathol 2007;19:1161-80. [DOI] [PubMed]
- 25.Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology 2008;33:73-83. [DOI] [PubMed]
- 26.Bath KG, Lee FS. Variant BDNF (Val66Met) impact on brain structure and function. Cogn Affect Behav Neurosci 2006;6:79-85. [DOI] [PubMed]
- 27.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry 2006;59:1116-27. [DOI] [PubMed]
- 28.Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nat Neurosci 2007;10:1089-93. [DOI] [PubMed]
- 29.Kim J-M, Stewart R, Kim S-W, et al. Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biol Psychiatry 2007;62:423-8. [DOI] [PubMed]
- 30.Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry 2006;59:673-80. [DOI] [PubMed]
- 31.Wichers M, Kenis G, Jacobs N, et al. The BDNF Val(66)Met x 5-HTTLPR x child adversity interaction and depressive symptoms: An attempt at replication. Am J Med Genet B Neuropsychiatr Genet 2008;147B:120-3. [DOI] [PubMed]
- 32.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry 2008;64: 527-32. [DOI] [PMC free article] [PubMed]
- 33.Lang UE, Hellweg R, Seifert F, et al. Correlation between serum brain-derived neurotrophic factor level and an in vivo marker of cortical integrity. Biol Psychiatry 2007;62:530-5. [DOI] [PubMed]
- 34.Suomi SJ. Risk, resilience, and gene x environment interactions in rhesus monkeys. Ann N Y Acad Sci 2006;1094:52-62. [DOI] [PubMed]
- 35.Tsankova N, Renthal W, Kumar A, et al. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007;8:355-67. [DOI] [PubMed]
- 36.Szyf M, McGowan P, Meaney MJ. The social environment and the epigenome. Environ Mol Mutagen 2008;49:46-60. [DOI] [PubMed]
- 37.de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci 2005;6:463-75. [DOI] [PubMed]
- 38.Abe H, Hidaka N, Kawagoe C, et al. Prenatal psychological stress causes higher emotionality, depression-like behavior, and elevated activity in the hypothalamo–pituitary–adrenal axis. Neurosci Res 2007;59:145-51. [DOI] [PubMed]
- 39.Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci U S A 2000; 97:325-30. [DOI] [PMC free article] [PubMed]
- 40.Makino S, Hashimoto K, Gold PW. Multiple feedback mechanisms activating corticotropin-releasing hormone system in the brain during stress. Pharmacol Biochem Behav 2002;73:147-58. [DOI] [PubMed]
- 41.Gold PW, Chrousos GP. The endocrinology of melancholic and atypical depression: relation to neurocircuitry and somatic consequences. Proc Assoc Am Physicians 1999;111:22-34. [DOI] [PubMed]
- 42.Pine DS. Developmental psychobiology and response to threats: relevance to trauma in children and adolescents. Biol Psychiatry 2003;53:796-808. [DOI] [PubMed]
- 43.Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology 2004;29:641-8. [DOI] [PubMed]
- 44.Dunn AJ, Swiergiel AH, Palamarchouk V. Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Ann N Y Acad Sci 2004; 1018:25-34. [DOI] [PubMed]
- 45.Bradley RG, Binder EB, Epstein MP, et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry 2008;65:190-200. [DOI] [PMC free article] [PubMed]
- 46.Frank E, Thase ME. Natural history and preventative treatment of recurrent mood disorders. Annu Rev Med 1999;50:453-68. [DOI] [PubMed]
- 47.Firk C, Markus CR. Review: Serotonin by stress interaction: a susceptibility factor for the development of depression? J Psychopharmacol 2007;21:538-44. [DOI] [PubMed]
- 48.Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 2006;59:1151-9. [DOI] [PubMed]
- 49.McClung CA, Nestler EJ. Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology 2008;33:3-17. [DOI] [PubMed]
- 50.Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry 2007;64:327-37. [DOI] [PubMed]
- 51.Elovainio M, Jokela M, Kivimaki M, et al. Genetic variants in the DRD2 gene moderate the relationship between stressful life events and depressive symptoms in adults: cardiovascular risk in young Finns study. Psychosom Med 2007;69:391-5. [DOI] [PubMed]
- 52.Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8. [DOI] [PubMed]
- 53.Campbell S, MacQueen G. An update on regional brain volume differences associated with mood disorders. Curr Opin Psychiatry 2006;19:25-33. [DOI] [PubMed]
- 54.Konarski JZ, McIntyre RS, Kennedy SH, et al. Volumetric neuroimaging investigations in mood disorders: bipolar disorder versus major depressive disorder. Bipolar Disord 2008;10:1-37. [DOI] [PubMed]
- 55.Hajek T, Kozeny J, Kopecek M, et al. Reduced subgenual cingulate volumes in mood disorders: a meta-analysis. J Psychiatry Neurosci 2008;33:91-9. [PMC free article] [PubMed]
- 56.Merens W, Booij L, Van Der Does AJ. Residual cognitive impairments in remitted depressed patients. Depress Anxiety. 2007. [DOI] [PubMed]
- 57.Segal ZV, Kennedy S, Gemar M, et al. Cognitive reactivity to sad mood provocation and the prediction of depressive relapse. Arch Gen Psychiatry 2006;63:749-55. [DOI] [PubMed]
- 58.Duman RS. Depression: A case of neuronal life and death? Biol Psychiatry 2004;56:140-5. [DOI] [PubMed]
- 59.Bouhuys AL, Bos EH, Geerts E, et al. The association between levels of cortisol secretion and fear perception in patients with remitted depression predicts recurrence. J Nerv Ment Dis 2006;194:478-84. [DOI] [PubMed]
- 60.Brown ES, Woolston DJ, Frol AB. Amygdala volume in patients receiving chronic corticosteroid therapy. Biol Psychiatry 2008;63:705-9. [DOI] [PMC free article] [PubMed]
- 61. Sanacora G, Zarate CA, Krystal JH, et al. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 2008;7:426-37. [DOI] [PMC free article] [PubMed]
- 62.Hasler G, van der Veen JW, Tumonis T, et al. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch Gen Psychiatry 2007;64:193-200. [DOI] [PubMed]
- 63.Hasler G, Drevets WC, Manji HK, et al. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004;29:1765-81. [DOI] [PubMed]
- 64.Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci 2007;10:1116-24. [DOI] [PMC free article] [PubMed]
- 65.Anand A, Li Y, Wang Y, et al. Activity and connectivity of brain mood regulating circuit in depression: a functional magnetic resonance study. Biol Psychiatry 2005;57:1079-88. [DOI] [PubMed]
- 66.Bremner JD, Vythilingam M, Ng CK, et al. Regional brain metabolic correlates of alpha-methylparatyrosine-induced depressive symptoms: implications for the neural circuitry of depression. JAMA 2003;289:3125-34. [DOI] [PMC free article] [PubMed]
- 67.Neumeister A, Nugent AC, Waldeck T, et al. Neural and behavioral responses to tryptophan depletion in unmedicated patients with remitted major depressive disorder and controls. Arch Gen Psychiatry 2004;61:765-73. [DOI] [PubMed]
- 68.Nemeroff CB. Prevalence and management of treatment-resistant depression. J Clin Psychiatry 2007;68(Suppl 8):17-25. [PubMed]
- 69.Penttila J, Kajander J, Aalto S, et al. Effects of fluoxetine on dopamine D2 receptors in the human brain: a positron emission tomography study with [11C]raclopride. Int J Neuropsychopharmacol 2004;7:431-9. [DOI] [PubMed]
- 70.Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology 2008;33:2080-92. [DOI] [PubMed]
- 71. Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry 2008;165:617-20. [DOI] [PubMed]
- 72.Zarate CA Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006;63:856-64. [DOI] [PubMed]
- 73.Muhonen LH, Lonnqvist J, Juva K, et al. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry 2008;69:392-9. [DOI] [PubMed]
- 74.Zarate CA Jr, Payne JL, Quiroz J, et al. An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 2004;161:171-4. [DOI] [PubMed]
- 75.Hu XZ, Rush AJ, Charney D, et al. Association between a functional serotonin transporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch Gen Psychiatry 2007;64:783-92. [DOI] [PubMed]
- 76.McMahon FJ, Buervenich S, Charney D, et al. Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet 2006;78:804-14. [DOI] [PMC free article] [PubMed]
- 77.Binder EB, Holsboer F. Pharmacogenomics and antidepressant drugs. Ann Med 2006;38:82-94. [DOI] [PubMed]
- 78.Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci USA 2003;100:14293-6. [DOI] [PMC free article] [PubMed]
- 79.Sackeim HA, Prudic J, Fuller R, et al. The cognitive effects of electroconvulsive therapy in community settings. Neuropsychopharmacology 2006;32:244-54. [DOI] [PubMed]
- 80.Post RM. Role of BDNF in bipolar and unipolar disorder: clinical and theoretical implications. J Psychiatr Res 2007;41:979-90. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.